PI3k and Stat3: Oncogenes that are Required for Gap Junctional, Intercellular Communication


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
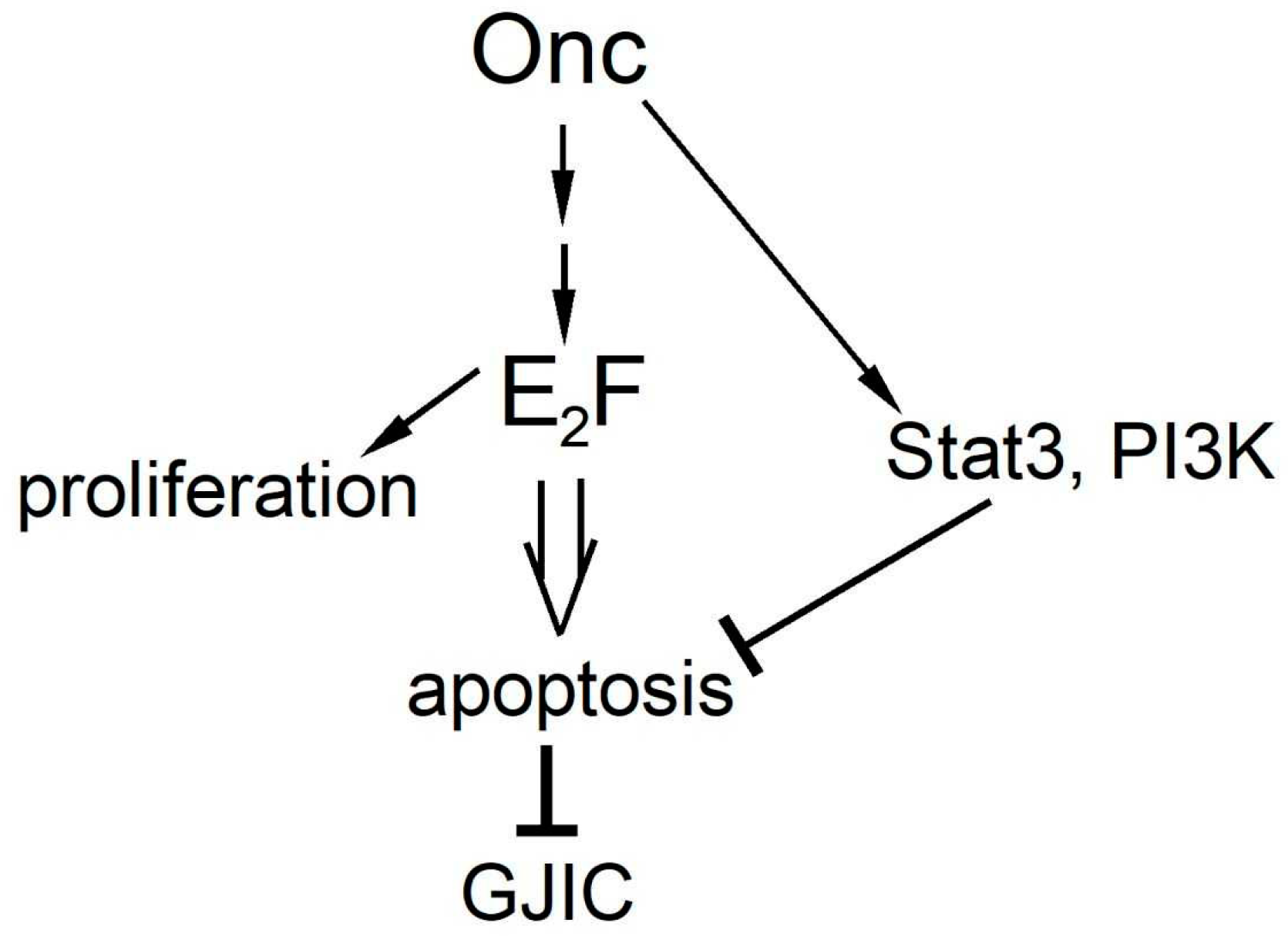
:1. Introduction
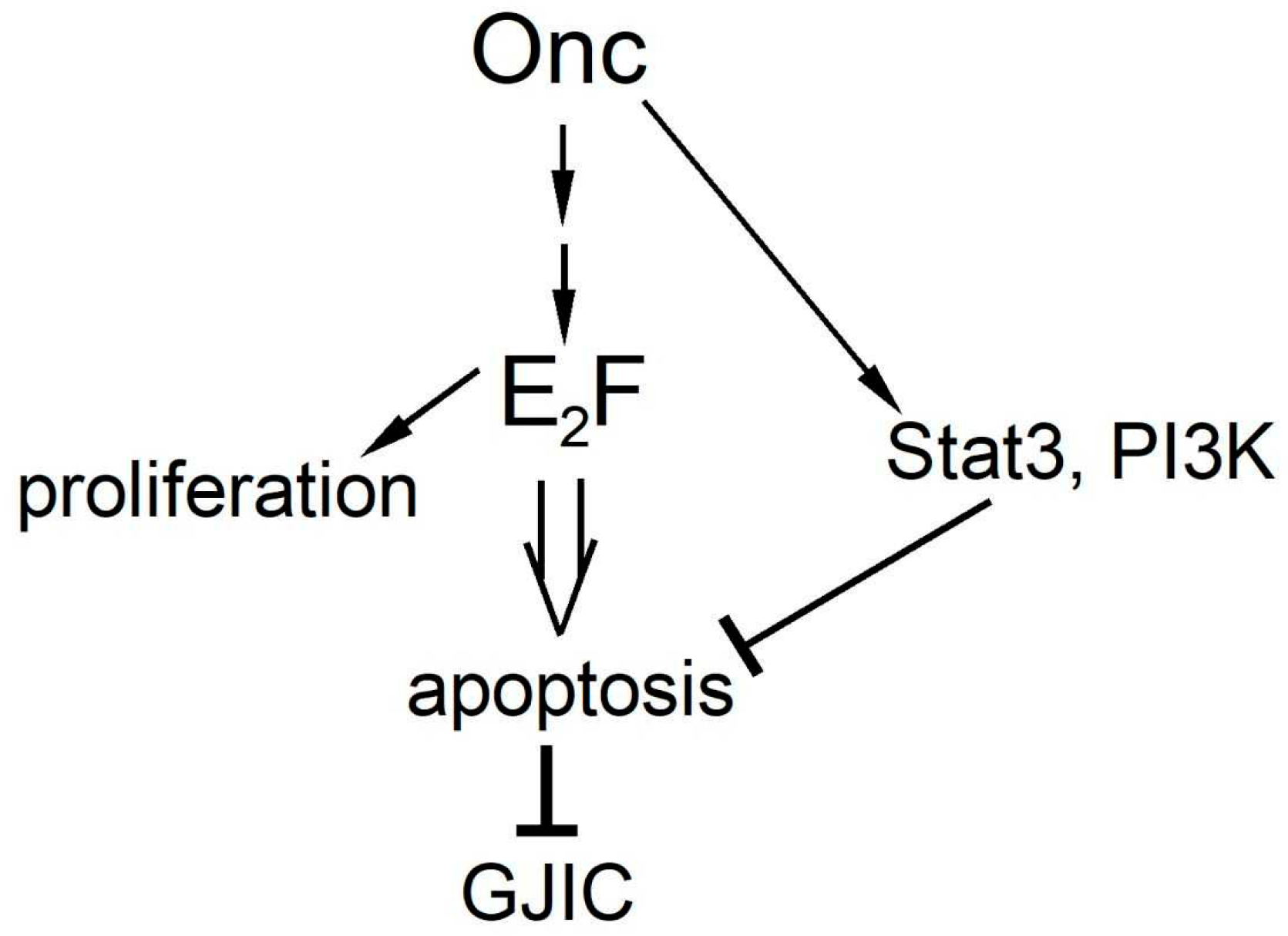
Oncogenes and Gap Junctional Communication
2. PI3k as a Positive Regulator of Gap Junctional Communication
2.1. The Phosphatidylinositol-3 Kinase (PI3k)
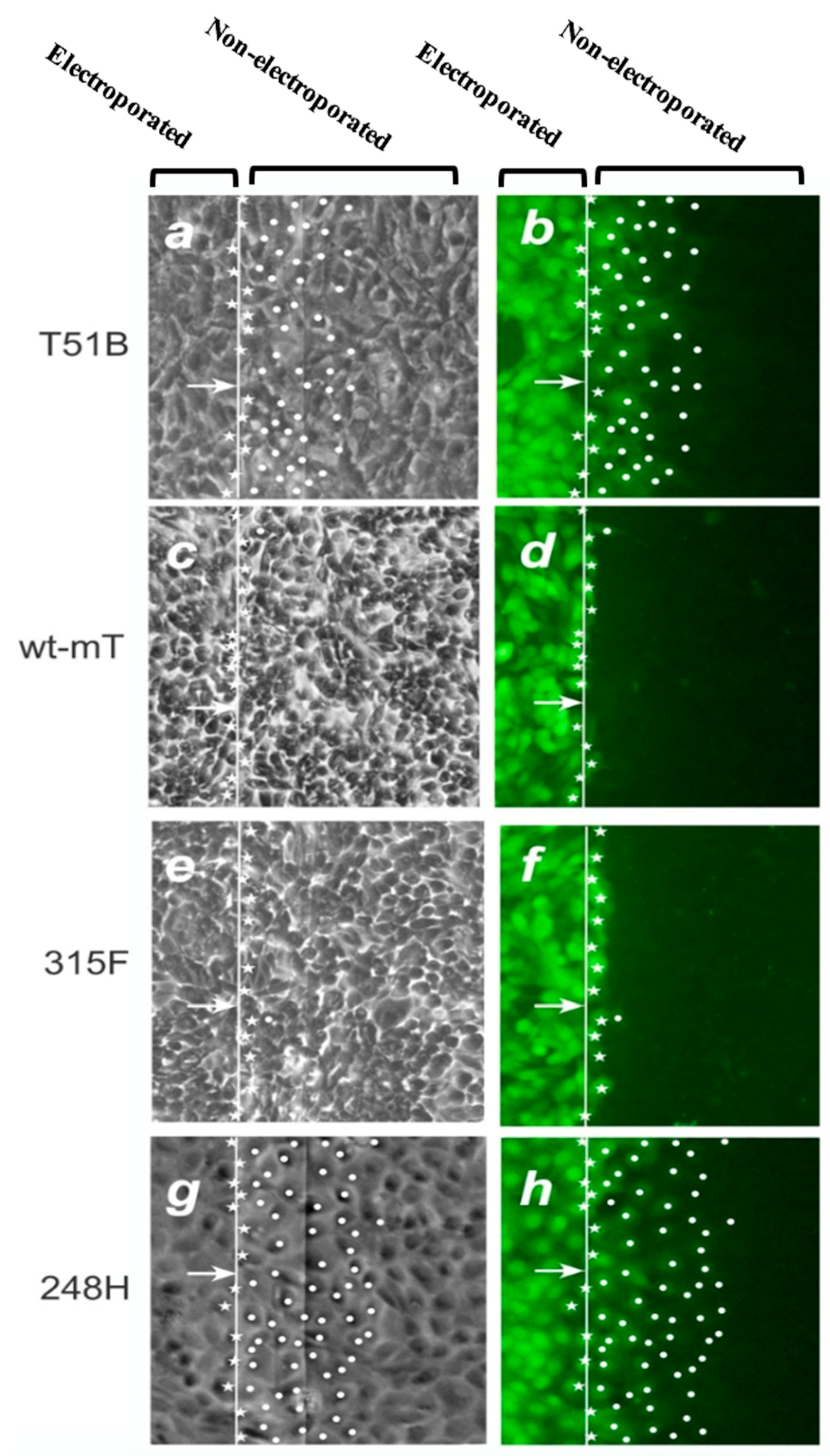
2.2. mT, PI3k and GJIC
2.3. PI3k Mutations, Membrane Translocation and GJIC
3. Stat3 as a Positive Regulator of Gap Junctional Communication
3.1. The Signal Transducer and Activator of Transcription-3 (Stat3)
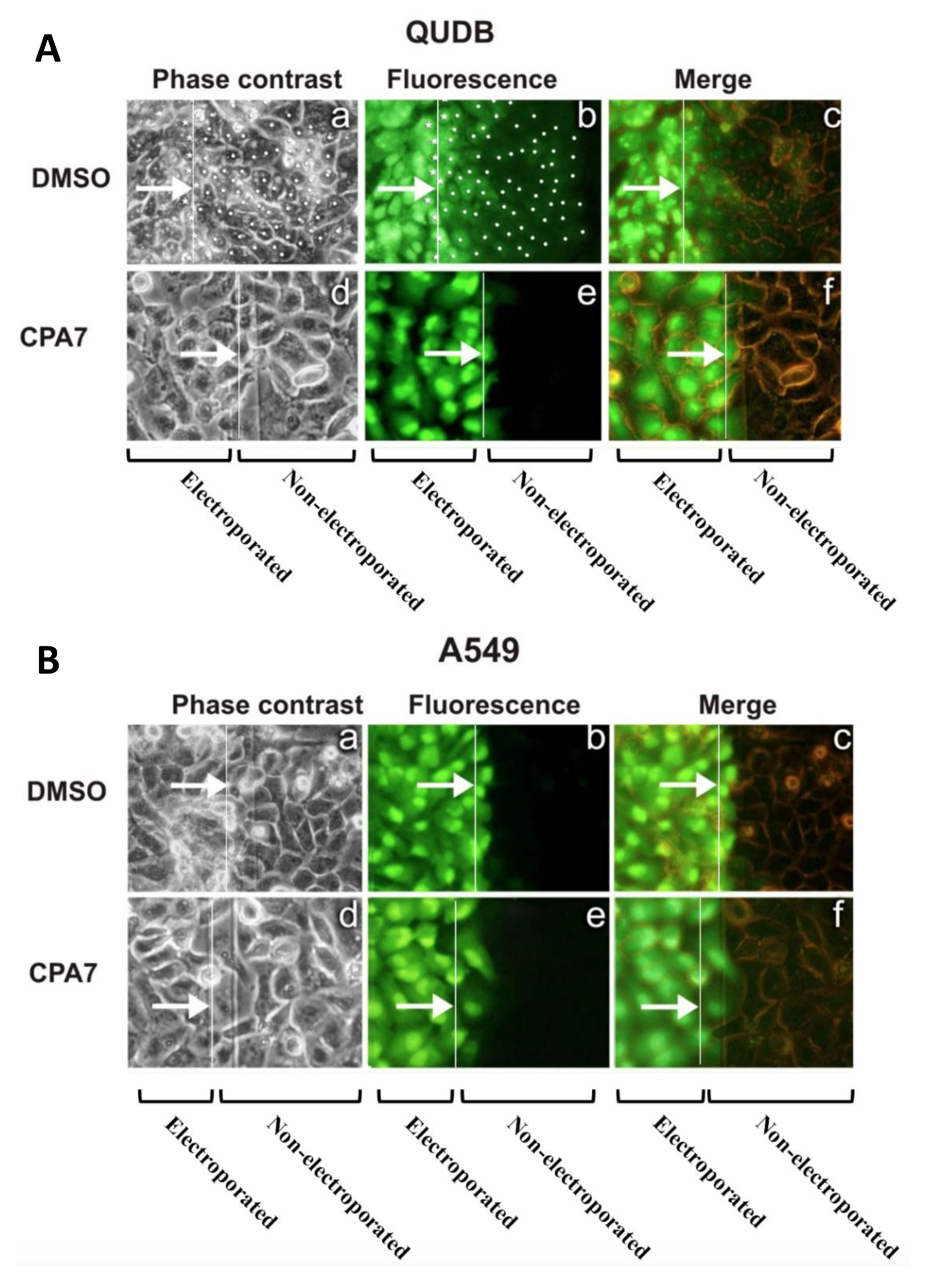
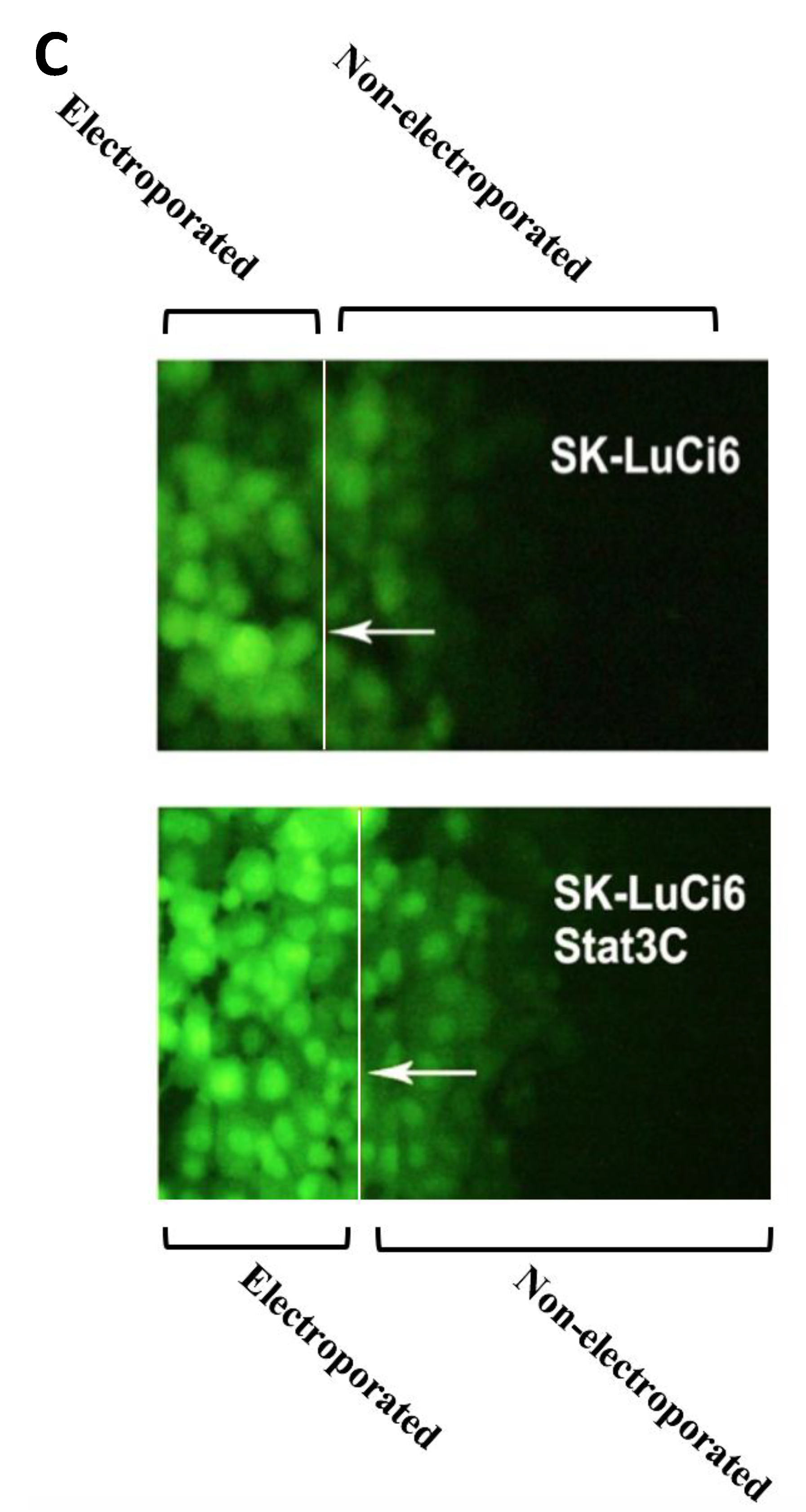
3.2. Stat3 Inhibition Eliminates GJIC While Stat3C Increases GJIC
4. Discussion
5. Conclusions
Funding
Acknowledgements
Conflicts of Interest
Abbreviations
| Erk1/2 | Extracellular signal regulated kinase 1 and 2 |
| GJIC | Gap junctional, intercellular communication |
| ITO | indium Tin Oxide |
| LY | Lucifer yellow |
| mT | middle tumor antigen of polyoma virus |
| myr-p110 | myristylated, p110 catalytic subunit of PI3k |
| PI3k | Phosphatidylinositol-3 kinase |
| p110 | catalytic subunit of PI3k |
| PDK1 | phosphoinositide-dependent kinase-1 |
| PH | plekstrin-homology |
| PKC | Protein kinase C |
| PLCγ | Phospholipase-C gamma |
| PTB | phosphotyrosine binding domain |
| Ras | Rat sarcoma |
| Ser | serine (or S: serine) |
| SH2 | Src-homology-2 domain |
| SH3 | Src-homology-3 domain |
| Shc | Src-homology-2 domain-containing transforming protein |
| TORC2 | Target of Rapamycin complex 2. |
| Tyr | tyrosine (or Y: tyrosine) |
| wt | Wild-type |
References
- Nielsen, M.S.; Nygaard, A.L.; Sorgen, P.L.; Verma, V.; Delmar, M.; Holstein-Rathlou, N.H. Gap junctions. Compr. Physiol. 2012, 2, 1981–2035. [Google Scholar] [PubMed]
- Su, V.; Lau, A.F. Connexins: Mechanisms regulating protein levels and intercellular communication. FEBS Lett. 2014, 588, 1212–1220. [Google Scholar] [CrossRef] [PubMed]
- Loewenstein, W.R.; Kanno, Y. Intercellular communication and the control of tissue growth: Lack of communication between cancer cells. Nature 1966, 209, 1248–1249. [Google Scholar] [CrossRef] [PubMed]
- Geletu, M.; Arulanandam, R.; Greer, S.; Trotman-Grant, A.; Tomai, E.; Raptis, L. Stat3 is a positive regulator of gap junctional intercellular communication in cultured, human lung carcinoma cells. BMC Cancer 2012, 12, 605. [Google Scholar] [CrossRef] [PubMed]
- Tomai, E.; Brownell, H.L.; Tufescu, T.; Reid, K.; Raptis, L. Gap junctional communication in lung carcinoma cells. Lung Cancer 1999, 23, 223–231. [Google Scholar] [CrossRef]
- Vinken, M.; Vanhaecke, T.; Papeleu, P.; Snykers, S.; Henkens, T.; Rogiers, V. Connexins and their channels in cell growth and cell death. Cell Signal. 2006, 18, 592–600. [Google Scholar] [CrossRef] [PubMed]
- Cronier, L.; Crespin, S.; Strale, P.O.; Defamie, N.; Mesnil, M. Gap junctions and cancer: New functions for an old story. Antioxid. Redox Signal. 2009, 11, 323–338. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.P.; Fan, Y.; Hossain, M.Z.; Peng, A.; Zeng, Z.L.; Boynton, A.L. Reversion of the neoplastic phenotype of human glioblastoma cells by connexin 43 (cx43). Cancer Res. 1998, 58, 5089–5096. [Google Scholar]
- Moorby, C.; Patel, M. Dual functions for connexins: Cx43 regulates growth independently of gap junction formation. Exp. Cell Res. 2001, 271, 238–248. [Google Scholar] [CrossRef]
