3D Mammary Epithelial Cell Models: A Goldmine of DCIS Biomarkers and Morphogenetic Mechanisms

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
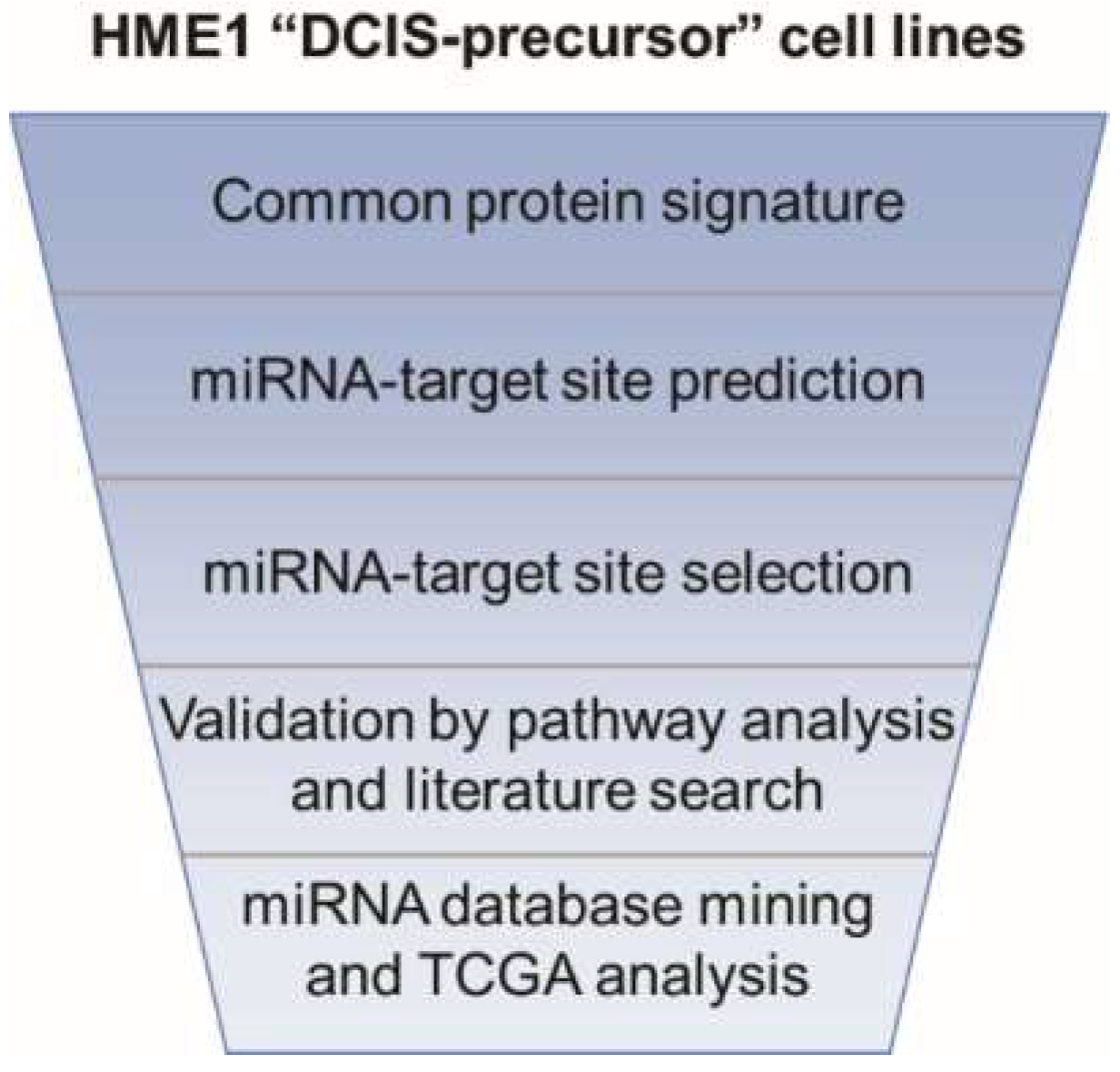
