A Simplified Genomic Profiling Approach Predicts Outcome in Metastatic Colorectal Cancer


 ,
, 
 , , , , ,
, , , , ,  and
and
Abstract
:1. Introduction
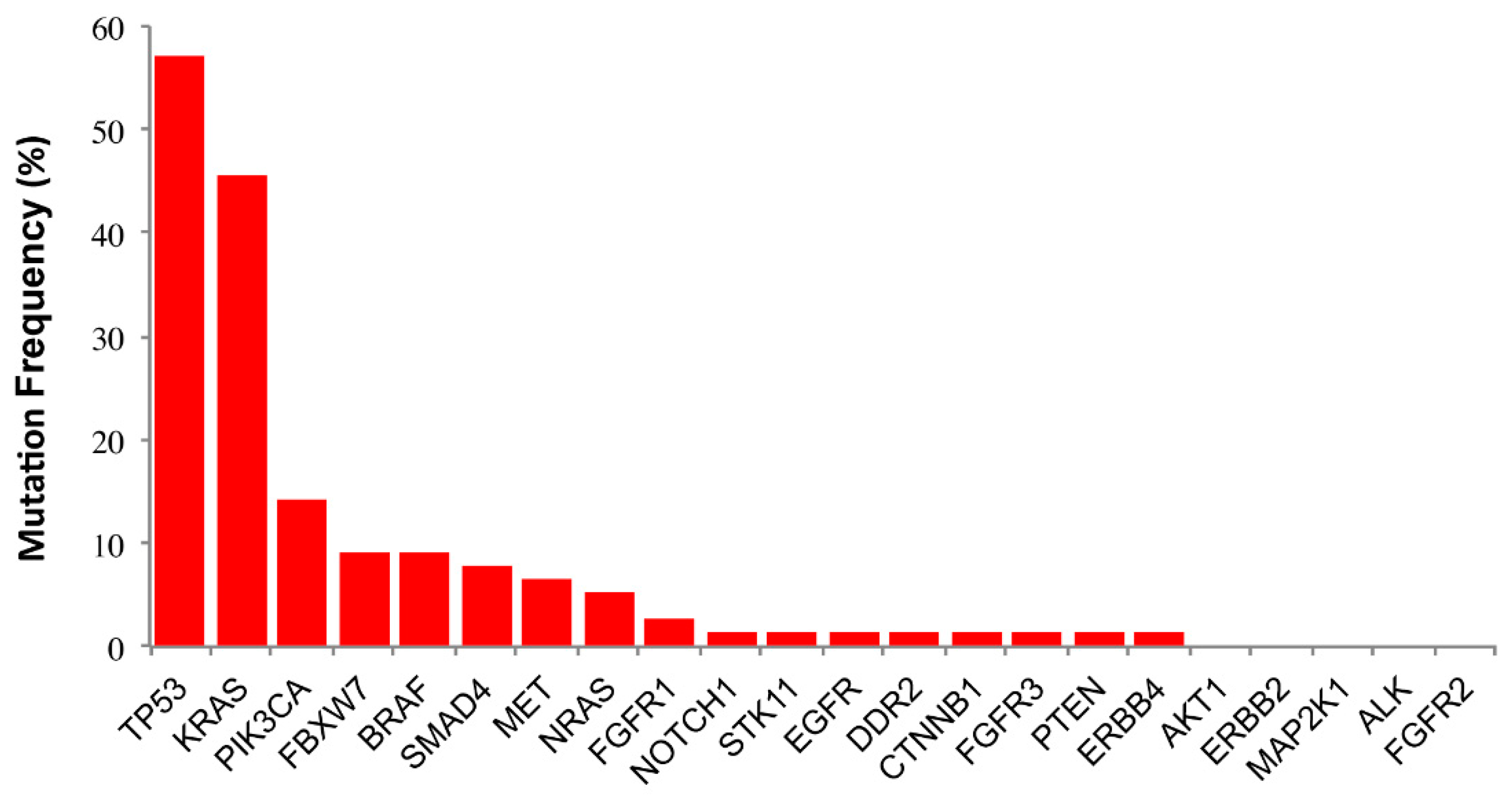
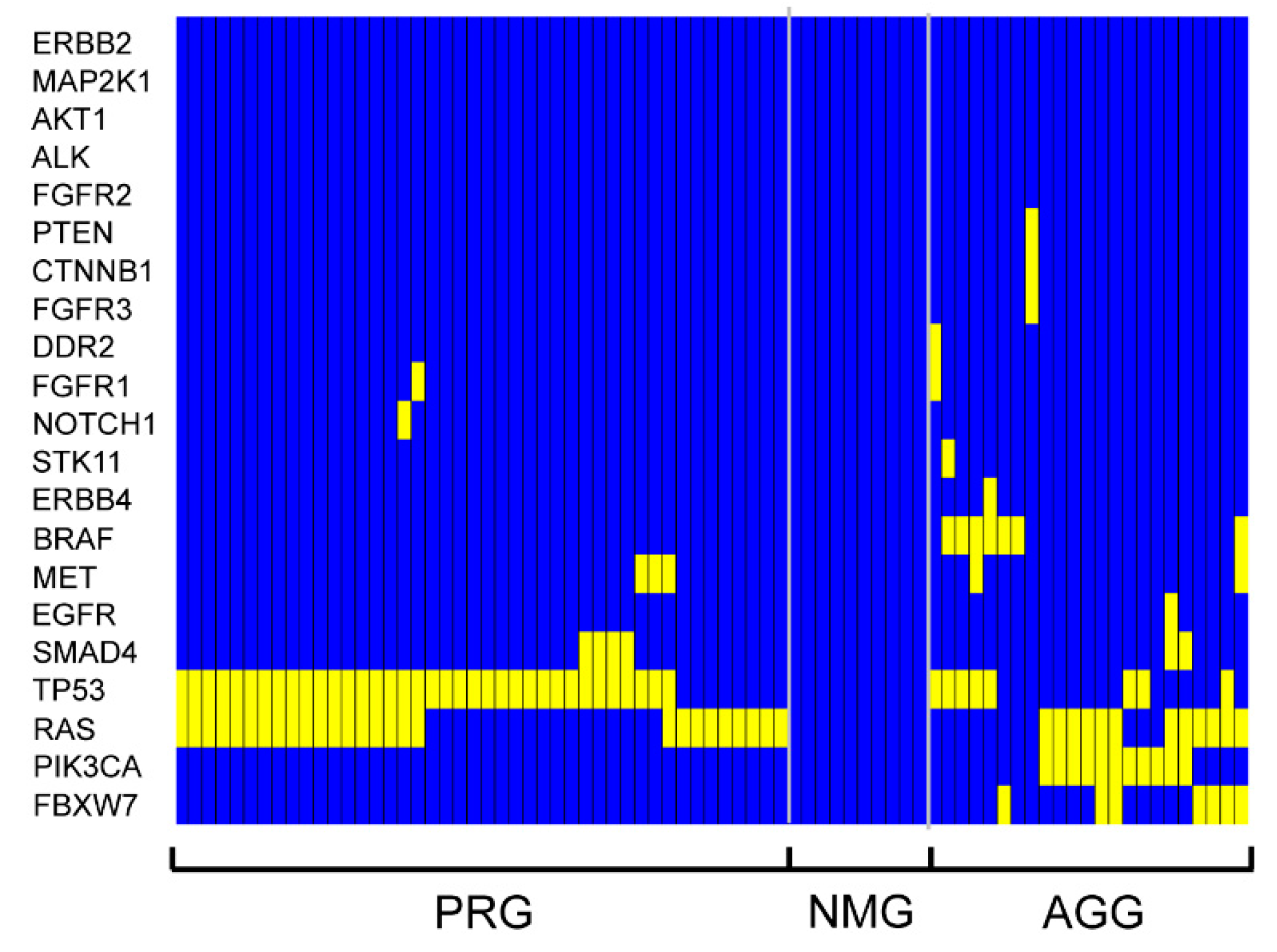
2. Results
3. Discussion
4. Patients and Methods
4.1. Patients and Data Collection
4.2. DNA Extraction
4.3. IT-PGM Sequencing and Variant Calling
4.4. Bioinformatic and Statistical Analysis
4.5. Determination of MSI-H phenotype
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fedewa, S.A.; Ahnen, D.J.; Meester, R.G.S.; Barzi, A.; Jemal, A. Colorectal cancer statistics. CA Cancer J. Clin. 2017, 67, 177–193. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef]
- Colucci, G.; Gebbia, V.; Paoletti, G.; Giuliani, F.; Caruso, M.; Gebbia, N.; Carteni, G.; Agostara, B.; Pezzella, G.; Manzione, L.; et al. Phase III randomized trial of FOLFIRI versus FOLFOX4 in the treatment of advanced colorectal cancer: A multicenter study of the Gruppo Oncologico Dell’Italia Meridionale. J. Clin. Oncol. 2005, 23, 4866–4875. [Google Scholar] [CrossRef] [PubMed]
- Tournigand, C.; Andre, T.; Achille, E.; Lledo, G.; Flesh, M.; Mery-Mignard, D.; Quinaux, E.; Couteau, C.; Buyse, M.; Ganem, G.; et al. FOLFIRI followed by FOLFOX6 or the reverse sequence in advanced colorectal cancer: A randomized GERCOR study. J. Clin. Oncol. 2004, 22, 229–237. [Google Scholar] [CrossRef]
- Golfinopoulos, V.; Salanti, G.; Pavlidis, N.; Ioannidis, J.P. Survival and disease-progression benefits with treatment regimens for advanced colorectal cancer: A meta-analysis. Lancet Oncol. 2007, 8, 898–911. [Google Scholar] [CrossRef]
- Bokemeyer, C.; Bondarenko, I.; Makhson, A.; Hartmann, J.T.; Aparicio, J.; de Braud, F.; Donea, S.; Ludwig, H.; Schuch, G.; Stroh, C.; et al. Fluorouracil, Leucovorin, and Oxaliplatin with and without cetuximab in the first-line treatment of metastatic colorectal cancer. J. Clin. Oncol. 2009, 27, 663–671. [Google Scholar] [CrossRef]