- Zhang, Y.W.; Kaneda, M.; Morita, I. The gap junction-independent tumor-suppressing effect of connexin 43. J. Biol. Chem. 2003, 278, 44852–44856. [Google Scholar] [CrossRef]
- Fu, C.T.; Bechberger, J.F.; Ozog, M.A.; Perbal, B.; Naus, C.C. Ccn3 (nov) interacts with connexin43 in c6 glioma cells: Possible mechanism of connexin-mediated growth suppression. J. Biol. Chem. 2004, 279, 36943–36950. [Google Scholar] [CrossRef] [PubMed]
- Gellhaus, A.; Dong, X.; Propson, S.; Maass, K.; Klein-Hitpass, L.; Kibschull, M.; Traub, O.; Willecke, K.; Perbal, B.; Lye, S.J.; et al. Connexin43 interacts with nov: A possible mechanism for negative regulation of cell growth in choriocarcinoma cells. J. Biol. Chem. 2004, 279, 36931–36942. [Google Scholar] [CrossRef] [PubMed]
- Pahujaa, M.; Anikin, M.; Goldberg, G.S. Phosphorylation of connexin43 induced by src: Regulation of gap junctional communication between transformed cells. Exp. Cell Res. 2007, 313, 4083–4090. [Google Scholar] [CrossRef] [PubMed]
- Brownell, H.L.; Narsimhan, R.; Corbley, M.J.; Mann, V.M.; Whitfield, J.F.; Raptis, L. Ras is involved in gap junction closure in mouse fibroblasts or preadipocytes but not in differentiated adipocytes. DNA Cell Biol. 1996, 15, 443–451. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, M.M.; Sheridan, J.D. Altered junctional permeability between cells transformed by v-ras, v-mos, or v-src. Am. J. Physiol. 1988, 255, C674–C683. [Google Scholar] [CrossRef] [PubMed]
- Brownell, H.L.; Whitfield, J.F.; Raptis, L. Cellular ras partly mediates gap junction closure by the polyoma virus middle tumor antigen. Cancer Lett. 1996, 103, 99–106. [Google Scholar] [CrossRef]
- Brownell, H.L.; Whitfield, J.F.; Raptis, L. Elimination of intercellular junctional communication requires lower rasleu61 levels than stimulation of anchorage-independent proliferation. Cancer Detect. Prev. 1997, 21, 289–294. [Google Scholar]
- Grammatikakis, N.; Vultur, A.; Ramana, C.V.; Siganou, A.; Schweinfest, C.W.; Raptis, L. The role of hsp90n, a new member of the hsp90 family, in signal transduction and neoplastic transformation. J. Biol. Chem. 2002, 277, 8312–8320. [Google Scholar] [CrossRef]
- Geletu, M.; Trotman-Grant, A.; Raptis, L. Mind the gap; regulation of gap junctional, intercellular communication by the src oncogene product and its effectors. Anticancer Res. 2012, 32, 4245–4250. [Google Scholar]
- Crow, D.S.; Beyer, E.C.; Paul, D.L.; Kobe, S.S.; Lau, A.F. Phosphorylation of connexin43 gap junction protein in uninfected and rous sarcoma virus-transformed mammalian fibroblasts. Mol. Cell. Biol. 1990, 10, 1754–1763. [Google Scholar] [CrossRef]
- Lin, R.; Warn-Cramer, B.J.; Kurata, W.E.; Lau, A.F. V-src phosphorylation of connexin 43 on tyr247 and tyr265 disrupts gap junctional communication. J. Cell Biol. 2001, 154, 815–827. [Google Scholar] [CrossRef] [PubMed]
- Swenson, L.I.; Piwnica-Worms, H.; McNamee, H.; Paul, D.L. Tyrosine phosphorylation of the gap junction protein connexin43 is required for the pp60v-src-induced inhibition of communication. Cell Regul. 1990, 1, 989–1002. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Kasperek, E.M.; Nicholson, B.J. Dissection of the molecular basis of pp60(v-src) induced gating of connexin 43 gap junction channels. J. Cell Biol. 1999, 144, 1033–1045. [Google Scholar] [CrossRef] [PubMed]
