A Novel Combination Cancer Therapy with Iron Chelator Targeting Cancer Stem Cells via Suppressing Stemness


 ,
,  , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
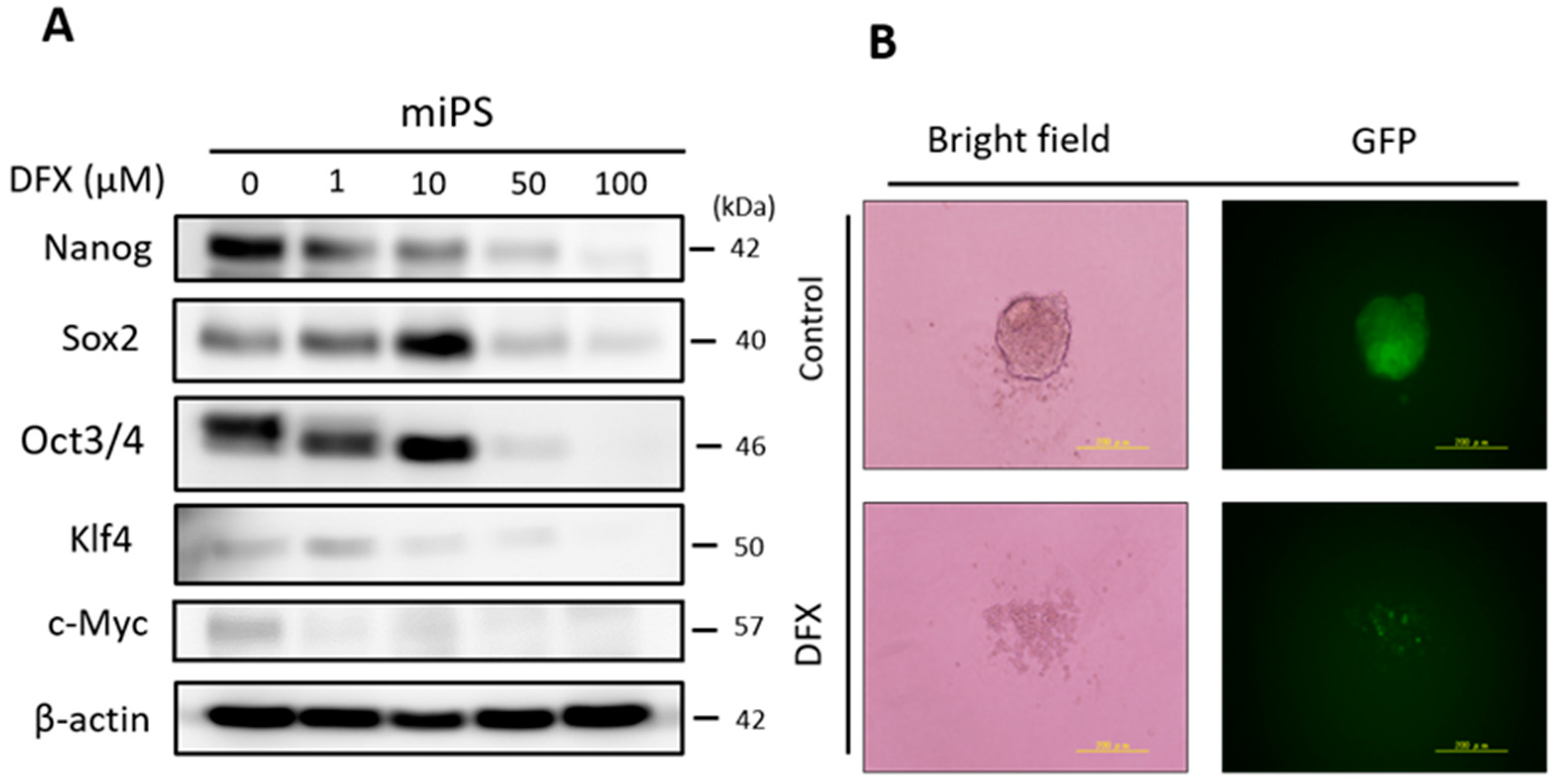
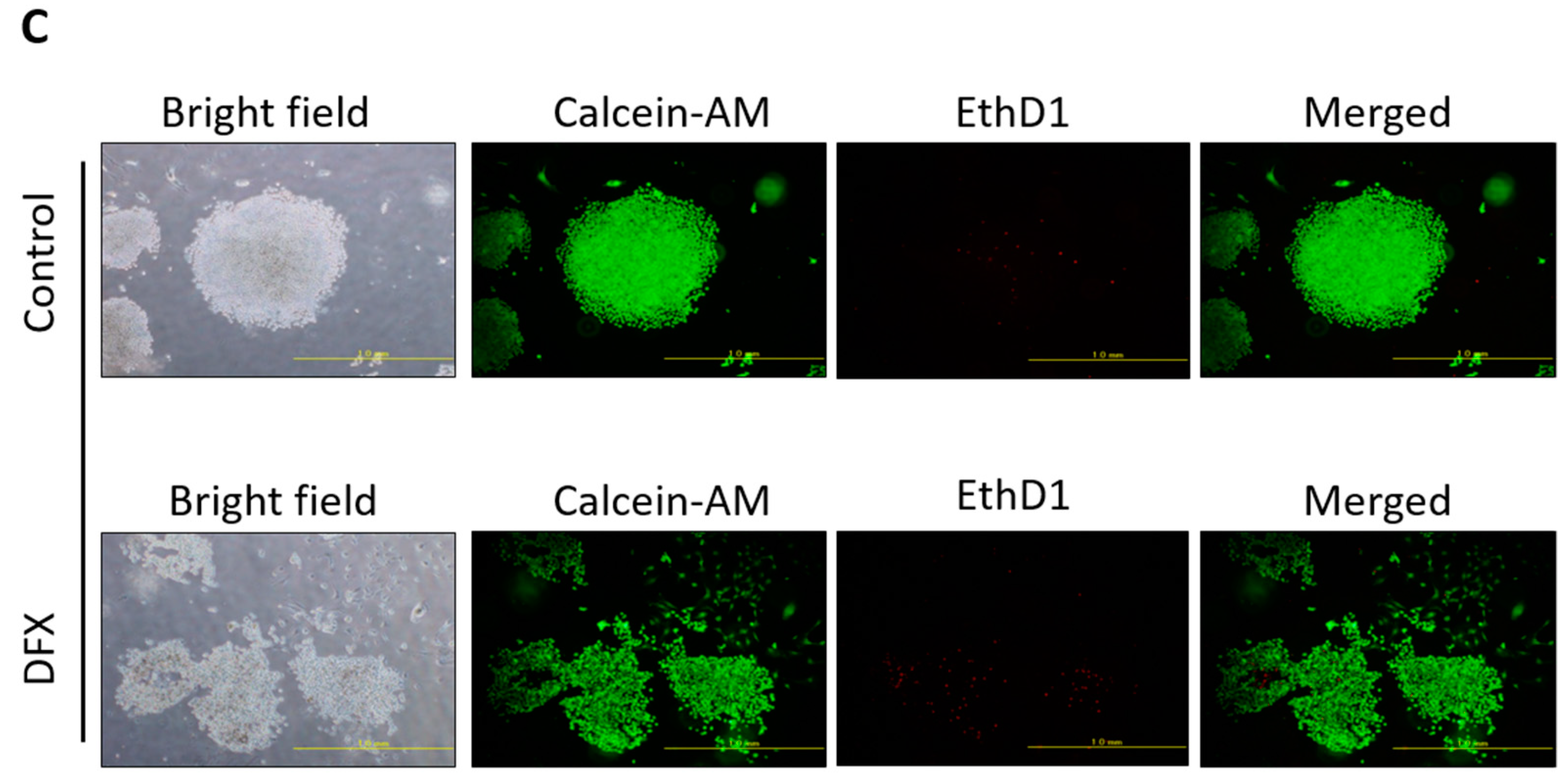
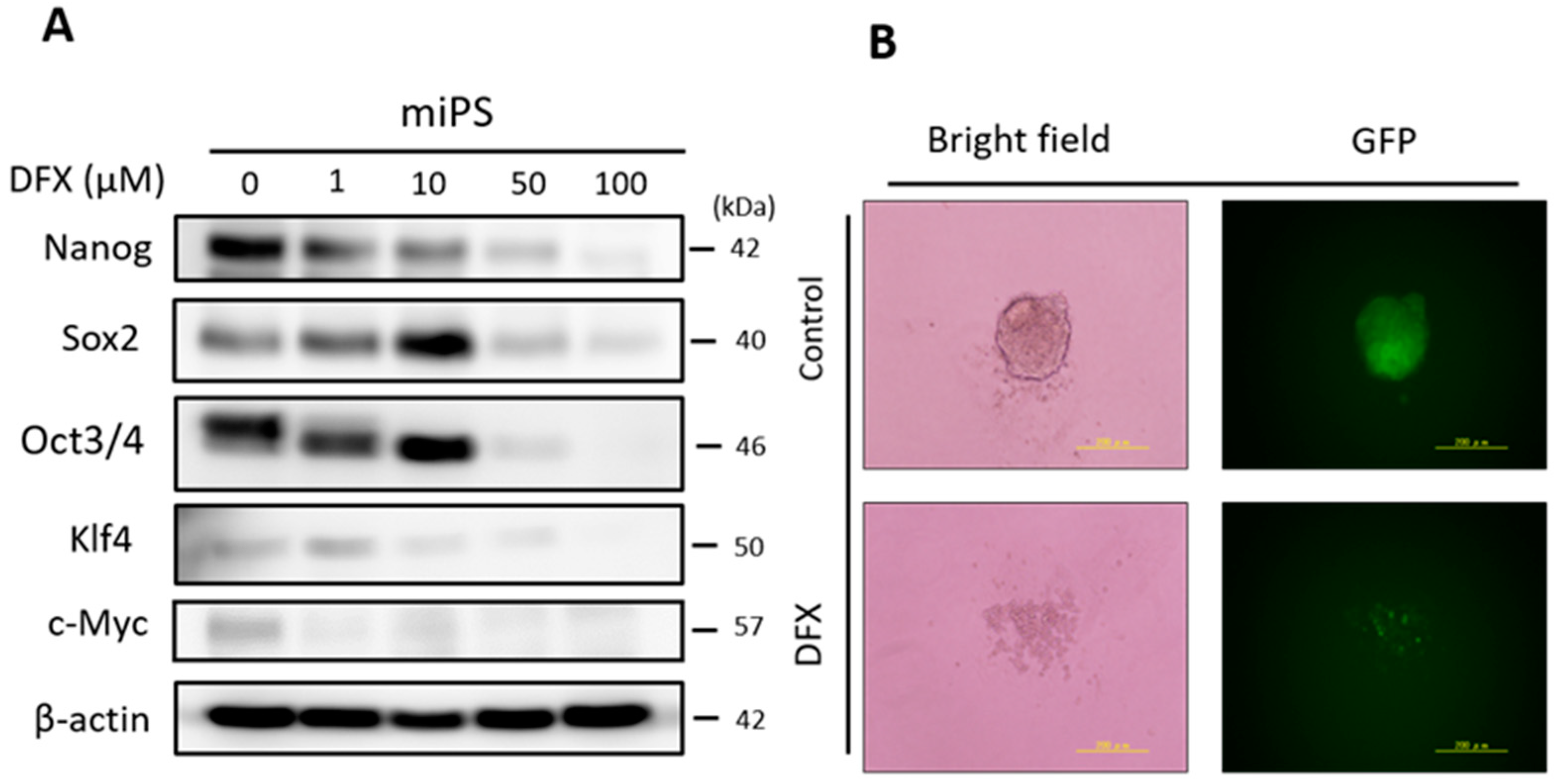
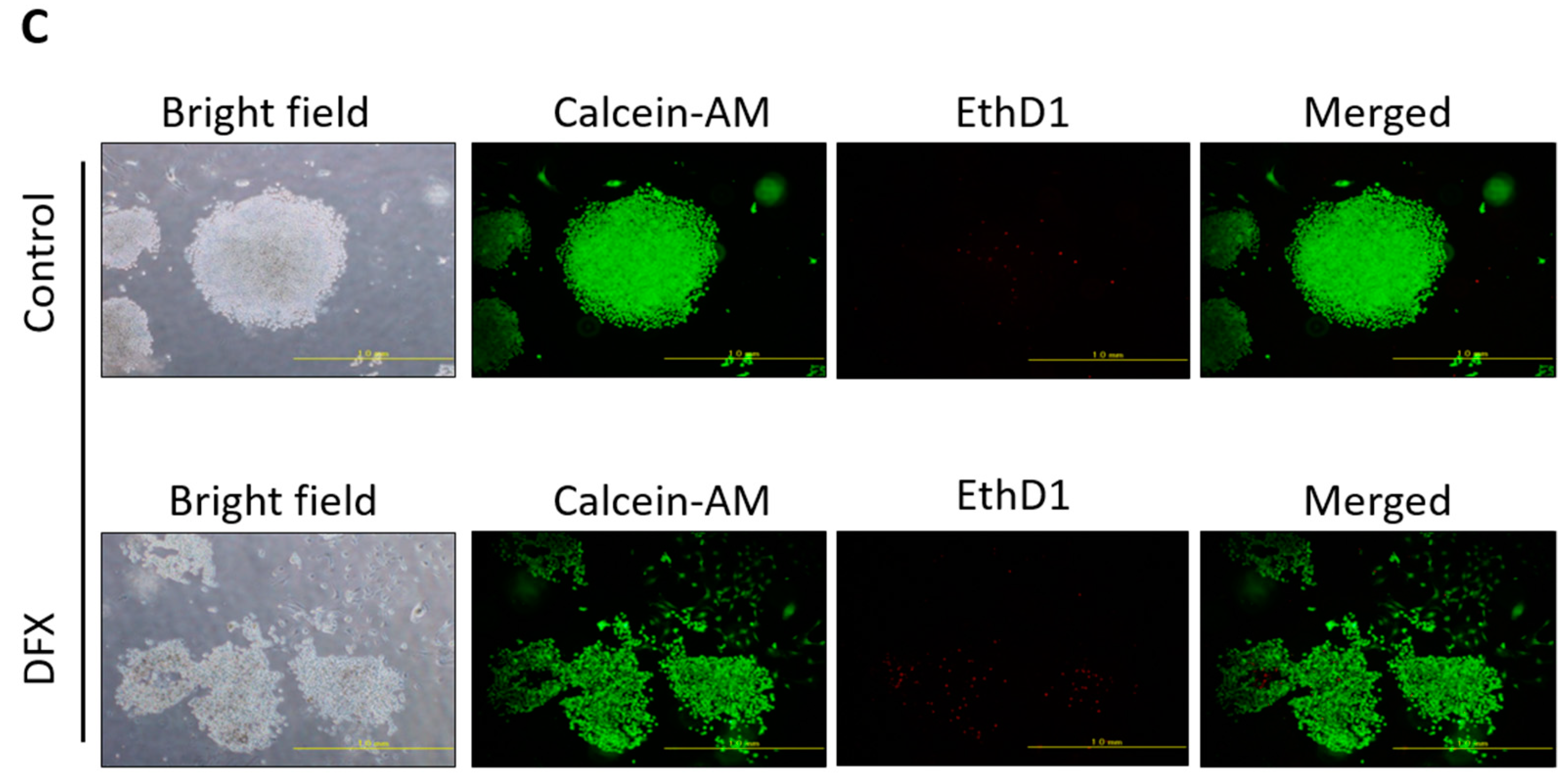
2.1. DFX Suppresses Expression of Stemness Markers and Spherogenicity of miPS Cells
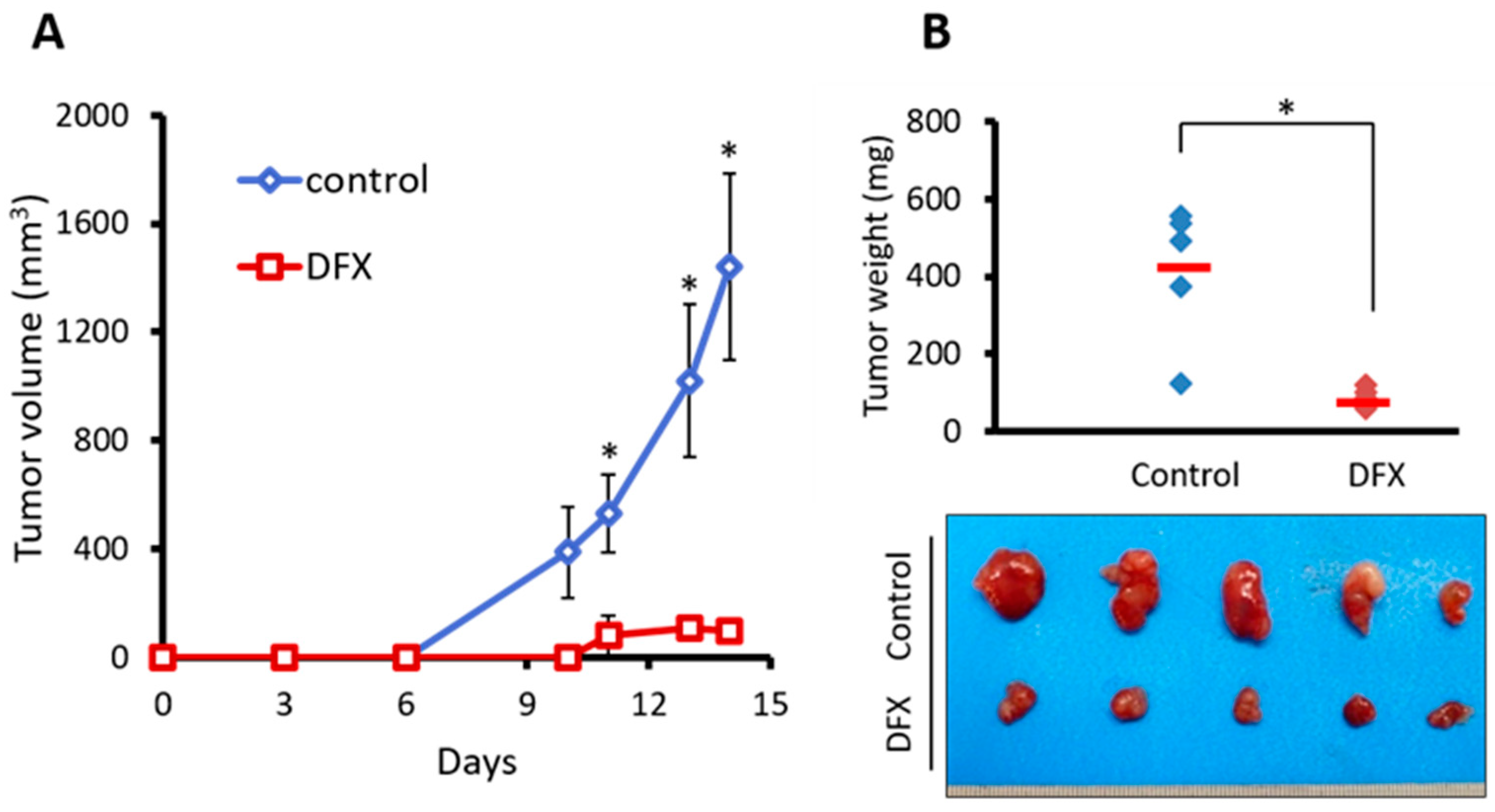
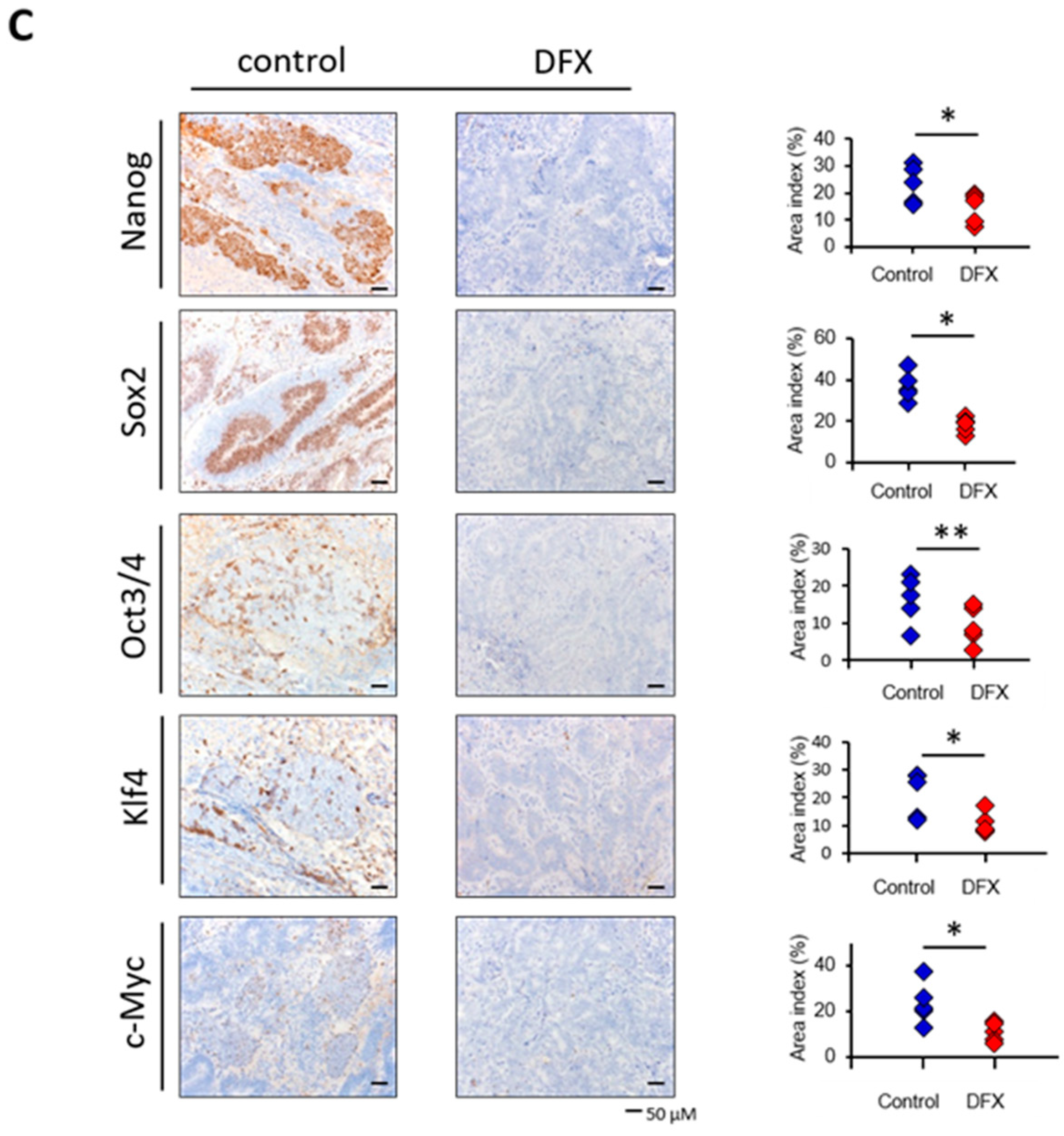
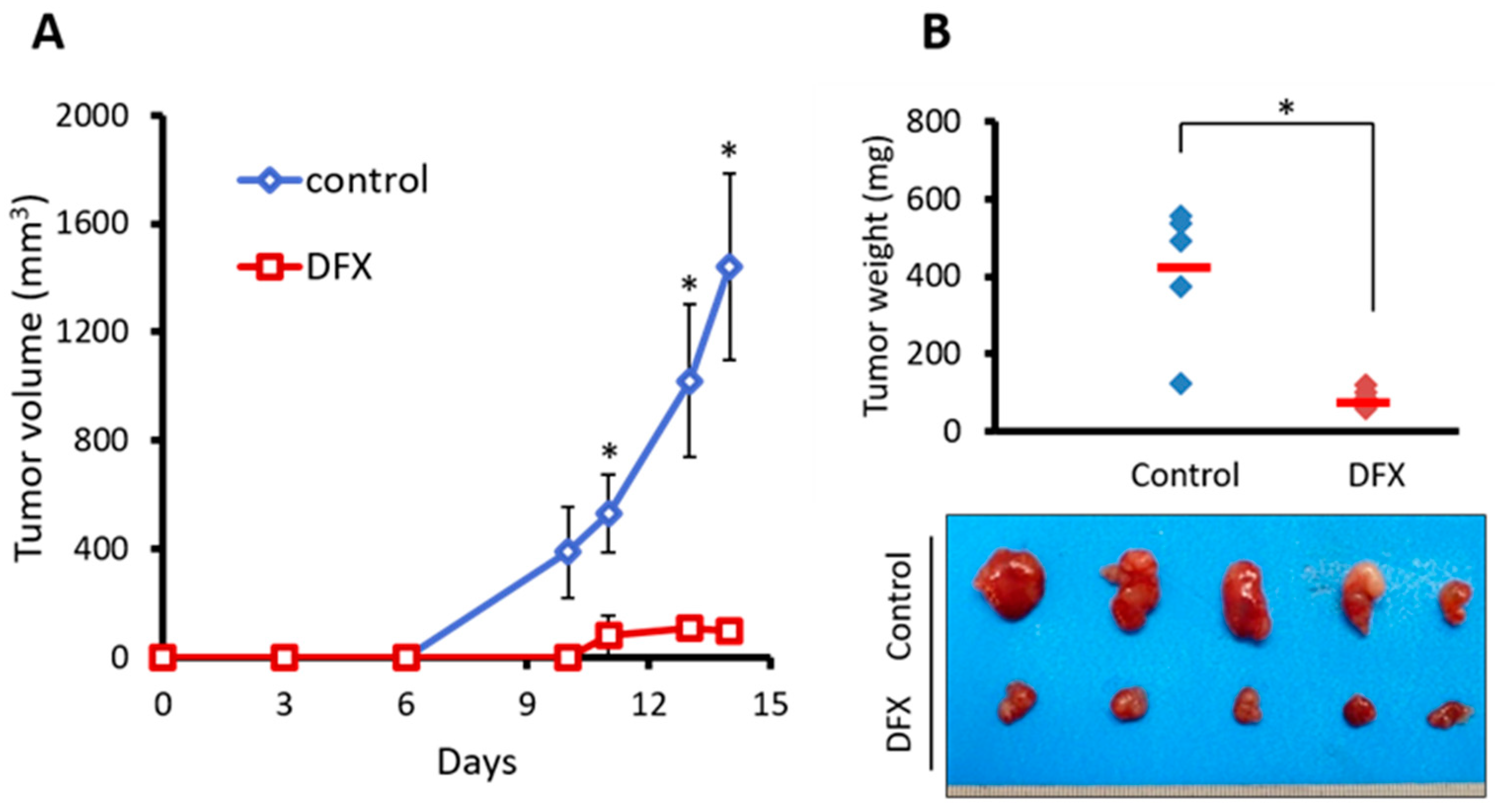
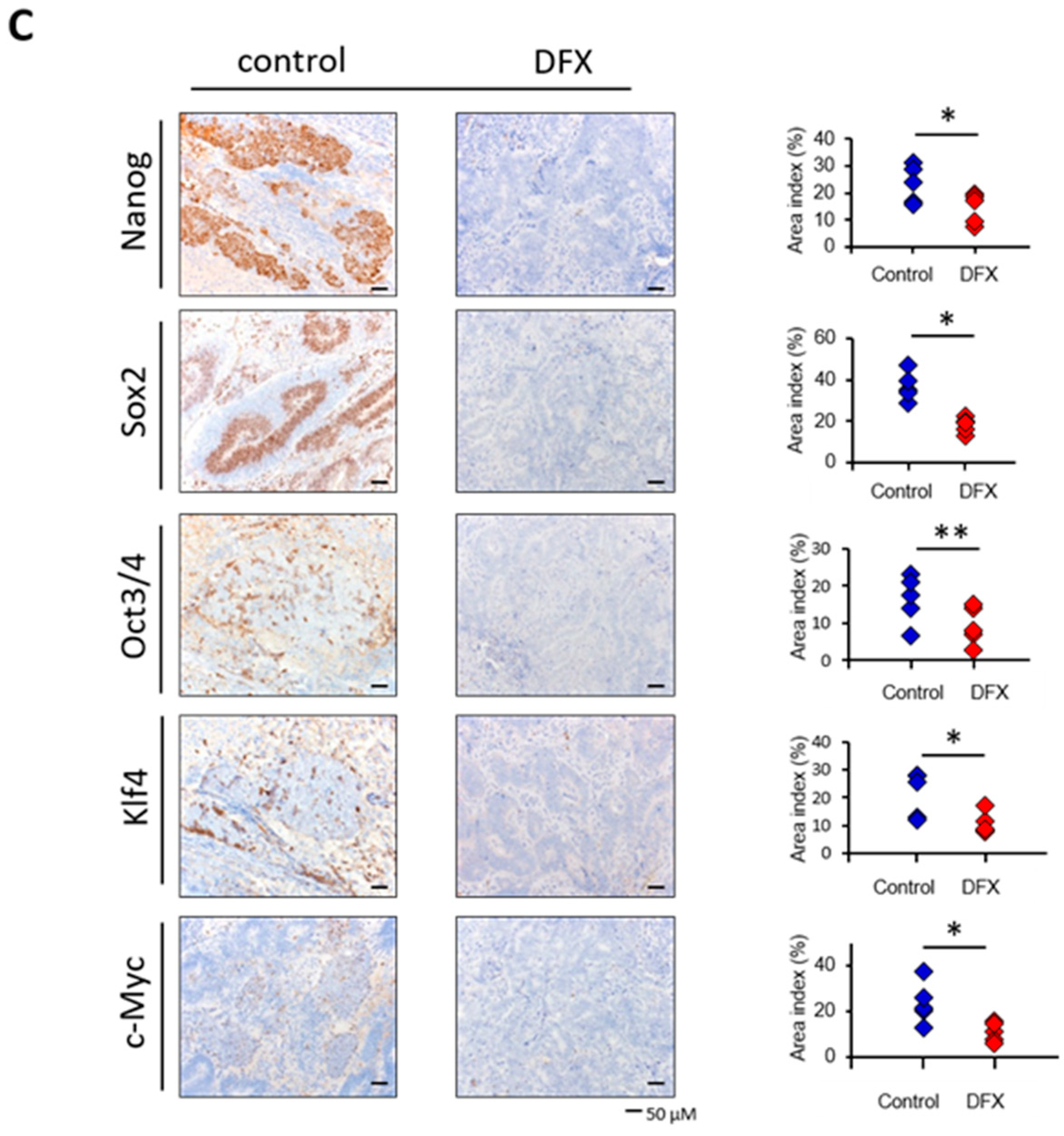
2.2. DFX Suppresses Tumorigenicity and Expression of Tumor Stemness Markers in miPS Cells In Vivo
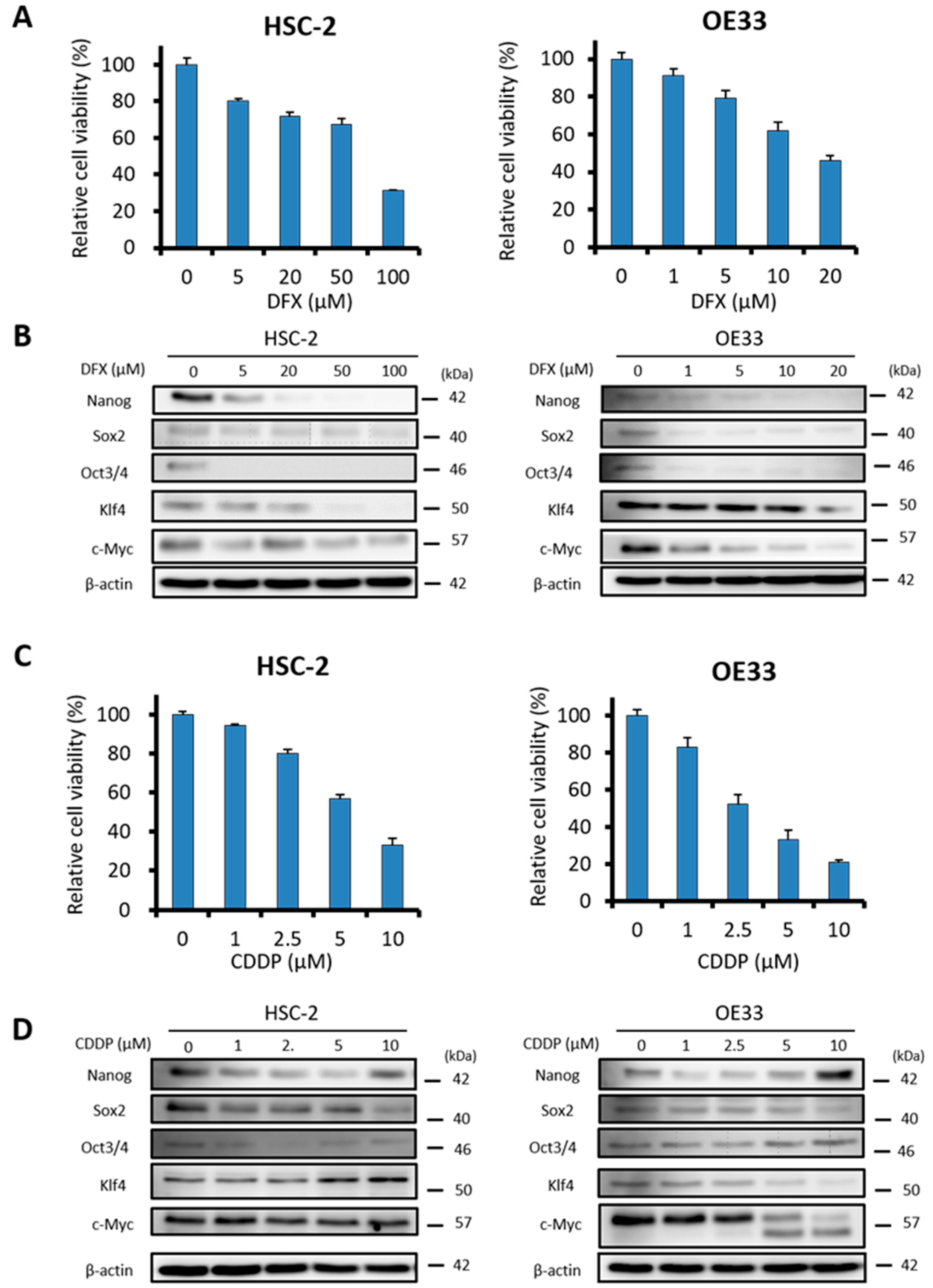
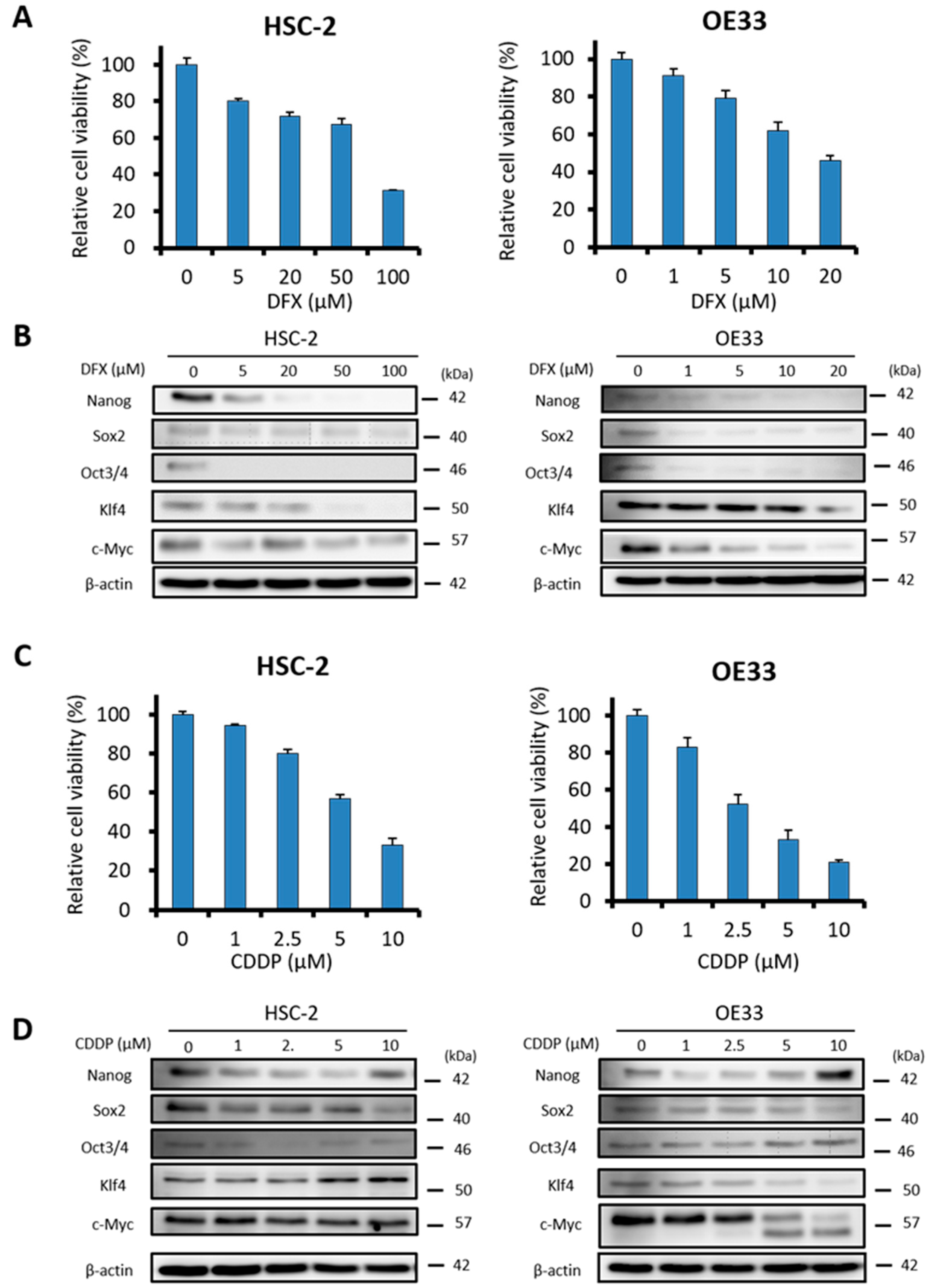
2.3. DFX Suppresses Proliferation and Expression of Stemness Markers in Human Cancer Cell Lines
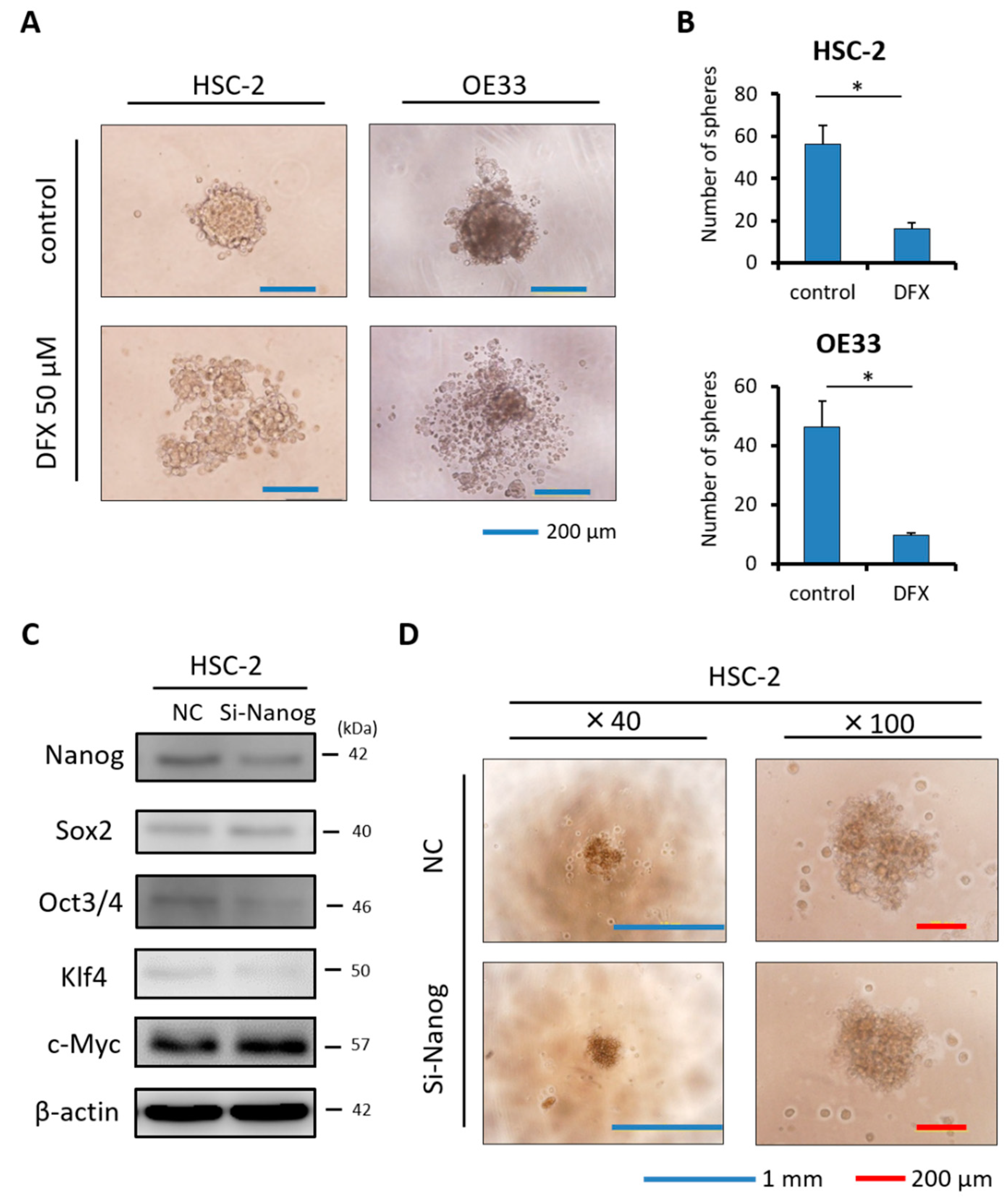
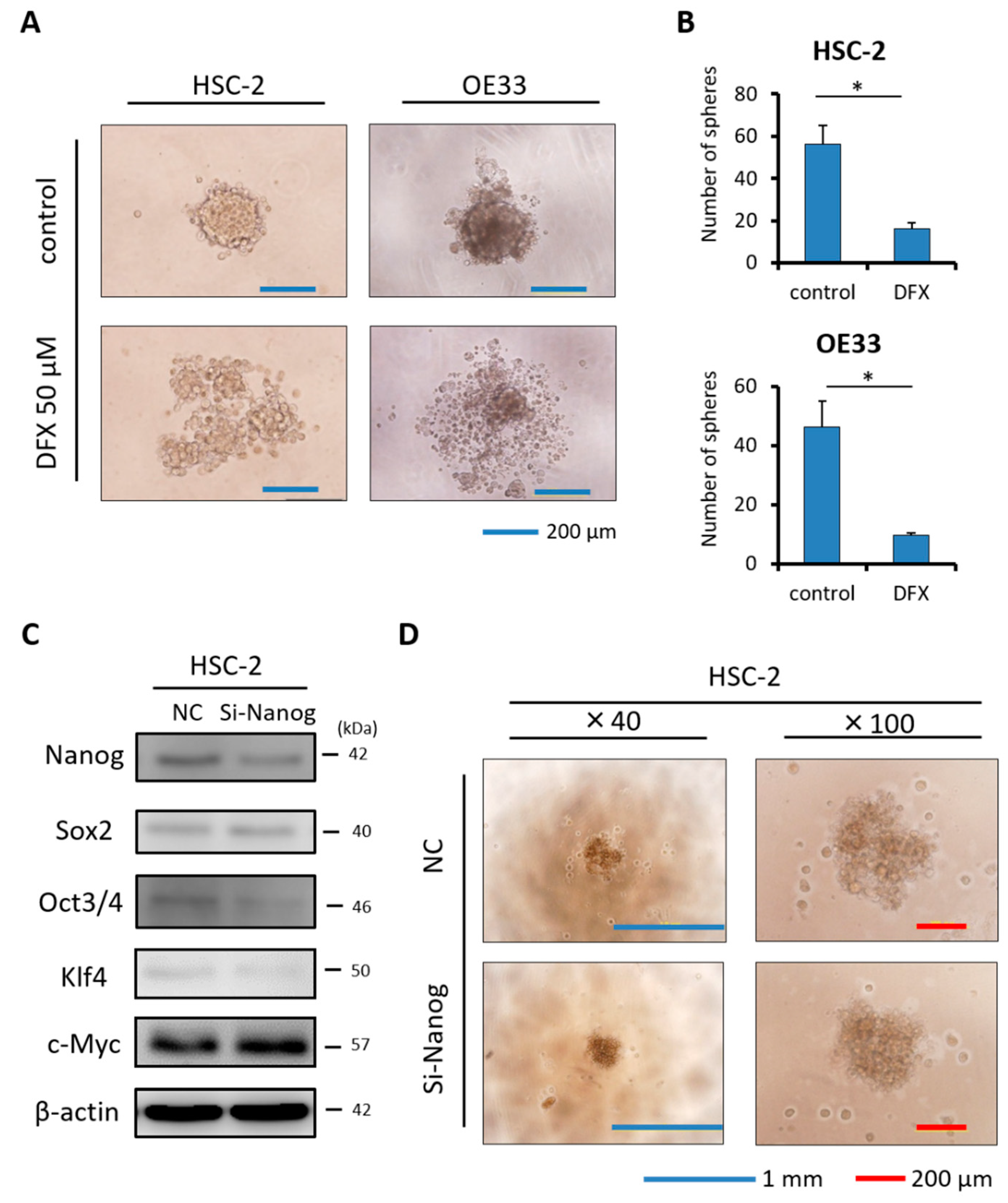
2.4. DFX Suppresses Spherogenicity in Human Cancer Cell Lines
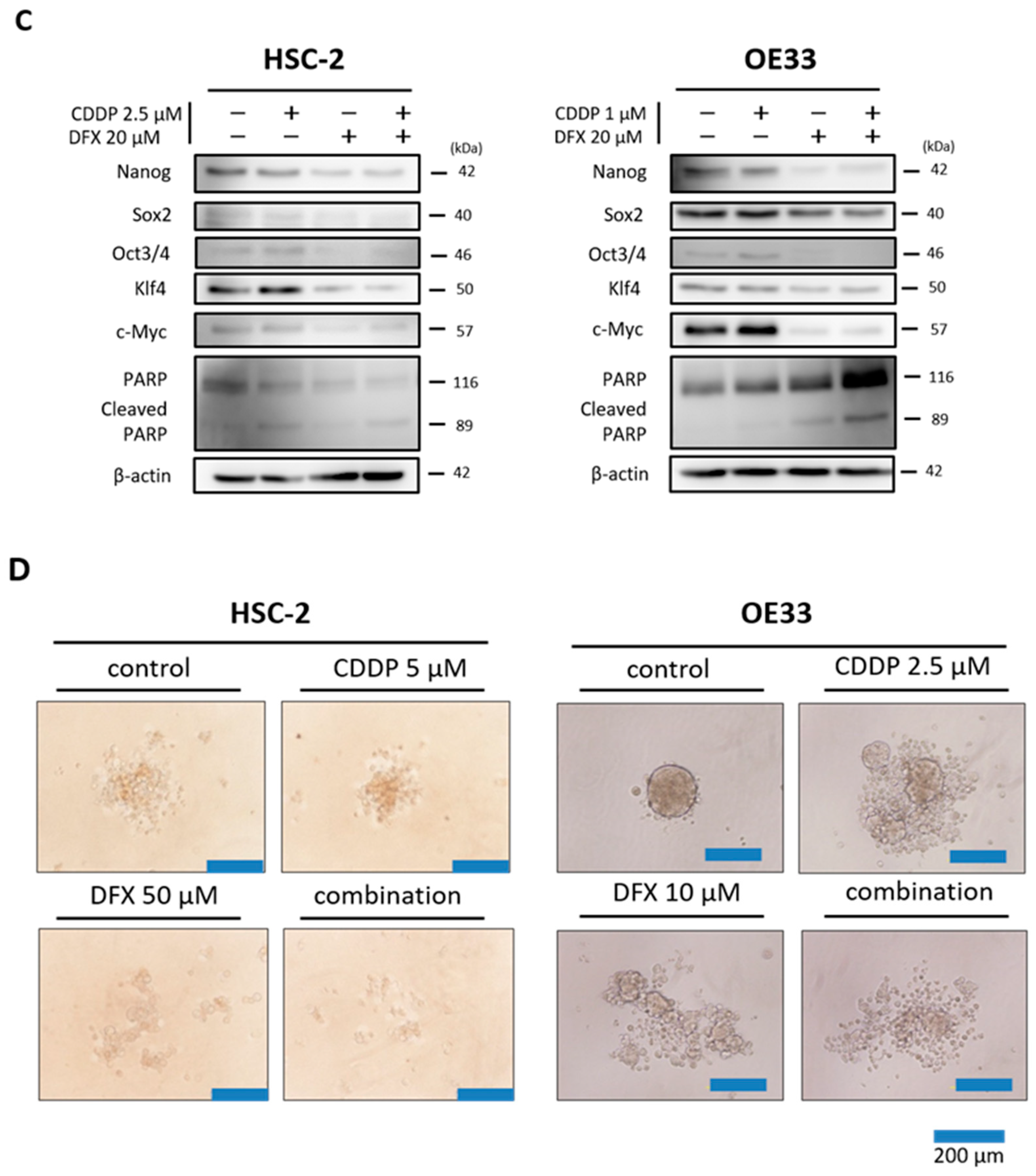
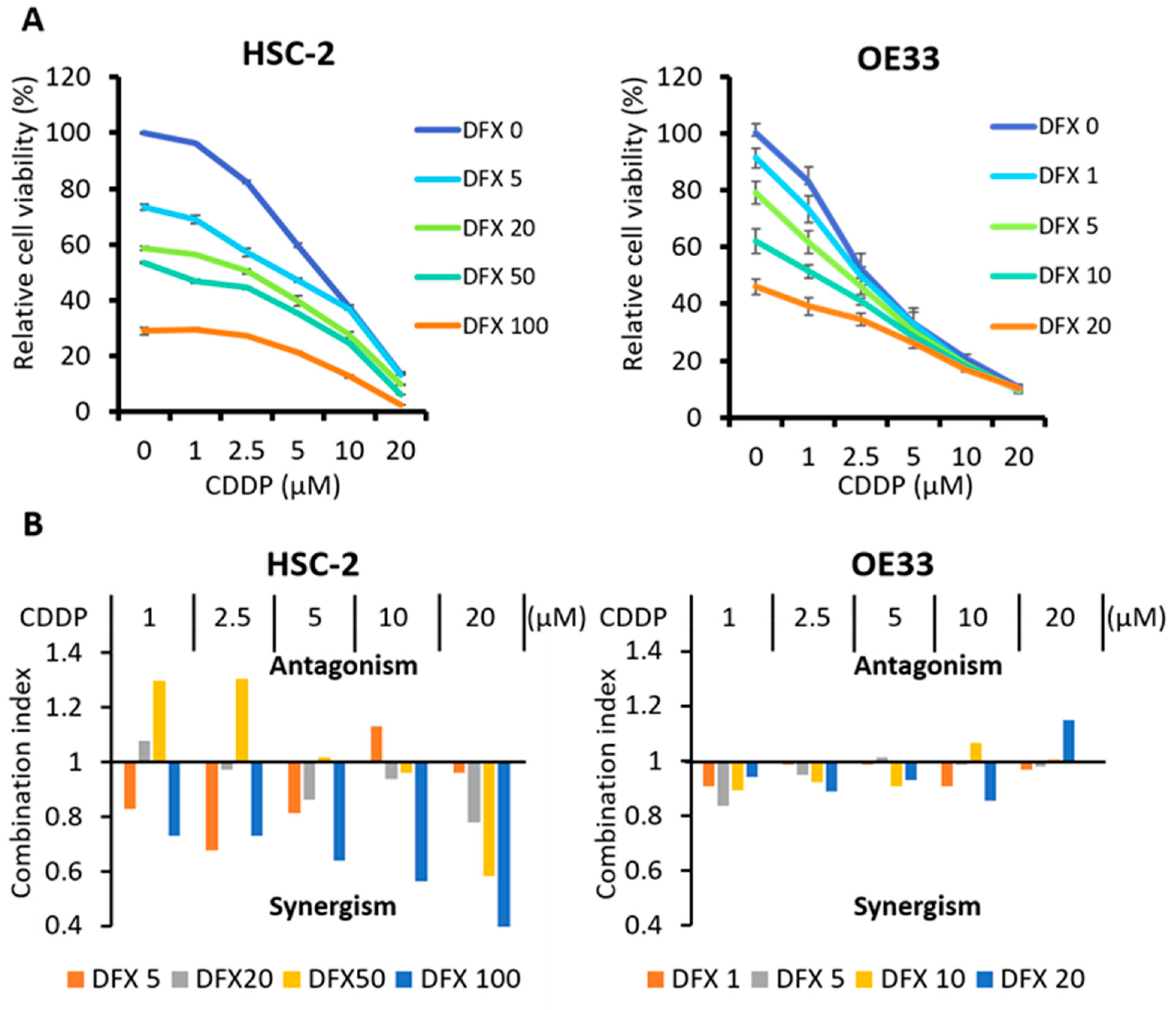
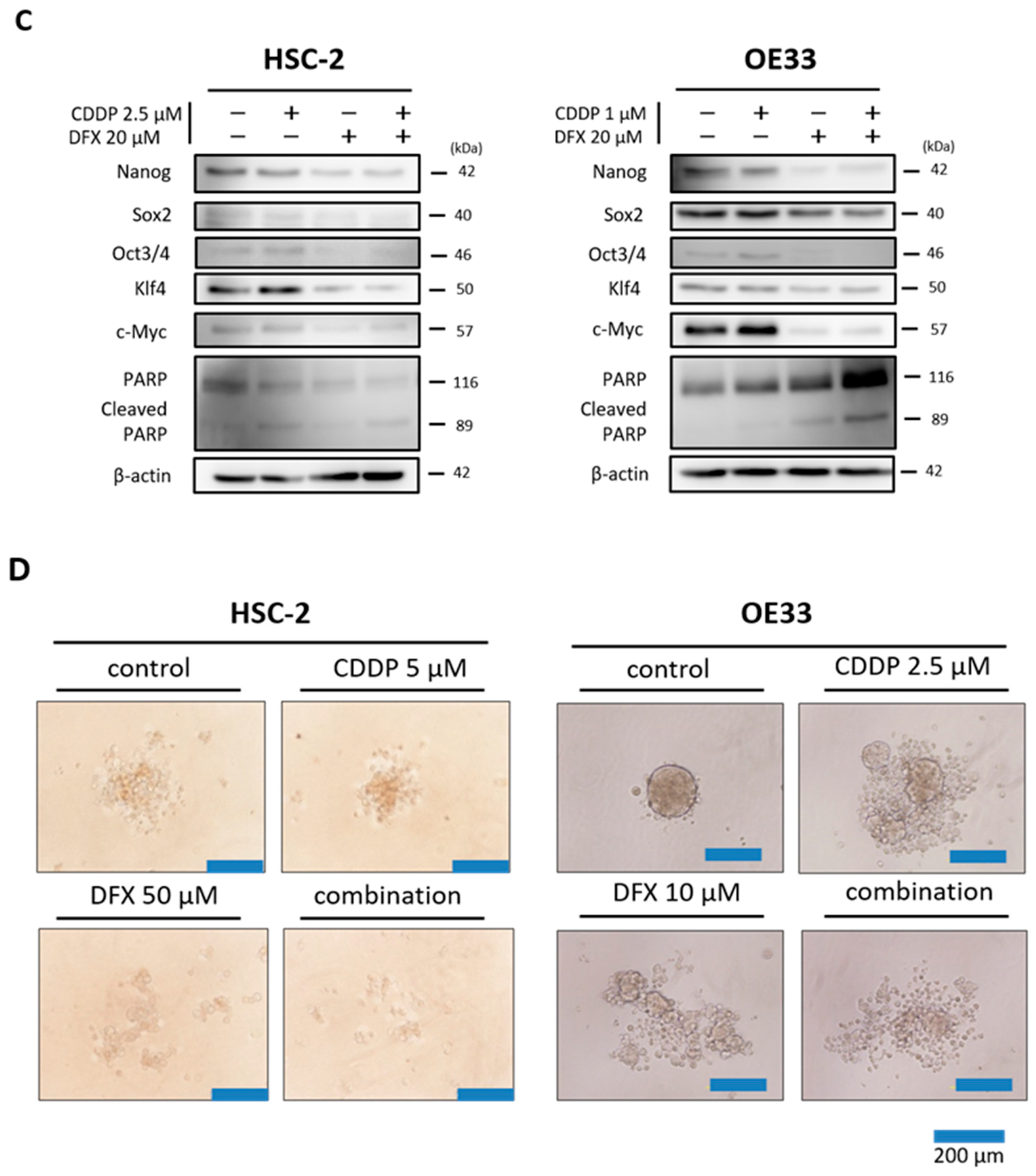
2.5. Combination Therapy with DFX and Chemotherapy Induces Synergistic Antitumor Effects in Human Cancer Cell Lines and Suppresses Expression of Stemness Markers and Function
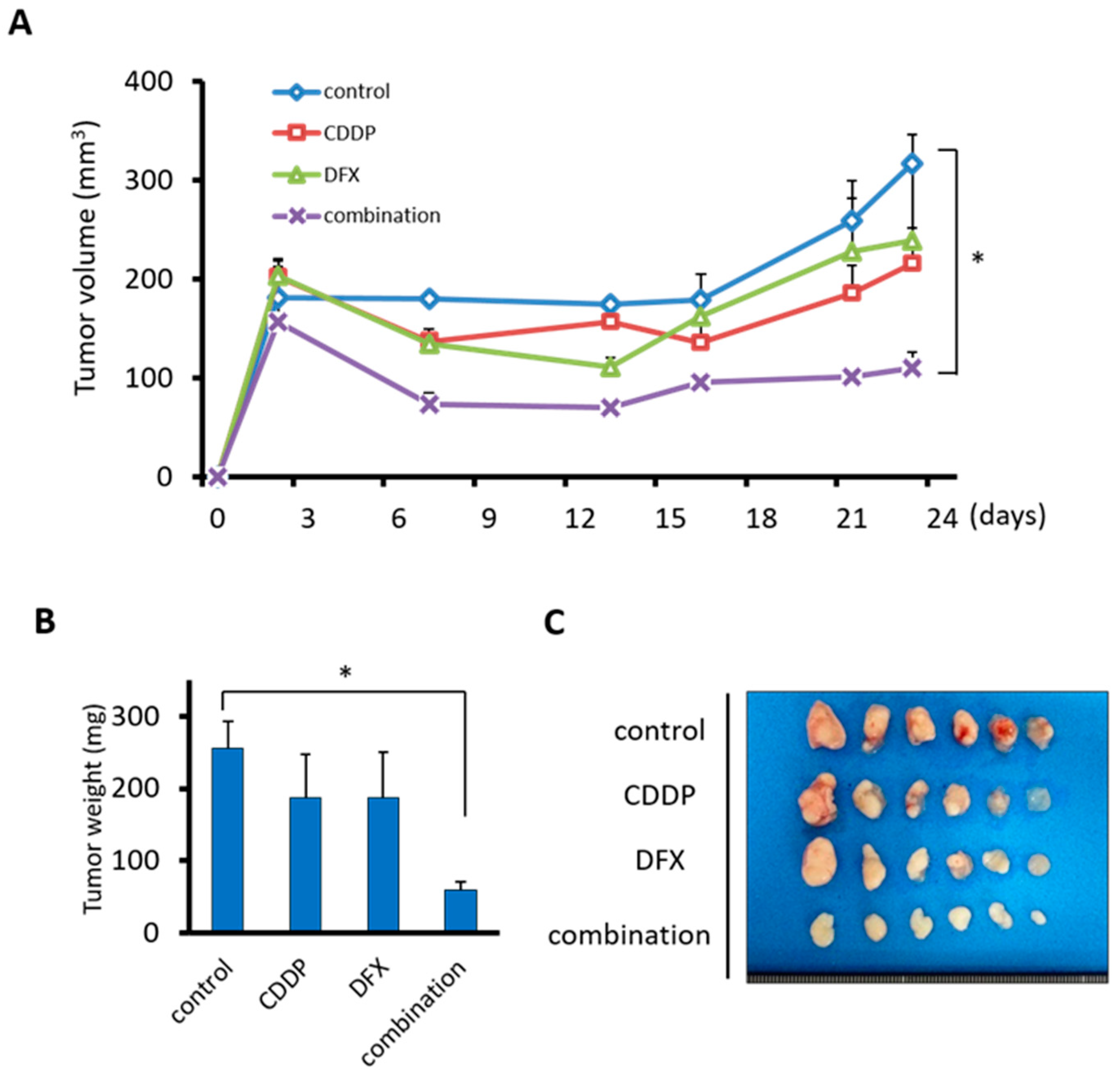
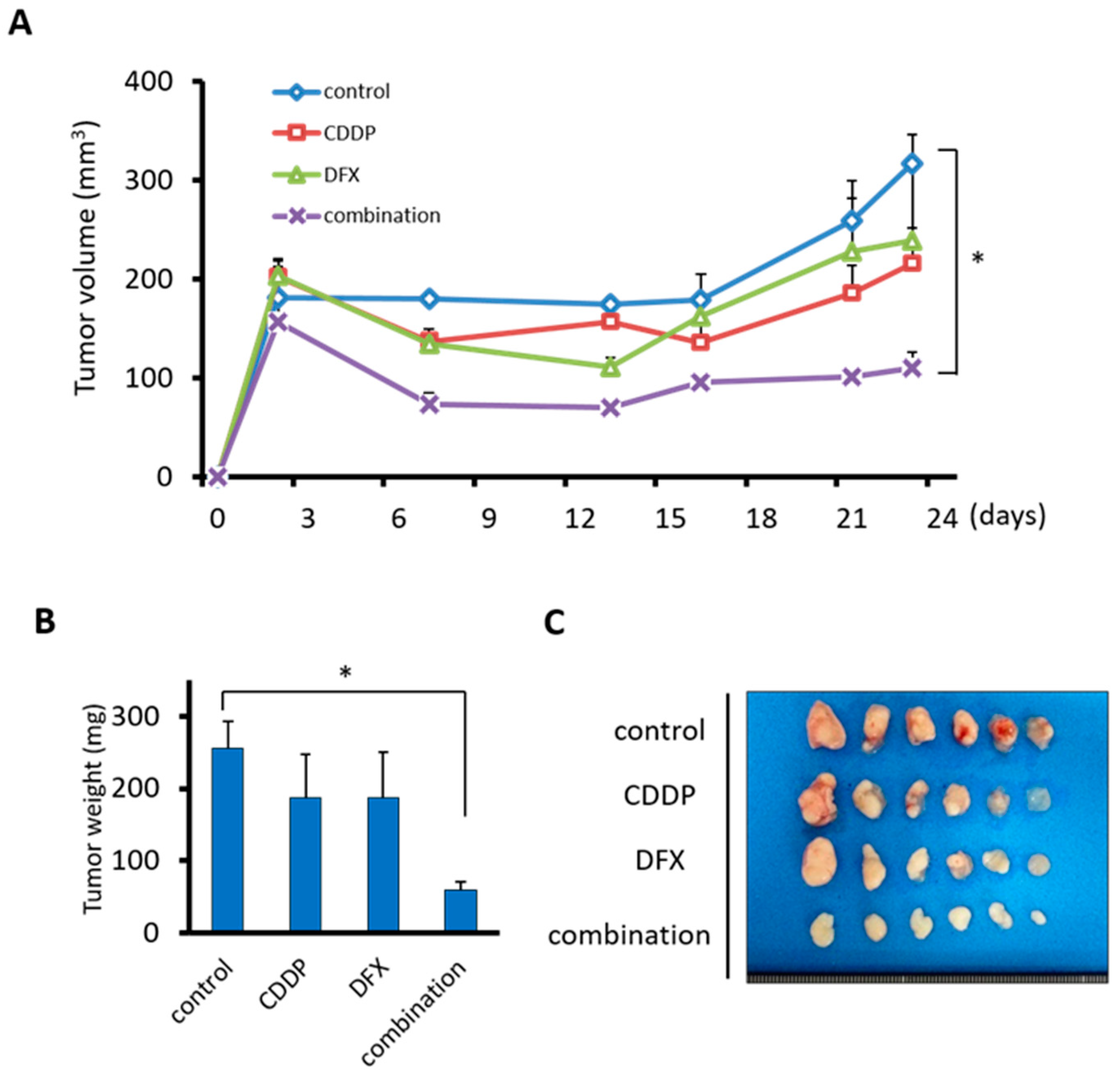
2.6. Combination Therapy with DFX and Chemotherapy Suppresses Tumor Growth of Human Oral Squamous Carcinoma In Vivo
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Cell Culture