- Hurwitz, H.; Fehrenbacher, L.; Novotny, W.; Cartwright, T.; Hainsworth, J.; Heim, W.; Berlin, J.; Baron, A.; Griffing, S.; Holmgren, E.; et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N. Engl. J. Med. 2004, 350, 2335–2342. [Google Scholar] [CrossRef]
- Saltz, L.B.; Clarke, S.; Diaz-Rubio, E.; Scheithauer, W.; Figer, A.; Wong, R.; Koski, S.; Lichinitser, M.; Yang, T.S.; Rivera, F.; et al. Bevacizumab in combination with oxaliplatin-based chemotherapy as first-line therapy in metastatic colorectal cancer: A randomized phase III study. J. Clin. Oncol. 2008, 26, 2013–2019. [Google Scholar] [CrossRef] [PubMed]
- Van Cutsem, E.; Kohne, C.H.; Hitre, E.; Zaluski, J.; Chien, C.R.C.; Makhson, A.; D’Haens, G.; Pinter, T.; Lim, R.; Bodoky, G.; et al. Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. N. Engl. J. Med. 2009, 360, 1408–1417. [Google Scholar] [CrossRef] [PubMed]
- Douillard, J.Y.; Oliner, K.S.; Siena, S.; Tabernero, J.; Burkes, R.; Barugel, M.; Humblet, Y.; Bodoky, G.; Cunningham, D.; Jassem, J.; et al. Panitumumab-FOLFOX4 treatment and RAS mutations in colorectal cancer. N. Engl. J. Med. 2013, 369, 1023–1034. [Google Scholar] [CrossRef]
- Van Cutsem, E.; Lenz, H.J.; Kohne, C.H.; Heinemann, V.; Tejpar, S.; Melezinek, I.; Beier, F.; Stroh, C.; Rougier, P.; van Krieken, J.H.; et al. Fluorouracil, leucovorin, and irinotecan plus cetuximab treatment and RAS mutations in colorectal cancer. J. Clin. Oncol. 2015, 33, 692–700. [Google Scholar] [CrossRef] [PubMed]
- Heinemann, V.; von Weikersthal, L.F.; Decker, T.; Kiani, A.; Vehling-Kaiser, U.; Al-Batran, S.E.; Heintges, T.; Lerchenmuller, C.; Kahl, C.; Seipelt, G.; et al. FOLFIRI plus cetuximab versus FOLFIRI plus bevacizumab as first-line treatment for patients with metastatic colorectal cancer (FIRE-3): A randomised, open-label, phase 3 trial. Lancet Oncol. 2014, 15, 1065–1075. [Google Scholar] [CrossRef]
- Rivera, F.; Karthaus, M.; Hecht, J.R.; Sevilla, I.; Forget, F.; Fasola, G.; Canon, J.L.; Guan, X.S.; Demonty, G.; Schwartzberg, L.S. Final analysis of the randomised PEAK trial: Overall survival and tumour responses during first-line treatment with mFOLFOX6 plus either panitumumab or bevacizumab in patients with metastatic colorectal carcinoma. Int. J. Colorectal Dis. 2017, 32, 1179–1190. [Google Scholar] [CrossRef] [PubMed]