- Lin, R.; Martyn, K.D.; Guyette, C.V.; Lau, A.F.; Warn-Cramer, B.J. V-src tyrosine phosphorylation of connexin43: Regulation of gap junction communication and effects on cell transformation. Cell Commun. Adhes. 2006, 13, 199–216. [Google Scholar] [CrossRef] [PubMed]
- Mitra, S.S.; Xu, J.; Nicholson, B.J. Coregulation of multiple signalling mechanisms in pp60v-src-induced closure of cx43 gap junction channels. J. Membr. Biol. 2012, 245, 495–506. [Google Scholar] [CrossRef] [PubMed]
- Solan, J.L.; Lampe, P.D. Specific cx43 phosphorylation events regulate gap junction turnover in vivo. FEBS Lett. 2014, 588, 1423–1429. [Google Scholar] [CrossRef]
- Axelsen, L.N.; Calloe, K.; Holstein-Rathlou, N.H.; Nielsen, M.S. Managing the complexity of communication: Regulation of gap junctions by post-translational modification. Front. Pharmacol. 2013, 4, 130. [Google Scholar] [CrossRef]
- Govindarajan, R.; Chakraborty, S.; Johnson, K.E.; Falk, M.M.; Wheelock, M.J.; Johnson, K.R.; Mehta, P.P. Assembly of connexin43 into gap junctions is regulated differentially by e-cadherin and n-cadherin in rat liver epithelial cells. Mol. Biol. Cell 2010, 21, 4089–4107. [Google Scholar] [CrossRef]
- Kovacs, E.M.; Ali, R.G.; McCormack, A.J.; Yap, A.S. E-cadherin homophilic ligation directly signals through rac and phosphatidylinositol 3-kinase to regulate adhesive contacts. J. Biol. Chem. 2002, 277, 6708–6718. [Google Scholar] [CrossRef]
- Geletu, M.; Guy, S.; Arulanandam, R.; Feracci, H.; Raptis, L. Engaged for survival; from cadherin ligation to stat3 activation. JAKS-STAT 2013, 2, e27361–e27363. [Google Scholar] [CrossRef]
- Geletu, M.; Guy, S.; Greer, S.; Raptis, L. Differential effects of polyoma virus middle tumor antigen mutants upon gap junctional, intercellular communication. Exp. Cell Res. 2015, 336, 223–231. [Google Scholar] [CrossRef] [PubMed]
- Geletu, M.; Chaize, C.; Arulanandam, R.; Vultur, A.; Kowolik, C.; Anagnostopoulou, A.; Jove, R.; Raptis, L. Stat3 activity is required for gap junctional permeability in normal epithelial cells and fibroblasts. DNA Cell Biol. 2009, 28, 319–327. [Google Scholar] [CrossRef] [PubMed]
- Whitman, M.; Downes, C.P.; Keeler, M.; Keller, T.; Cantley, L. Type i phosphatidylinositol kinase makes a novel inositol phospholipid, phosphatidylinositol-3-phosphate. Nature 1988, 332, 644–646. [Google Scholar] [CrossRef] [PubMed]
- Utermark, T.; Schaffhausen, B.S.; Roberts, T.M.; Zhao, J.J. The p110alpha isoform of phosphatidylinositol 3-kinase is essential for polyomavirus middle T antigen-mediated transformation. J. Virol. 2007, 81, 7069–7076. [Google Scholar] [CrossRef] [PubMed]
- Webster, M.A.; Hutchinson, J.N.; Rauh, M.J.; Muthuswamy, S.K.; Anton, M.; Tortorice, C.G.; Cardiff, R.D.; Graham, F.L.; Hassell, J.A.; Muller, W.J. Requirement for both shc and phosphatidylinositol 3′ kinase signalling pathways in polyomavirus middle t-mediated mammary tumorigenesis. Mol. Cell. Biol. 1998, 18, 2344–2359. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Knapp, S.; Ahmed, A.A. The structural basis of pi3k cancer mutations: From mechanism to therapy. Cancer Res. 2014, 74, 641–646. [Google Scholar] [CrossRef] [PubMed]
- Manning, B.D.; Cantley, L.C. Akt/pkb signalling: Navigating downstream. Cell 2007, 129, 1261–1274. [Google Scholar] [CrossRef]
- Franke, T.; Kaplan, D.R.; Cantley, L.C. Pi3k:Downstream aktion blocks apoptosis. Cell 1997, 88, 435–437. [Google Scholar] [CrossRef]
- Franke, T.F.; Cantley, L.C. Apoptosis. A bad kinase makes good. Nature 1997, 390, 116–117. [Google Scholar] [CrossRef]