4.2. Reagents
4.3. Cell Viability Assay
4.4. Sphere Formation Assay
4.5. Live/Dead Assay
4.6. Nanog Small Interfering RNA Transfection
4.7. Western blotting
4.8. Flow Cytometry Analysis
4.9. Real-Time Quantitative PCR
4.10. VEGF ELISA Assay
4.11. Tumor Xenograft Model and Experiment
4.12. Immunohistochemistry of In Vivo-Derived Tumor Tissues
4.13. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Shen, J.; Sheng, X.; Chang, Z.; Wu, Q.; Wang, S.; Xuan, Z.; Li, D.; Wu, Y.; Shang, Y.; Kong, X.; et al. Iron metabolism regulates p53 signaling through direct heme-p53 interaction and modulation of p53 localization, stability, and function. Cell Rep. 2014, 7, 180–193. [Google Scholar] [CrossRef] [PubMed]
- Torti, S.V.; Torti, F.M. Iron and cancer: More ore to be mined. Nat. Rev. Cancer 2013, 13, 342–355. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Zhang, F. Iron homeostasis and tumorigenesis: Molecular mechanisms and therapeutic opportunities. Protein Cell 2015, 6, 88–100. [Google Scholar] [CrossRef] [PubMed]
- Yamasaki, T.; Terai, S.; Sakaida, I. Deferoxamine for advanced hepatocellular carcinoma. N. Engl. J. Med. 2011, 365, 576–578. [Google Scholar] [CrossRef] [PubMed]
- Harima, H.; Kaino, S.; Takami, T.; Shinoda, S.; Matsumoto, T.; Fujisawa, K.; Yamamoto, N.; Yamasaki, T.; Sakaida, I. Deferasirox, a novel oral iron chelator, shows antiproliferative activity against pancreatic cancer in vitro and in vivo. BMC Cancer 2016, 16, 702. [Google Scholar] [CrossRef] [PubMed]