- Cremolini, C.; Schirripa, M.; Antoniotti, C.; Moretto, R.; Salvatore, L.; Masi, G.; Falcone, A.; Loupakis, F. First-line chemotherapy for mCRC—A review and evidence-based algorithm. Nat. Rev. Clin. Oncol. 2015, 12, 607–619. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guinney, J.; Dienstmann, R.; Wang, X.; de Reynies, A.; Schlicker, A.; Soneson, C.; Marisa, L.; Roepman, P.; Nyamundanda, G.; Angelino, P.; et al. The consensus molecular subtypes of colorectal cancer. Nat. Med. 2015, 21, 1350–1356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Punt, C.J.; Koopman, M.; Vermeulen, L. From tumour heterogeneity to advances in precision treatment of colorectal cancer. Nat. Rev. Clin. Oncol. 2017, 14, 235–246. [Google Scholar] [CrossRef]
- Raimondi, C.; Nicolazzo, C.; Gradilone, A.; Giannini, G.; De Falco, E.; Chimenti, I.; Varriale, E.; Hauch, S.; Plappert, L.; Cortesi, E.; et al. Circulating tumor cells: Exploring intratumor heterogeneity of colorectal cancer. Cancer Biol. Ther. 2014, 15, 496–503. [Google Scholar] [CrossRef]
- Janku, F. Phosphoinositide 3-kinase (PI3K) pathway inhibitors in solid tumors: From laboratory to patients. Cancer Treat. Rev. 2017, 59, 93–101. [Google Scholar] [CrossRef]
- Capalbo, C.; Marchetti, P.; Coppa, A.; Calogero, A.; Anastasi, E.; Buffone, A.; Belardinilli, F.; Gulino, M.; Frati, P.; Catalano, C.; et al. Vemurafenib and panitumumab combination tailored therapy in BRAF-mutated metastatic colorectal cancer: A case report. Cancer Biol. Ther. 2014, 15, 826–831. [Google Scholar] [CrossRef]
- Corcoran, R.B.; Andre, T.; Atreya, C.E.; Schellens, J.H.M.; Yoshino, T.; Bendell, J.C.; Hollebecque, A.; McRee, A.J.; Siena, S.; Middleton, G.; et al. Combined BRAF, EGFR, and MEK Inhibition in Patients with BRAF(V600E)-Mutant Colorectal Cancer. Cancer Discov. 2018, 8, 428–443. [Google Scholar] [CrossRef] [PubMed]