- Marcotte, R.; Muller, W.J. Signal transduction in transgenic mouse models of human breast cancer--implications for human breast cancer. J. Mammary Gland. Biol. Neoplasia 2008, 13, 323–335. [Google Scholar] [CrossRef]
- Dilworth, S.M. Polyoma virus middle t antigen and its role in identifying cancer-related molecules. Nat. Rev. Cancer 2002, 2, 951–956. [Google Scholar] [CrossRef] [PubMed]
- Ichaso, N.; Dilworth, S.M. Cell transformation by the middle t-antigen of polyoma virus. Oncogene 2001, 20, 7908–7916. [Google Scholar] [CrossRef] [PubMed]
- Campbell, K.S.; Ogris, E.; Burke, B.; Su, W.; Auger, K.R.; Druker, B.J.; Schaffhausen, B.S.; Roberts, T.M.; Pallas, D.C. Polyoma middle tumor antigen interacts with shc protein via the npty (asn-pro-thr-tyr) motif in middle tumor antigen. Proc. Nat. Acad. Sci. USA 1994, 91, 6344–6348. [Google Scholar] [CrossRef] [PubMed]
- Dilworth, S.M.; Brewster, C.E.; Jones, M.D.; Lanfrancone, L.; Pelicci, G.; Pelicci, P.G. Transformation by polyoma virus middle t-antigen involves the binding and tyrosine phosphorylation of shc. Nature 1994, 367, 87–90. [Google Scholar] [CrossRef]
- Courtneidge, S.A.; Heber, A. An 81 kd protein complexed with middle t antigen and pp60c-src: A possible phosphatidylinositol kinase. Cell 1987, 50, 1031–1037. [Google Scholar] [CrossRef]
- Kaplan, D.R.; Whitman, M.; Schaffhausen, B.S.; Pallas, D.C.; White, M.; Cantley, L.; Roberts, T.M. Common elements in growth factor stimulation and oncogenic transformation: 85 kd phosphoprotein and phosphatidylinositol kinase activity. Cell 1987, 50, 1021–1029. [Google Scholar] [CrossRef]
- Whitman, M.; Kaplan, D.R.; Schaffhausen, B.S.; Cantley, L.; Roberts, T.M. Association of phosphatidylinositol kinase activity with polyoma mt competent for transformation. Nature 1985, 315, 239–242. [Google Scholar] [CrossRef]
- Talmage, D.A.; Freund, R.; Young, A.T.; Dahl, J.; Dawe, C.J.; Benjamin, T.L. Phosphorylation of middle t by pp60c-src: A switch for binding of phosphatidylinositol 3-kinase and optimal tumorigenesis. Cell 1989, 59, 55–65. [Google Scholar] [CrossRef]
- Yoakim, M.; Hou, W.; Liu, Y.; Carpenter, C.L.; Kapeller, R.; Schaffhausen, B.S. Interactions of polyomavirus middle t with the sh2 domains of the pp85 subunit of phosphatidylinositol-3-kinase. J. Virol. 1992, 66, 5485–5491. [Google Scholar]
- Songyang, Z.; Shoelson, S.E.; Chaudhuri, M.; Gish, G.; Pawson, T.; Haser, W.G.; King, F.; Roberts, T.; Ratnofsky, S.; Lechleider, R.J.; et al. Sh2 domains recognize specific phosphopeptide sequences. Cell 1993, 72, 767–778. [Google Scholar] [PubMed]
- Kaplan, D.R.; Whitman, M.; Schaffhausen, B.; Raptis, L.; Garcea, R.L.; Pallas, D.; Roberts, T.M.; Cantley, L. Phosphatidylinositol metabolism and polyoma-mediated transformation. Proc. Nat. Acad. Sci. USA 1986, 83, 3624–3628. [Google Scholar] [CrossRef] [PubMed]
- Smolar, N.; Griffin, B.E. DNA sequences of polyoma virus early deletion mutants. J. Virol. 1981, 38, 958–967. [Google Scholar] [PubMed]
- Raptis, L.; Brownell, H.L.; Firth, K.L.; MacKenzie, L.W. A novel technique for the study of intercellular, junctional communication; electroporation of adherent cells on a partly conductive slide. DNA Cell Biol. 1994, 13, 963–975. [Google Scholar] [CrossRef] [PubMed]
- Azarnia, R.; Loewenstein, W.R. Polyomavirus middle t antigen downregulates junctional cell-to-cell communication. Mol. Cell. Biol 1987, 7, 946–950. [Google Scholar] [CrossRef] [PubMed]