- Kalinowski, D.S.; Richardson, D.R. The evolution of iron chelators for the treatment of iron overload disease and cancer. Pharmacol. Rev. 2005, 57, 547–583. [Google Scholar] [CrossRef] [PubMed]
- Ohara, T.; Noma, K.; Urano, S.; Watanabe, S.; Nishitani, S.; Tomono, Y.; Kimura, F.; Kagawa, S.; Shirakawa, Y.; Fujiwara, T. A novel synergistic effect of iron depletion on antiangiogenic cancer therapy. Int. J. Cancer 2013, 132, 2705–2713. [Google Scholar] [CrossRef]
- Bonnet, D.; Dick, J.E. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat. Med. 1997, 3, 730–737. [Google Scholar] [CrossRef]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef]
- O’Brien, C.A.; Pollett, A.; Gallinger, S.; Dick, J.E. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature 2007, 445, 106–110. [Google Scholar] [CrossRef]
- Prince, M.E.; Sivanandan, R.; Kaczorowski, A.; Wolf, G.T.; Kaplan, M.J.; Dalerba, P.; Weissman, I.L.; Clarke, M.F.; Ailles, L.E. Identification of a subpopulation of cells with cancer stem cell properties in head and neck squamous cell carcinoma. Proc. Natl. Acad. Sci. USA 2007, 104, 973–978. [Google Scholar] [CrossRef] [PubMed]
- Chiou, S.H.; Yu, C.C.; Huang, C.Y.; Lin, S.C.; Liu, C.J.; Tsai, T.H.; Chou, S.H.; Chien, C.S.; Ku, H.H.; Lo, J.F. Positive correlations of Oct-4 and Nanog in oral cancer stem-like cells and high-grade oral squamous cell carcinoma. Clin. Cancer Res. 2008, 14, 4085–4095. [Google Scholar] [CrossRef] [PubMed]