- Richman, S.D.; Seymour, M.T.; Chambers, P.; Elliott, F.; Daly, C.L.; Meade, A.M.; Taylor, G.; Barrett, J.H.; Quirke, P. KRAS and BRAF mutations in advanced colorectal cancer are associated with poor prognosis but do not preclude benefit from oxaliplatin or irinotecan: Results from the MRC FOCUS trial. J. Clin. Oncol. 2009, 27, 5931–5937. [Google Scholar] [CrossRef] [PubMed]
- Bardelli, A.; Siena, S. Molecular mechanisms of resistance to cetuximab and panitumumab in colorectal cancer. J. Clin. Oncol. 2010, 28, 1254–1261. [Google Scholar] [CrossRef] [PubMed]
- Zarkavelis, G.; Boussios, S.; Papadaki, A.; Katsanos, K.H.; Christodoulou, D.K.; Pentheroudakis, G. Current and future biomarkers in colorectal cancer. Ann. Gastroenterol 2017, 30, 613–621. [Google Scholar] [CrossRef] [PubMed]
- Belardinilli, F.; Capalbo, C.; Buffone, A.; Petroni, M.; Colicchia, V.; Ferraro, S.; Zani, M.; Nicolussi, A.; D’Inzeo, S.; Coppa, A.; et al. Validation of the Ion Torrent PGM sequencing for the prospective routine molecular diagnostic of colorectal cancer. Clin. Biochem. 2015, 48, 908–910. [Google Scholar] [CrossRef]
- D’Haene, N.; Fontanges, Q.; De Neve, N.; Blanchard, O.; Melendez, B.; Delos, M.; Dehou, M.F.; Maris, C.; Nagy, N.; Rousseau, E.; et al. Clinical application of targeted next-generation sequencing for colorectal cancer patients: A multicentric Belgian experience. Oncotarget 2018, 9, 20761–20768. [Google Scholar] [CrossRef]
- De Nicola, F.; Goeman, F.; Pallocca, M.; Sperati, F.; Pizzuti, L.; Melucci, E.; Casini, B.; Amoreo, C.A.; Gallo, E.; Diodoro, M.G.; et al. Deep sequencing and pathway-focused analysis revealed multigene oncodriver signatures predicting survival outcomes in advanced colorectal cancer. Oncogenesis 2018, 7, 55. [Google Scholar] [CrossRef]
- Gao, X.H.; Yu, G.Y.; Hong, Y.G.; Lian, W.; Chouhan, H.; Xu, Y.; Liu, L.J.; Bai, C.G.; Zhang, W. Clinical significance of multiple gene detection with a 22-gene panel in formalin-fixed paraffin-embedded specimens of 207 colorectal cancer patients. Int. J. Clin. Oncol. 2019. [Google Scholar] [CrossRef]