- Markland, W.; Cheng, S.H.; Oostra, B.A.; Smith, A.E. In vitro mutagenesis of the putative membrane-binding domain of polyoma virus middle t antigen. J. Virol. 1986, 59, 82–89. [Google Scholar] [PubMed]
- Druker, B.J.; Sibert, L.; Roberts, T.M. Polyomavirus middle t-antigen npty mutants. J. Virol. 1992, 66, 5770–5776. [Google Scholar] [PubMed]
- Hong, Y.K.; Mikami, A.; Schaffhausen, B.; Jun, T.; Roberts, T.M. A new class of mutations reveals a novel function for the original phosphatidylinositol 3-kinase binding site. Proc. Natl. Acad. Sci. USA 2003, 100, 9434–9439. [Google Scholar] [CrossRef] [PubMed]
- Royal, I.; Gourdeau, H.; Blouin, R.; Marceau, N. Down-regulation of cytokeratin 14 mrna in polyoma virus middle t- transformed rat liver epithelial cells. Cell Growth Differ. 1992, 3, 589–596. [Google Scholar] [PubMed]
- Geletu, M.; Guy, S.; Firth, K.; Raptis, L. A functional assay for gap junctional examination; electroporation of adherent cells on indium-tin oxide. J. Vis. Exp. 2014, 92, e51710. [Google Scholar] [CrossRef] [PubMed]
- Auger, K.R.; Wang, J.; Narsimhan, R.P.; Holcombe, T.; Roberts, T.M. Constitutive cellular expression of pi 3-kinase is distinct from transient expression. Biochem. Biophys. Res. Commun. 2000, 272, 822–829. [Google Scholar] [CrossRef] [PubMed]
- Dunn, C.A.; Su, V.; Lau, A.F.; Lampe, P.D. Activation of akt, not connexin 43 protein ubiquitination, regulates gap junction stability. J. Biol. Chem. 2012, 287, 2600–2607. [Google Scholar] [CrossRef] [PubMed]
- Su, V.; Lau, A.F. Ubiquitination, intracellular trafficking, and degradation of connexins. Arch. Biochem. Biophys. 2012, 524, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Su, V.; Cochrane, K.; Lau, A.F. Degradation of connexins through the proteasomal, endolysosomal and phagolysosomal pathways. J. Membr. Biol. 2012, 245, 389–400. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharjee, R.; Kaneda, M.; Nakahama, K.; Morita, I. The steady-state expression of connexin43 is maintained by the pi3k/akt in osteoblasts. Biochem. Biophys. Res. Commun. 2009, 382, 440–444. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Hyodo, T.; Hasegawa, H.; Yuan, H.; Hamaguchi, M.; Senga, T. Pi3k/akt signalling is involved in the disruption of gap junctional communication caused by v-src and tnf-alpha. Biochem. Biophys. Res. Commun. 2010, 400, 230–235. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Fang, Z.; Wang, G.; Wu, S. Gap junctional intercellular communication dysfunction mediates the cognitive impairment induced by cerebral ischemia-reperfusion injury: Pi3k/akt pathway involved. Am. J. Transl. Res. 2017, 9, 5442–5451. [Google Scholar] [PubMed]
- Martinez, J.M.; Wang, H.Z.; Lin, R.Z.; Brink, P.R.; White, T.W. Differential regulation of connexin50 and connexin46 by pi3k signalling. FEBS Lett. 2015, 589, 1340–1345. [Google Scholar] [CrossRef]
- Xia, X.; Batra, N.; Shi, Q.; Bonewald, L.F.; Sprague, E.; Jiang, J.X. Prostaglandin promotion of osteocyte gap junction function through transcriptional regulation of connexin 43 by glycogen synthase kinase 3/beta-catenin signalling. Mol. Cell. Biol. 2010, 30, 206–219. [Google Scholar] [CrossRef]
- Yu, H.; Pardoll, D.; Jove, R. Stats in cancer inflammation and immunity: A leading role for stat3. Nat. Rev. Cancer 2009, 9, 798–809. [Google Scholar] [CrossRef]
- Niu, G.; Wright, K.L.; Ma, Y.; Wright, G.M.; Huang, M.; Irby, R.; Briggs, J.; Karras, J.; Cress, W.D.; Pardoll, D.; et al. Role of stat3 in regulating p53 expression and function. Mol. Cell. Biol. 2005, 25, 7432–7440. [Google Scholar] [CrossRef]