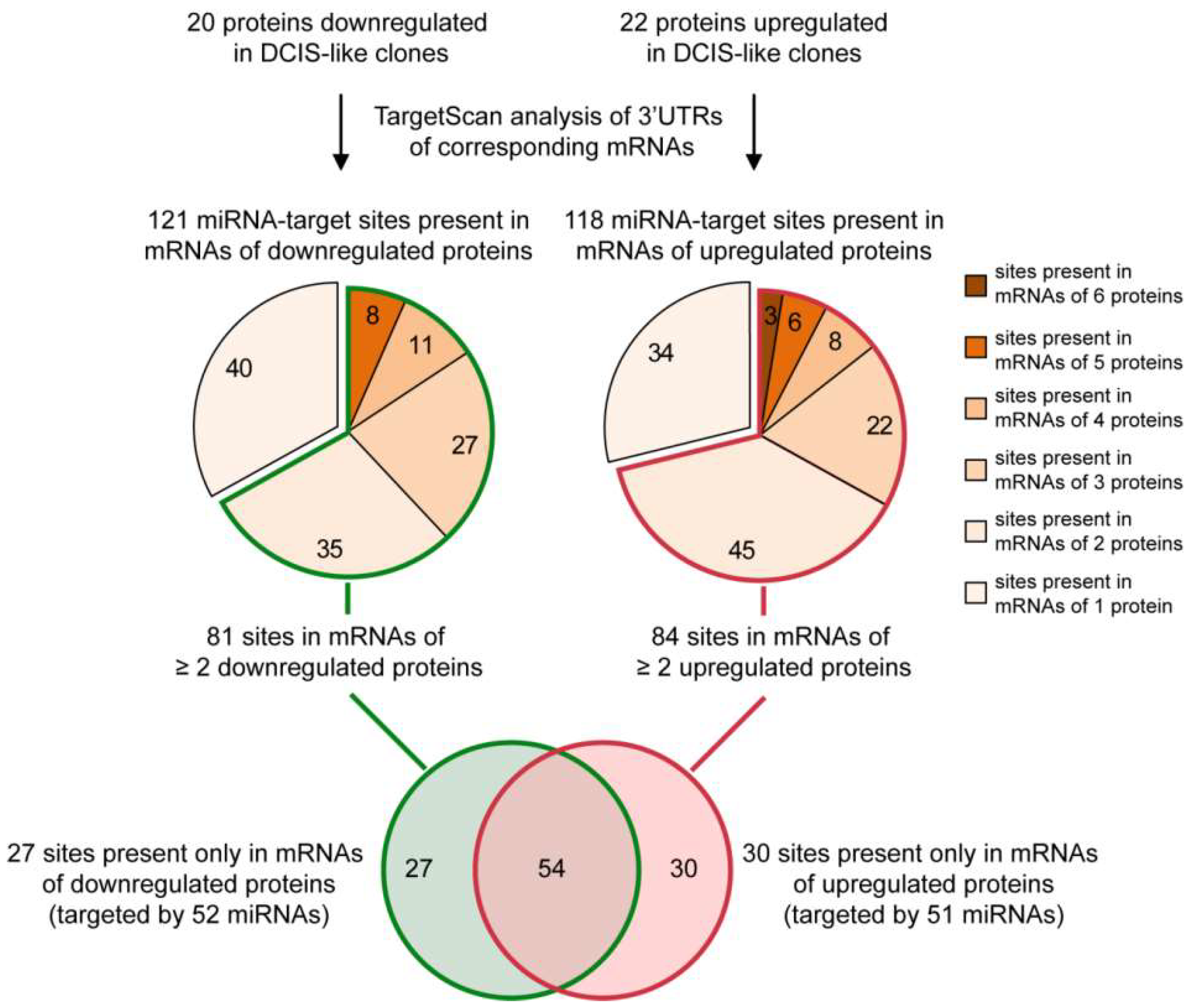
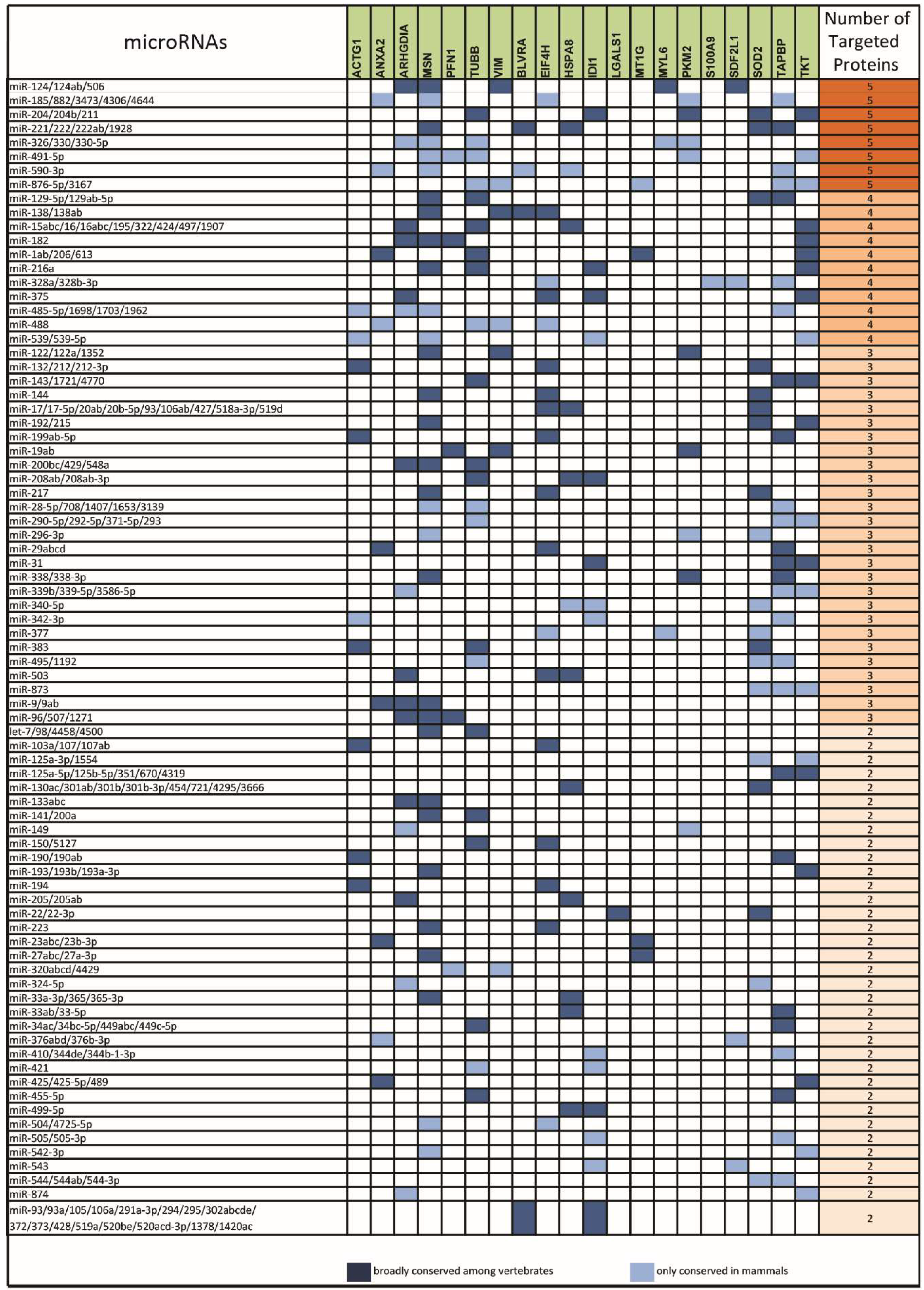