- Wong, D.J.; Liu, H.; Ridky, T.W.; Cassarino, D.; Segal, E.; Chang, H.Y. Module map of stem cell genes guides creation of epithelial cancer stem cells. Cell Stem Cell 2008, 2, 333–344. [Google Scholar] [CrossRef] [PubMed]
- Somervaille, T.C.; Matheny, C.J.; Spencer, G.J.; Iwasaki, M.; Rinn, J.L.; Witten, D.M.; Chang, H.Y.; Shurtleff, S.A.; Downing, J.R.; Cleary, M.L. Hierarchical maintenance of MLL myeloid leukemia stem cells employs a transcriptional program shared with embryonic rather than adult stem cells. Cell Stem Cell 2009, 4, 129–140. [Google Scholar] [CrossRef]
- Yu, F.; Li, J.; Chen, H.; Fu, J.; Ray, S.; Huang, S.; Zheng, H.; Ai, W. Kruppel-like factor 4 (KLF4) is required for maintenance of breast cancer stem cells and for cell migration and invasion. Oncogene 2011, 30, 2161–2172. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.C.; Shieh, G.S.; Wu, P.; Lin, C.C.; Shiau, A.L.; Wu, C.L. Oct-3/4 expression reflects tumor progression and regulates motility of bladder cancer cells. Cancer Res. 2008, 68, 6281–6291. [Google Scholar] [CrossRef] [PubMed]
- Wang, J. c-Myc Is Required for Maintenance of Glioma Cancer Stem Cells. PloS ONE 2008, 3, e3769. [Google Scholar] [CrossRef] [PubMed]
- Torres-Padilla, M.E.; Chambers, I. Transcription factor heterogeneity in pluripotent stem cells: A stochastic advantage. Development 2014, 141, 2173–2181. [Google Scholar] [CrossRef] [PubMed]
- Hadjimichael, C.; Chanoumidou, K.; Papadopoulou, N.; Arampatzi, P.; Papamatheakis, J.; Kretsovali, A. Common stemness regulators of embryonic and cancer stem cells. World J. Stem Cells 2015, 7, 1150–1184. [Google Scholar] [CrossRef]