- Malapelle, U.; Vigliar, E.; Sgariglia, R.; Bellevicine, C.; Colarossi, L.; Vitale, D.; Pallante, P.; Troncone, G. Ion Torrent next-generation sequencing for routine identification of clinically relevant mutations in colorectal cancer patients. J. Clin. Pathol. 2015, 68, 64–68. [Google Scholar] [CrossRef]
- Chakravarty, D.; Gao, J.; Phillips, S.M.; Kundra, R.; Zhang, H.; Wang, J.; Rudolph, J.E.; Yaeger, R.; Soumerai, T.; Nissan, M.H.; et al. OncoKB: A precision oncology knowledge base. JCO Precis Oncol. 2017. [Google Scholar] [CrossRef]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Del Vecchio, F.; Mastroiaco, V.; Di Marco, A.; Compagnoni, C.; Capece, D.; Zazzeroni, F.; Capalbo, C.; Alesse, E.; Tessitore, A. Next-generation sequencing: Recent applications to the analysis of colorectal cancer. J. Transl. Med. 2017, 15, 246. [Google Scholar] [CrossRef] [PubMed]
- Schell, M.J.; Yang, M.; Teer, J.K.; Lo, F.Y.; Madan, A.; Coppola, D.; Monteiro, A.N.; Nebozhyn, M.V.; Yue, B.; Loboda, A.; et al. A multigene mutation classification of 468 colorectal cancers reveals a prognostic role for APC. Nat. Commun. 2016, 7, 11743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yaeger, R.; Chatila, W.K.; Lipsyc, M.D.; Hechtman, J.F.; Cercek, A.; Sanchez-Vega, F.; Jayakumaran, G.; Middha, S.; Zehir, A.; Donoghue, M.T.A.; et al. Clinical Sequencing Defines the Genomic Landscape of Metastatic Colorectal Cancer. Cancer Cell 2018, 33, 125–136. [Google Scholar] [CrossRef] [PubMed]
- Capece, D.; Verzella, D.; Tessitore, A.; Alesse, E.; Capalbo, C.; Zazzeroni, F. Cancer secretome and inflammation: The bright and the dark sides of NF-kappaB. Semin Cell Dev. Biol. 2018, 78, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Dallol, A.; Buhmeida, A.; Al-Ahwal, M.S.; Al-Maghrabi, J.; Bajouh, O.; Al-Khayyat, S.; Alam, R.; Abusanad, A.; Turki, R.; Elaimi, A.; et al. Clinical significance of frequent somatic mutations detected by high-throughput targeted sequencing in archived colorectal cancer samples. J. Transl. Med. 2016, 14, 118. [Google Scholar] [CrossRef]