- Gritsko, T.; Williams, A.; Turkson, J.; Kaneko, S.; Bowman, T.; Huang, M.; Nam, S.; Eweis, I.; Diaz, N.; Sullivan, D.; et al. Persistent activation of stat3 signalling induces survivin gene expression and confers resistance to apoptosis in human breast cancer cells. Clin. Cancer Res. 2006, 12, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Demaria, M.; Poli, V. From the nucleus to the mitochondria and back: The odyssey of a multitask stat3. Cell Cycle 2011, 10, 3221–3222. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Wang, H.; Yang, J.J.; Liu, X.; Liu, Z.R. Pyruvate kinase m2 regulates gene transcription by acting as a protein kinase. Mol. Cell 2012, 45, 598–609. [Google Scholar] [CrossRef] [PubMed]
- Turkson, J.; Bowman, T.; Adnane, J.; Zhang, Y.; Djeu, J.Y.; Sekharam, M.; Frank, D.A.; Holzman, L.B.; Wu, J.; Sebti, S.; et al. Requirement for ras/rac1-mediated p38 and c-jun n-terminal kinase signalling in stat3 transcriptional activity induced by the src oncoprotein. Mol. Cell. Biol. 1999, 19, 7519–7528. [Google Scholar] [CrossRef] [PubMed]
- Wegrzyn, J.; Potla, R.; Chwae, Y.J.; Sepuri, N.B.; Zhang, Q.; Koeck, T.; Derecka, M.; Szczepanek, K.; Szelag, M.; Gornicka, A.; et al. Function of mitochondrial stat3 in cellular respiration. Science 2009, 323, 793–797. [Google Scholar] [CrossRef] [PubMed]
- Gough, D.J.; Corlett, A.; Schlessinger, K.; Wegrzyn, J.; Larner, A.C.; Levy, D.E. Mitochondrial stat3 supports ras-dependent oncogenic transformation. Science 2009, 324, 1713–1716. [Google Scholar] [CrossRef]
- Banerjee, K.; Resat, H. Constitutive activation of stat3 in breast cancer cells: A review. Int J. Cancer 2016, 138, 2570–2578. [Google Scholar] [CrossRef]
- Turkson, J.; Bowman, T.; Garcia, R.; Caldenhoven, E.; de Groot, R.P.; Jove, R. Stat3 activation by src induces specific gene regulation and is required for cell transformation. Mol. Cell. Biol. 1998, 18, 2545–2552. [Google Scholar] [CrossRef]
- Bromberg, J.F.; Horvath, C.M.; Besser, D.; Lathem, W.W.; Darnell, J.E., Jr. Stat3 activation is required for cellular transformation by v-src. Mol. Cell. Biol. 1998, 18, 2553–2558. [Google Scholar] [CrossRef]
- Bromberg, J.F.; Wrzeszczynska, M.H.; Devgan, G.; Zhao, Y.; Pestell, R.G.; Albanese, C.; Darnell, J.E., Jr. Stat3 as an oncogene. Cell 1999, 98, 295–303. [Google Scholar] [CrossRef]
- Vultur, A.; Cao, J.; Arulanandam, R.; Turkson, J.; Jove, R.; Greer, P.; Craig, A.; Elliott, B.E.; Raptis, L. Cell to cell adhesion modulates stat3 activity in normal and breast carcinoma cells. Oncogene 2004, 23, 2600–2616. [Google Scholar] [CrossRef]
- Vultur, A.; Arulanandam, R.; Turkson, J.; Niu, G.; Jove, R.; Raptis, L. Stat3 is required for full neoplastic transformation by the simian virus 40 large tumor antigen. Mol. Biol. Cell 2005, 16, 3832–3846. [Google Scholar] [CrossRef]
- Su, H.W.; Yeh, H.H.; Wang, S.W.; Shen, M.R.; Chen, T.L.; Kiela, P.R.; Ghishan, F.K.; Tang, M.J. Cell confluence-induced activation of signal transducer and activator of transcription-3 (stat3) triggers epithelial dome formation via augmentation of sodium hydrogen exchanger-3 (nhe3) expression. J. Biol. Chem. 2007, 282, 9883–9894. [Google Scholar] [CrossRef]
- Kreis, S.; Munz, G.A.; Haan, S.; Heinrich, P.C.; Behrmann, I. Cell density dependent increase of constitutive signal transducers and activators of transcription 3 activity in melanoma cells is mediated by janus kinases. Mol. Cancer Res. 2007, 5, 1331–1341. [Google Scholar] [CrossRef]