- Rizzino, A. Concise review: The Sox2-Oct4 connection: Critical players in a much larger interdependent network integrated at multiple levels. Stem Cells 2013, 31, 1033–1039. [Google Scholar] [CrossRef]
- Saunders, A.; Faiola, F.; Wang, J. Concise review: Pursuing self-renewal and pluripotency with the stem cell factor Nanog. Stem Cells 2013, 31, 1227–1236. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Rogoff, H.A.; Keates, S.; Gao, Y.; Murikipudi, S.; Mikule, K.; Leggett, D.; Li, W.; Pardee, A.B.; Li, C.J. Suppression of cancer relapse and metastasis by inhibiting cancer stemness. Proc. Natl. Acad. Sci. USA 2015, 112, 1839–1844. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Wang, X.; Chen, G.Y.; Dalerba, P.; Gurney, A.; Hoey, T.; Sherlock, G.; Lewicki, J.; Shedden, K.; Clarke, M.F. The prognostic role of a gene signature from tumorigenic breast-cancer cells. N. Engl. J. Med. 2007, 356, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Liao, R.; Li, D.; Sun, J. Induced cancer stem cells generated by radiochemotherapy and their therapeutic implications. Oncotarget 2017, 8, 17301–17312. [Google Scholar] [CrossRef] [PubMed]
- Jandial, R.; Waters, D.J.; Chen, M.Y. Cancer stem cells can arise from differentiated neoplastic cells. Neurosurgery 2011, 69, N22. [Google Scholar] [CrossRef]
- Carrier, F. Chromatin Modulation by Histone Deacetylase Inhibitors: Impact on Cellular Sensitivity to Ionizing Radiation. Mol. Cell. Pharmacol. 2013, 5, 51–59. [Google Scholar] [PubMed]