- Shimada, Y.; Kameyama, H.; Nagahashi, M.; Ichikawa, H.; Muneoka, Y.; Yagi, R.; Tajima, Y.; Okamura, T.; Nakano, M.; Sakata, J.; et al. Comprehensive genomic sequencing detects important genetic differences between right-sided and left-sided colorectal cancer. Oncotarget 2017, 8, 93567–93579. [Google Scholar] [CrossRef] [Green Version]
- Tejpar, S.; Stintzing, S.; Ciardiello, F.; Tabernero, J.; Van Cutsem, E.; Beier, F.; Esser, R.; Lenz, H.J.; Heinemann, V. Prognostic and predictive relevance of primary tumor location in patients with RAS wild-type metastatic colorectal cancer: Retrospective analyses of the CRYSTAL and FIRE-3 Trials. JAMA Oncol. 2016. [Google Scholar] [CrossRef]
- Willekens, F. Statistical Packages for Multistate Life History Analysis; Springer: Cham, Switzerlnd, 2014. [Google Scholar]
- Rozanska-Kudelska, M.; Walenczak, I.; Pepinski, W.; Sieskiewicz, A.; Skawronska, M.; Rogowski, M. Evaluation of tumor microsatellite instability in laryngeal cancer using five quasimonomorphic mononucleotide repeats and pentaplex PCR. Adv. Med. Sci. Pol. 2008, 53, 59–63. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| Characteristics | n |
| Sex | |
| Males | 41 |
| Females | 36 |
| Age | |
| Mean | 64 |
| Range | 38–81 |
| Site | |
| Right Colon | 28 |
| Left Colon | 31 |
| Rectum | 18 |
| Metastatic site (n) | |
| Liver | 38 |
| Peritoneum | 12 |
| Lung | 17 |
| Limph nodes | 7 |
| Bone | 2 |
| First Line Therapy | |
| Chemotherapy/anti-VEGF | 47 |
| Chemotherapy/anti-EGFR | 18 |
| Chemotherapy | 12 |
| Performance Status ECOG | |
| 0–1 | 65 |
| ≥2 | 12 |
| Gene a | Status | No. of pts (%) | No. of pts (%) with Additional Mutation |
|---|---|---|---|
| RAS | WT | 38 (49.4) | 28 (73.7) |
| Mut | 39 (50.6) | 31 (79.5) | |
| BRAF | WT | 70 (90.9) | 60 (77.9) |
| Mut | 7 (9.1) | 6 (85.7) | |
| EGFR | WT | 76 (98.7) | 66 (85.7) |
| Mut | 1 (1.3) | 1 (100.0) | |
| PIK3CA | WT | 66 (85.7) | 56 (84.8) |
| Mut | 11 (14.3) | 10 (90.9) | |
| MET | WT | 72 (93.5) | 62 (80.5) |
| Mut | 5 (6.5) | 5 (100.0) | |