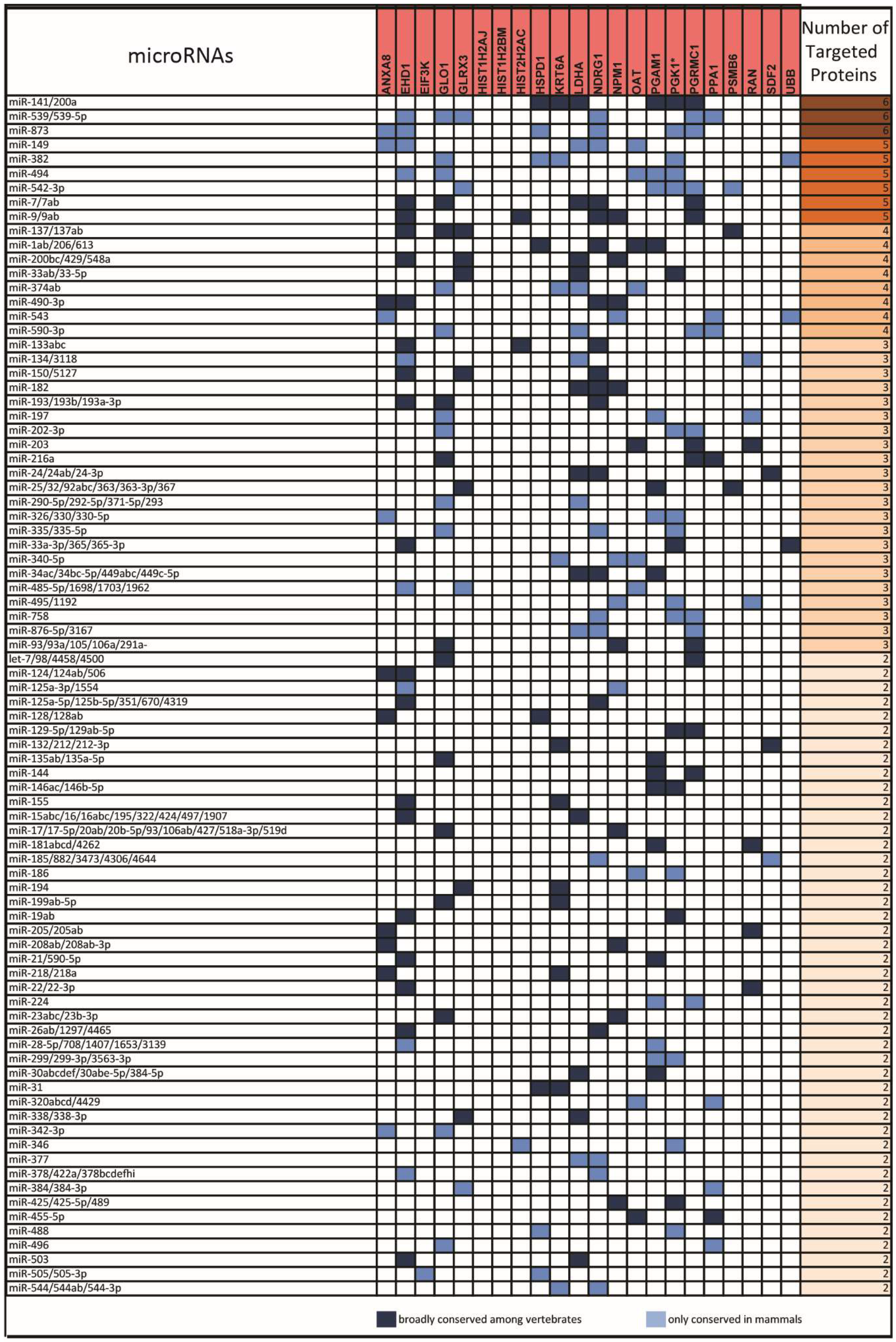
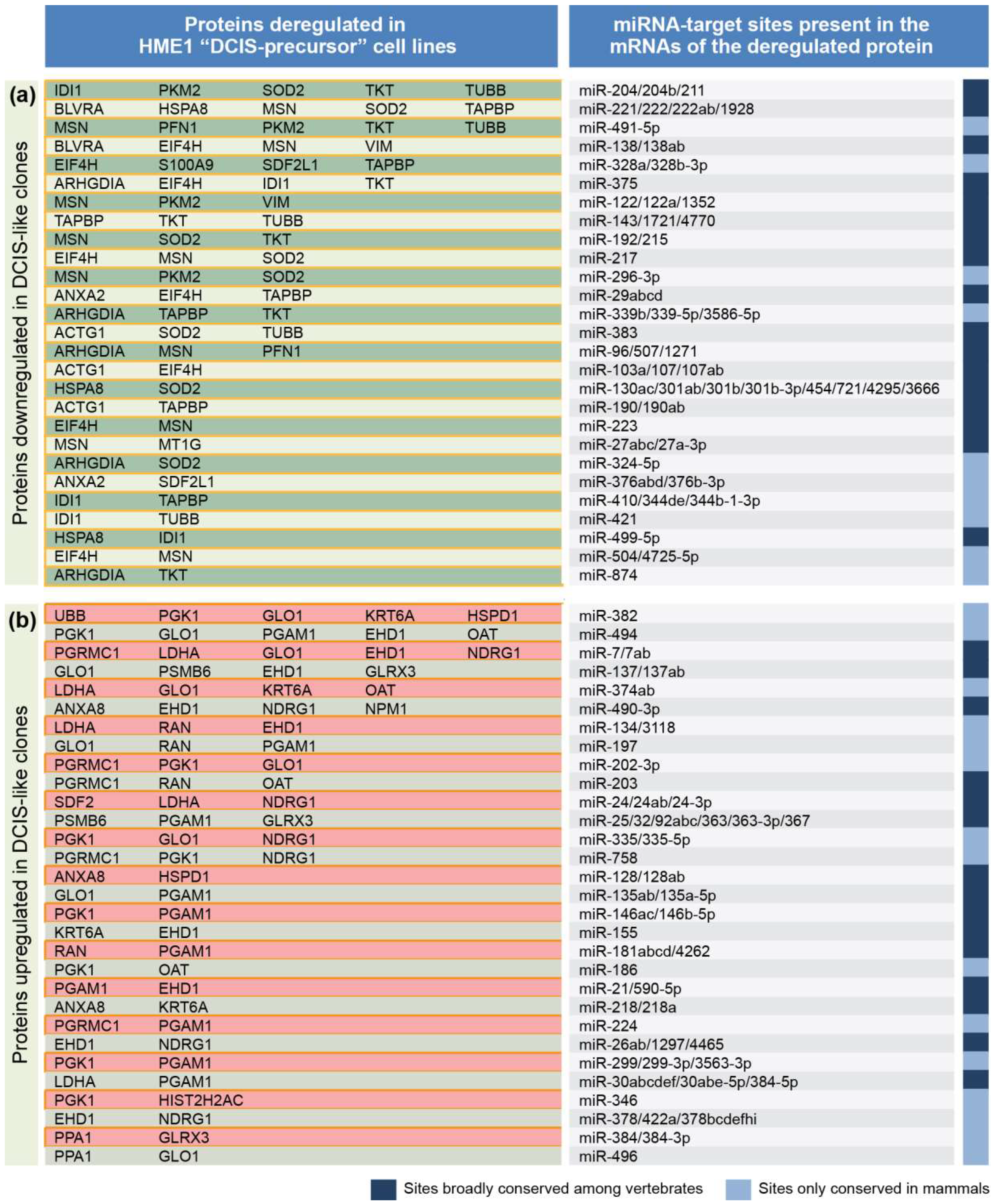
2.1. From a Common Protein Signature