- Debeb, B.G.; Lacerda, L.; Xu, W.; Larson, R.; Solley, T.; Atkinson, R.; Sulman, E.P.; Ueno, N.T.; Krishnamurthy, S.; Reuben, J.M.; et al. Histone deacetylase inhibitors stimulate dedifferentiation of human breast cancer cells through WNT/beta-catenin signaling. Stem Cells 2012, 30, 2366–2377. [Google Scholar] [CrossRef]
- Liang, Y.; Zhong, Z.; Huang, Y.; Deng, W.; Cao, J.; Tsao, G.; Liu, Q.; Pei, D.; Kang, T.; Zeng, Y.X. Stem-like cancer cells are inducible by increasing genomic instability in cancer cells. J. Biol. Chem. 2010, 285, 4931–4940. [Google Scholar] [CrossRef]
- Kim, M.S.; Cho, H.I.; Yoon, H.J.; Ahn, Y.H.; Park, E.J.; Jin, Y.H.; Jang, Y.K. JIB-04, A Small Molecule Histone Demethylase Inhibitor, Selectively Targets Colorectal Cancer Stem Cells by Inhibiting the Wnt/beta-Catenin Signaling Pathway. Sci. Rep. 2018, 8, 6611. [Google Scholar] [CrossRef] [PubMed]
- Ninomiya, T.; Ohara, T.; Noma, K.; Katsura, Y.; Katsube, R.; Kashima, H.; Kato, T.; Tomono, Y.; Tazawa, H.; Kagawa, S.; et al. Iron depletion is a novel therapeutic strategy to target cancer stem cells. Oncotarget 2017, 8, 98405–98416. [Google Scholar] [CrossRef]
- Bourguignon, L.Y.; Earle, C.; Wong, G.; Spevak, C.C.; Krueger, K. Stem cell marker (Nanog) and Stat-3 signaling promote MicroRNA-21 expression and chemoresistance in hyaluronan/CD44-activated head and neck squamous cell carcinoma cells. Oncogene 2012, 31, 149–160. [Google Scholar] [CrossRef] [PubMed]