| PTEN | WT | 76 (98.7) | 66 (85.7) |
| Mut | 1 (1.3) | 1 (100.0) | |
| MSI-H b | No | 72 (93.5) | 62 (80.5) |
| Yes | 5 (6.5) | 5 (100.0) |
| PFS | Univariate Cox Regression | Multivariable Cox Regression | ||
|---|---|---|---|---|
| HR (95%CI) | p-Value | HR (95%CI) | p-Value | |
| Gene stratification | ||||
| PRG | 1.00 | 1.00 | ||
| AGG | 1.91 (1.16–3.15) | 0.011 | 1.84 (1.03–3.28) | 0.039 |
| NMG | 2.63 (1.26–5.47) | 0.010 | 1.81 (0.65–5.01) | 0.253 |
| Age (>65y vs. ≤65y) | 1.30 (0.74–2.29) | 0.364 | ||
| Gender (F vs. M) | 2.02 (1.11–3.67) | 0.021 | ||
| Grading (G1 vs. G2 vs. G3) | 1.05 (0.65–1.70) | 0.837 | ||
| Primay tumor location (RCT vs. RC vs. LC) | 0.78 (0.54–1.13) | 0.192 | ||
| Adjuvant therapy (Y vs. N) | 1.38 (0.83−2.28) | 0.210 | ||
| Metastatic site (liver and/or other vs. liver) | 1.57 (0.92–2.68) | 0.100 | ||
| ECOG PS (≥2 vs. 0–1) | 0.22 (0.04–1.17) | 0.077 | ||
| Surgery for primiry tumor (Y vs. N) | 0.66 (0.18–2.44) | 0.537 | ||
| Therapy (CHT vs. CHT+antiVEGF vs. CHT+antiEGFR) | 0.64 (0.30–1.36) | 0.251 | ||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Capalbo, C.; Belardinilli, F.; Raimondo, D.; Milanetti, E.; Malapelle, U.; Pisapia, P.; Magri, V.; Prete, A.; Pecorari, S.; Colella, M.; et al. A Simplified Genomic Profiling Approach Predicts Outcome in Metastatic Colorectal Cancer. Cancers 2019, 11, 147. https://doi.org/10.3390/cancers11020147
Capalbo C, Belardinilli F, Raimondo D, Milanetti E, Malapelle U, Pisapia P, Magri V, Prete A, Pecorari S, Colella M, et al. A Simplified Genomic Profiling Approach Predicts Outcome in Metastatic Colorectal Cancer. Cancers. 2019; 11(2):147. https://doi.org/10.3390/cancers11020147
Chicago/Turabian StyleCapalbo, Carlo, Francesca Belardinilli, Domenico Raimondo, Edoardo Milanetti, Umberto Malapelle, Pasquale Pisapia, Valentina Magri, Alessandra Prete, Silvia Pecorari, Mariarosaria Colella, and et al. 2019. "A Simplified Genomic Profiling Approach Predicts Outcome in Metastatic Colorectal Cancer" Cancers 11, no. 2: 147. https://doi.org/10.3390/cancers11020147