Shared by HME1 “DCIS-Precursor” Cell Lines to a Subset of Potential Morphoregulatory miRNAs
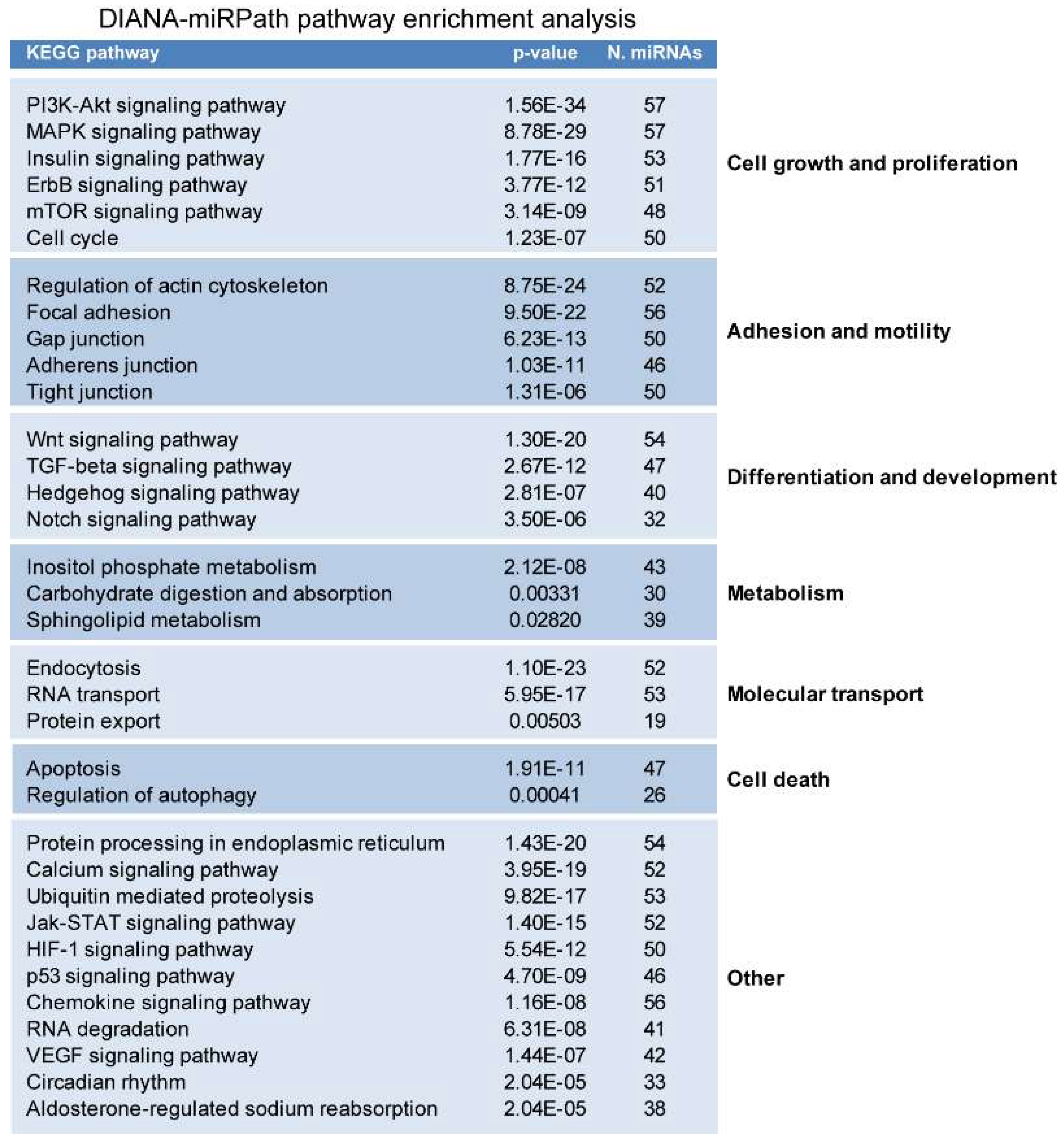
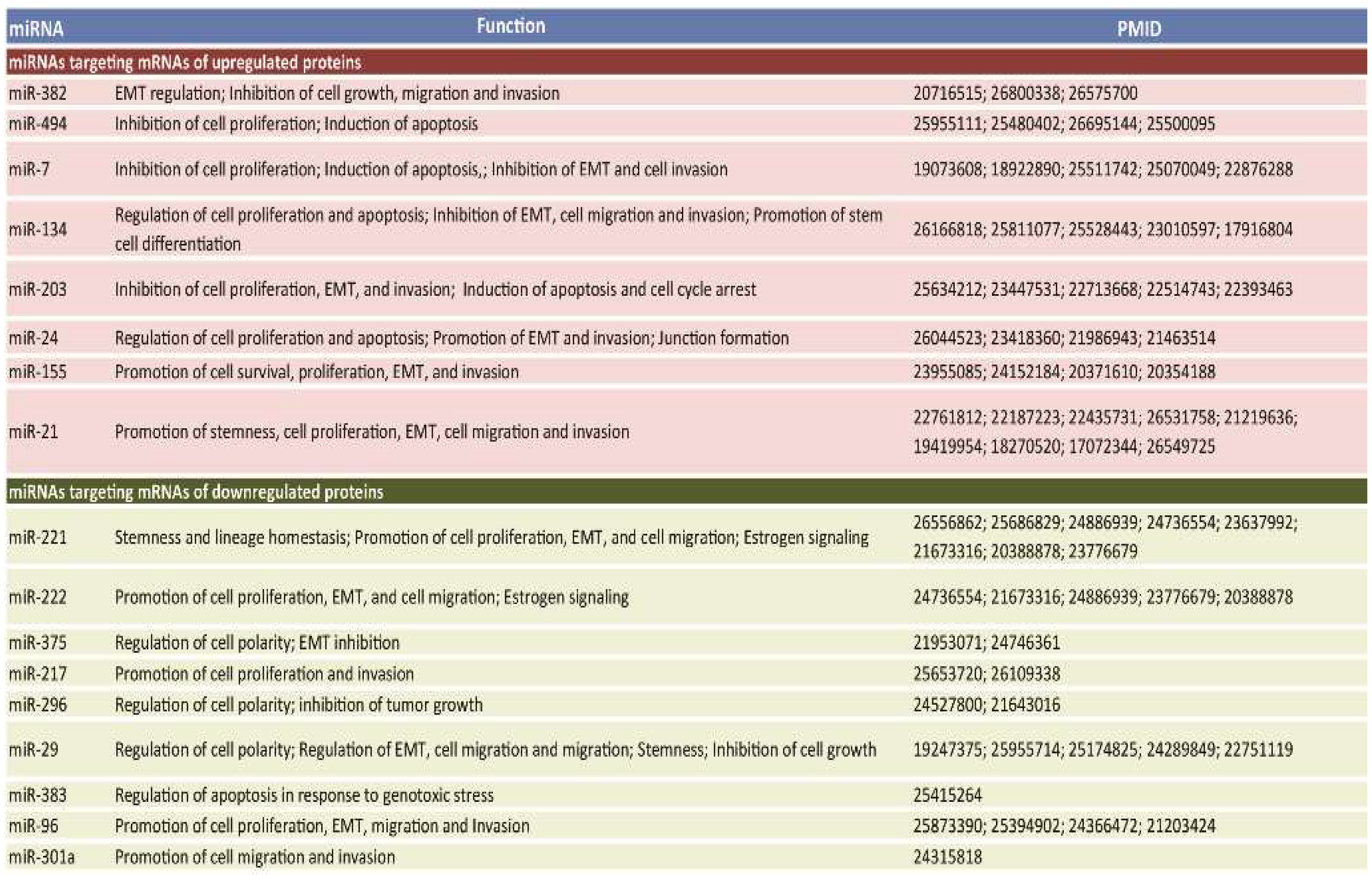
2.2. miRNAs