- Gong, S.; Li, Q.; Jeter, C.R.; Fan, Q.; Tang, D.G.; Liu, B. Regulation of NANOG in cancer cells. Mol. Carcinog 2015, 54, 679–687. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.L.; Chiou, S.H.; Wu, C.W. Targeting cancer stem cells: Emerging role of Nanog transcription factor. Onco Targets Ther. 2013, 6, 1207–1220. [Google Scholar] [CrossRef] [PubMed]
- Raggi, C.; Gammella, E.; Correnti, M.; Buratti, P.; Forti, E.; Andersen, J.B.; Alpini, G.; Glaser, S.; Alvaro, D.; Invernizzi, P.; et al. Dysregulation of Iron Metabolism in Cholangiocarcinoma Stem-like Cells. Sci. Rep. 2017, 7, 17667. [Google Scholar] [CrossRef] [PubMed]
- Vela, D. Iron Metabolism in Prostate Cancer; From Basic Science to New Therapeutic Strategies. Front. Oncol. 2018, 8, 547. [Google Scholar] [CrossRef] [PubMed]
- El Hout, M.; Dos Santos, L.; Hamai, A.; Mehrpour, M. A promising new approach to cancer therapy: Targeting iron metabolism in cancer stem cells. Semin. Cancer Biol. 2018, 53, 125–138. [Google Scholar] [CrossRef] [PubMed]
- Manz, D.H.; Blanchette, N.L.; Paul, B.T.; Torti, F.M.; Torti, S.V. Iron and cancer: Recent insights. Ann. N. Y. Acad. Sci. 2016, 1368, 149–161. [Google Scholar] [CrossRef]
- Nishitani, S.; Noma, K.; Ohara, T.; Tomono, Y.; Watanabe, S.; Tazawa, H.; Shirakawa, Y.; Fujiwara, T. Iron depletion-induced downregulation of N-cadherin expression inhibits invasive malignant phenotypes in human esophageal cancer. Int. J. Oncol. 2016, 49, 1351–1359. [Google Scholar] [CrossRef]