- Onishi, A.; Chen, Q.; Humtsoe, J.O.; Kramer, R.H. Stat3 signalling is induced by intercellular adhesion in squamous cell carcinoma cells. Exp. Cell Res. 2008, 314, 377–386. [Google Scholar] [CrossRef]
- Arulanandam, R.; Vultur, A.; Cao, J.; Carefoot, E.; Truesdell, P.; Elliott, B.; Larue, L.; Feracci, H.; Raptis, L. Cadherin-cadherin engagement promotes survival via rac/cdc42 and stat3. Mol. Cancer Res. 2009, 17, 1310–1327. [Google Scholar] [CrossRef]
- Geletu, M.; Arulanandam, R.; Chevalier, S.; Saez, B.; Larue, L.; Feracci, H.; Raptis, L. Classical cadherins control survival through the gp130/stat3 axis. BBA-Mol. Cell Res. 2013, 1833, 1947–1959. [Google Scholar] [CrossRef]
- Raptis, L.; Arulanandam, R.; Vultur, A.; Geletu, M.; Chevalier, S.; Feracci, H. Beyond structure, to survival: Stat3 activation by cadherin engagement. Biochem. Cell Biol. 2009, 87, 835–843. [Google Scholar] [CrossRef]
- Arulanandam, R.; Geletu, M.; Feracci, H.; Raptis, L. Racv12 requires gp130 for stat3 activation, cell proliferation and migration. Exp. Cell Res. 2010, 316, 875–886. [Google Scholar] [CrossRef]
- Noren, N.K.; Niessen, C.M.; Gumbiner, B.M.; Burridge, K. Cadherin engagement regulates rho family gtpases. J. Biol. Chem. 2001, 276, 33305–33308. [Google Scholar] [CrossRef]
- Charrasse, S.; Comunale, F.; Fortier, M.; Portales-Casamar, E.; Debant, A.; Gauthier-Rouviere, C. M-cadherin activates rac1 gtpase through the rho-gef trio during myoblast fusion. Mol. Biol. Cell 2007, 18, 1734–1743. [Google Scholar] [CrossRef]
- Anagnostopoulou, A.; Vultur, A.; Arulanandam, R.; Cao, J.; Turkson, J.; Jove, R.; Kim, J.S.; Glenn, M.; Hamilton, A.D.; Raptis, L. Differential effects of stat3 inhibition in sparse vs confluent normal and breast cancer cells. Cancer Lett. 2006, 242, 120–132. [Google Scholar] [CrossRef]
- von Manstein, V.; Groner, B. Tumor cell resistance against targeted therapeutics: The density of cultured glioma tumor cells enhances stat3 activity and offers protection against the tyrosine kinase inhibitor canertinib. Medchemcomm 2017, 8, 96–102. [Google Scholar] [CrossRef]
- Ito, S.; Ito, Y.; Senga, T.; Hattori, S.; Matsuo, S.; Hamaguchi, M. V-src requires ras signalling for the suppression of gap junctional intercellular communication. Oncogene 2006, 25, 2420–2424. [Google Scholar] [CrossRef]
- Turkson, J.; Zhang, S.; Palmer, J.; Kay, H.; Stanko, J.; Mora, L.B.; Sebti, S.; Yu, H.; Jove, R. Inhibition of constitutive signal transducer and activator of transcription 3 activation by novel platinum complexes with potent antitumor activity. Mol. Cancer Ther. 2004, 3, 1533–1542. [Google Scholar]
- Beardslee, M.A.; Laing, J.G.; Beyer, E.C.; Saffitz, J.E. Rapid turnover of connexin43 in the adult rat heart. Circ. Res. 1998, 83, 629–635. [Google Scholar] [CrossRef]
- Geletu, M.; Guy, S.; Raptis, L. Effects of src and stat3 upon gap junctional, intercellular communication in lung cancer lines. Anticancer Res. 2013, 33, 4401–4410. [Google Scholar]
- Lampe, P.D.; Lau, A.F. The effects of connexin phosphorylation on gap junctional communication. Int. J. Biochem. Cell Biol. 2004, 36, 1171–1186. [Google Scholar] [CrossRef]
- Warn-Cramer, B.J.; Lau, A.F. Regulation of gap junctions by tyrosine protein kinases. Biochim. Biophys. Acta 2004, 1662, 81–95. [Google Scholar] [CrossRef]
- Griner, E.M.; Kazanietz, M.G. Protein kinase c and other diacylglycerol effectors in cancer. Nat. Rev. Cancer 2007, 7, 281–294. [Google Scholar] [CrossRef]