Potentially Relevant to Mammary Epithelial Cell Morphogenesis
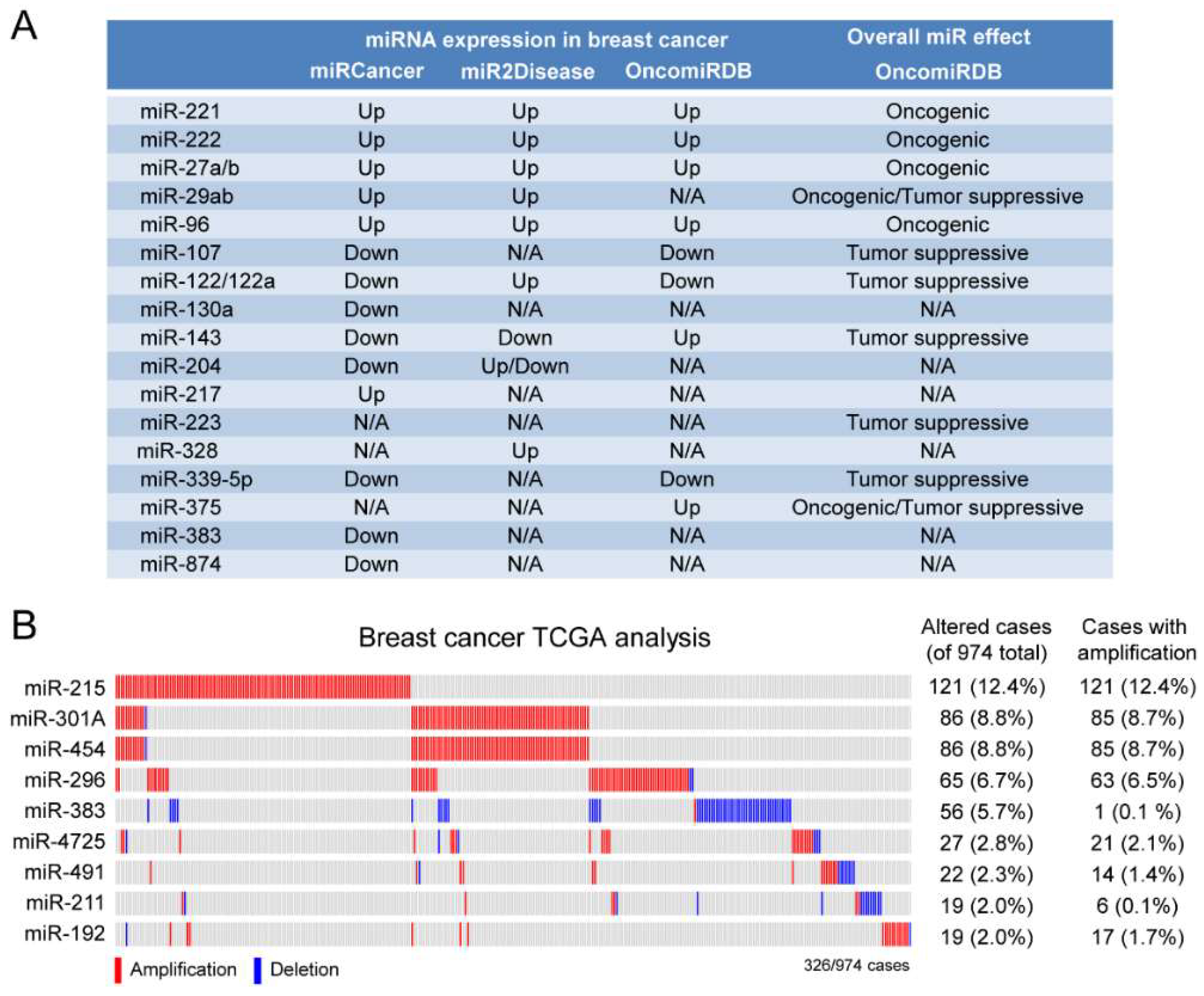