- Noma, K.; Smalley, K.S.; Lioni, M.; Naomoto, Y.; Tanaka, N.; El-Deiry, W.; King, A.J.; Nakagawa, H.; Herlyn, M. The essential role of fibroblasts in esophageal squamous cell carcinoma-induced angiogenesis. Gastroenterology 2008, 134, 1981–1993. [Google Scholar] [CrossRef]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Katsura, Y.; Ohara, T.; Noma, K.; Ninomiya, T.; Kashima, H.; Kato, T.; Sato, H.; Komoto, S.; Narusaka, T.; Tomono, Y.; et al. A Novel Combination Cancer Therapy with Iron Chelator Targeting Cancer Stem Cells via Suppressing Stemness. Cancers 2019, 11, 177. https://doi.org/10.3390/cancers11020177
Katsura Y, Ohara T, Noma K, Ninomiya T, Kashima H, Kato T, Sato H, Komoto S, Narusaka T, Tomono Y, et al. A Novel Combination Cancer Therapy with Iron Chelator Targeting Cancer Stem Cells via Suppressing Stemness. Cancers. 2019; 11(2):177. https://doi.org/10.3390/cancers11020177
Chicago/Turabian StyleKatsura, Yuki, Toshiaki Ohara, Kazuhiro Noma, Takayuki Ninomiya, Hajime Kashima, Takuya Kato, Hiroaki Sato, Satoshi Komoto, Toru Narusaka, Yasuko Tomono, and et al. 2019. "A Novel Combination Cancer Therapy with Iron Chelator Targeting Cancer Stem Cells via Suppressing Stemness" Cancers 11, no. 2: 177. https://doi.org/10.3390/cancers11020177
APA StyleKatsura, Y., Ohara, T., Noma, K., Ninomiya, T., Kashima, H., Kato, T., Sato, H., Komoto, S., Narusaka, T., Tomono, Y., Xing, B., Chen, Y., Tazawa, H., Kagawa, S., Shirakawa, Y., Kasai, T., Seno, M., Matsukawa, A., & Fujiwara, T. (2019). A Novel Combination Cancer Therapy with Iron Chelator Targeting Cancer Stem Cells via Suppressing Stemness. Cancers, 11(2), 177. https://doi.org/10.3390/cancers11020177