- Shah, M.M.; Martinez, A.M.; Fletcher, W.H. The connexin43 gap junction protein is phosphorylated by protein kinase a and protein kinase c: In vivo and in vitro studies. Mol. Cell Biochem. 2002, 238, 57–68. [Google Scholar] [CrossRef] [PubMed]
- Bao, X.; Reuss, L.; Altenberg, G.A. Regulation of purified and reconstituted connexin 43 hemichannels by protein kinase c-mediated phosphorylation of serine 368. J. Biol. Chem. 2004, 279, 20058–20066. [Google Scholar] [CrossRef] [PubMed]
- Lampe, P.D.; TenBroek, E.M.; Burt, J.M.; Kurata, W.E.; Johnson, R.G.; Lau, A.F. Phosphorylation of connexin43 on serine368 by protein kinase c regulates gap junctional communication. J. Cell Biol. 2000, 149, 1503–1512. [Google Scholar] [CrossRef] [PubMed]
- Royal, I.; Raptis, L.; Druker, B.J.; Marceau, N. Downregulation of cytokeratin 14 gene expression by the polyoma virus middle t antigen is dependent on c-src association but independent of full transformation in rat liver nonparenchymal epithelial cells. Cell Growth Differ. 1996, 7, 737–743. [Google Scholar]
- Theiss, C.; Mazur, A.; Meller, K.; Mannherz, H.G. Changes in gap junction organization and decreased coupling during induced apoptosis in lens epithelial and nih-3t3 cells. Exp. Cell Res. 2007, 313, 38–52. [Google Scholar] [CrossRef]
- Datta, S.R.; Ranger, A.M.; Lin, M.Z.; Sturgill, J.F.; Ma, Y.C.; Cowan, C.W.; Dikkes, P.; Korsmeyer, S.J.; Greenberg, M.E. Survival factor-mediated bad phosphorylation raises the mitochondrial threshold for apoptosis. Dev. Cell 2002, 3, 631–643. [Google Scholar] [CrossRef]
- Datta, S.R.; Dudek, H.; Tao, X.; Masters, S.; Fu, H.; Gotoh, Y.; Greenberg, M.E. Akt phosphorylation of bad couples survival signals to the cell-intrinsic death machinery. Cell 1997, 91, 231–241. [Google Scholar] [CrossRef]
- Yu, H.; Jove, R. The stats of cancer--new molecular targets come of age. Nat. Rev. Cancer 2004, 4, 97–105. [Google Scholar] [CrossRef]
- Sulkowska, U.; Febp, A.W.; Sulkowski, S. Association of stat3 with cx26 and cx43 in human uterine endometrioid adenocarcinoma. Oncol. Lett. 2016, 11, 4134–4138. [Google Scholar] [CrossRef]
- Young, A.P.; Nagarajan, R.; Longmore, G.D. Mechanisms of transcriptional regulation by rb-e2f segregate by biological pathway. Oncogene 2003, 22, 7209–7217. [Google Scholar] [CrossRef]
- Sears, R.C.; Nevins, J.R. Signaling networks that link cell proliferation and cell fate. J. Biol. Chem. 2002, 277, 11617–11620. [Google Scholar] [CrossRef] [PubMed]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Geletu, M.; Taha, Z.; Gunning, P.T.; Raptis, L. PI3k and Stat3: Oncogenes that are Required for Gap Junctional, Intercellular Communication. Cancers 2019, 11, 167. https://doi.org/10.3390/cancers11020167
Geletu M, Taha Z, Gunning PT, Raptis L. PI3k and Stat3: Oncogenes that are Required for Gap Junctional, Intercellular Communication. Cancers. 2019; 11(2):167. https://doi.org/10.3390/cancers11020167
Chicago/Turabian StyleGeletu, Mulu, Zaid Taha, Patrick T. Gunning, and Leda Raptis. 2019. "PI3k and Stat3: Oncogenes that are Required for Gap Junctional, Intercellular Communication" Cancers 11, no. 2: 167. https://doi.org/10.3390/cancers11020167
APA StyleGeletu, M., Taha, Z., Gunning, P. T., & Raptis, L. (2019). PI3k and Stat3: Oncogenes that are Required for Gap Junctional, Intercellular Communication. Cancers, 11(2), 167. https://doi.org/10.3390/cancers11020167