2.3. A Subset of Deregulated miRNA in Breast Cancer
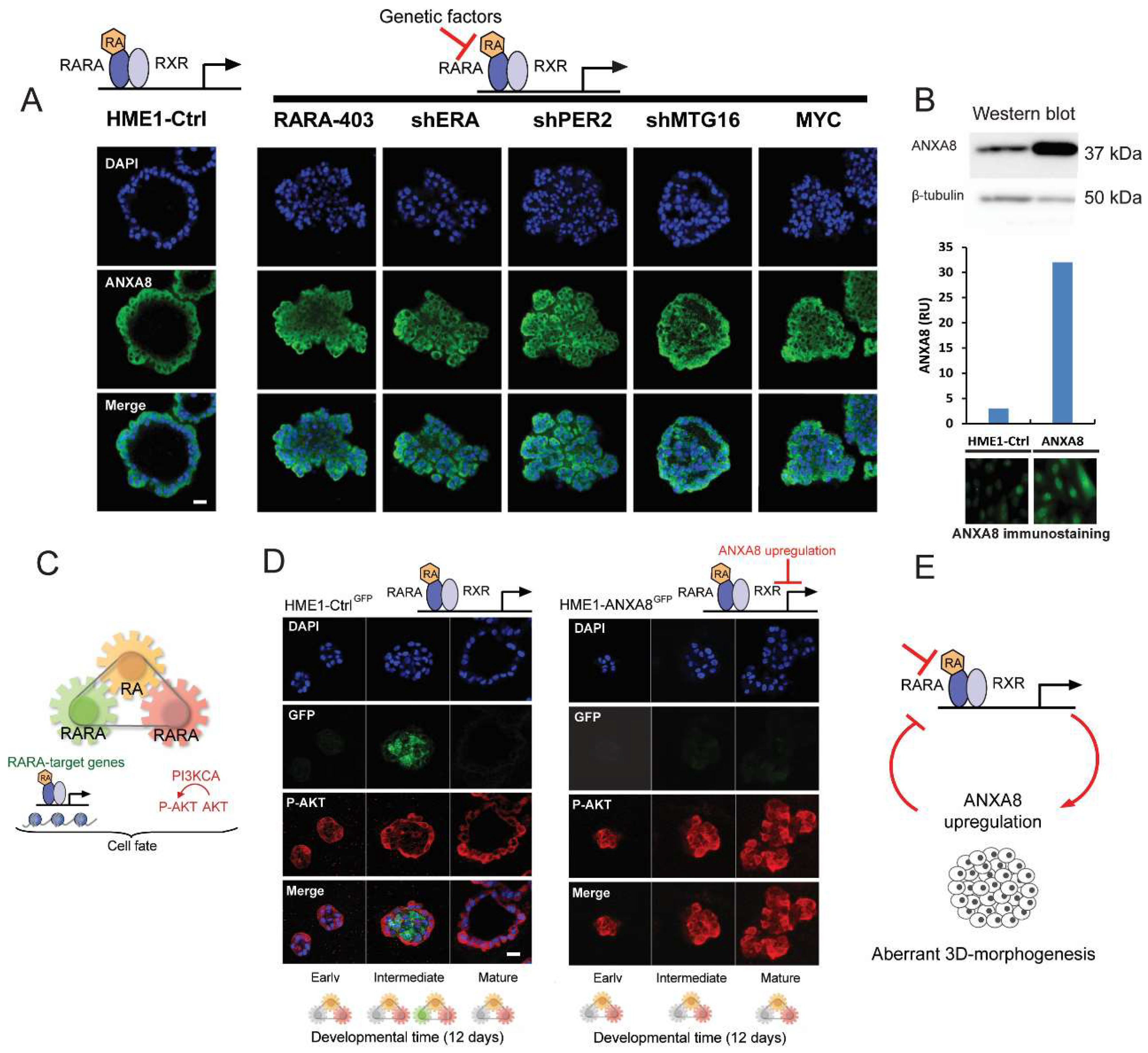
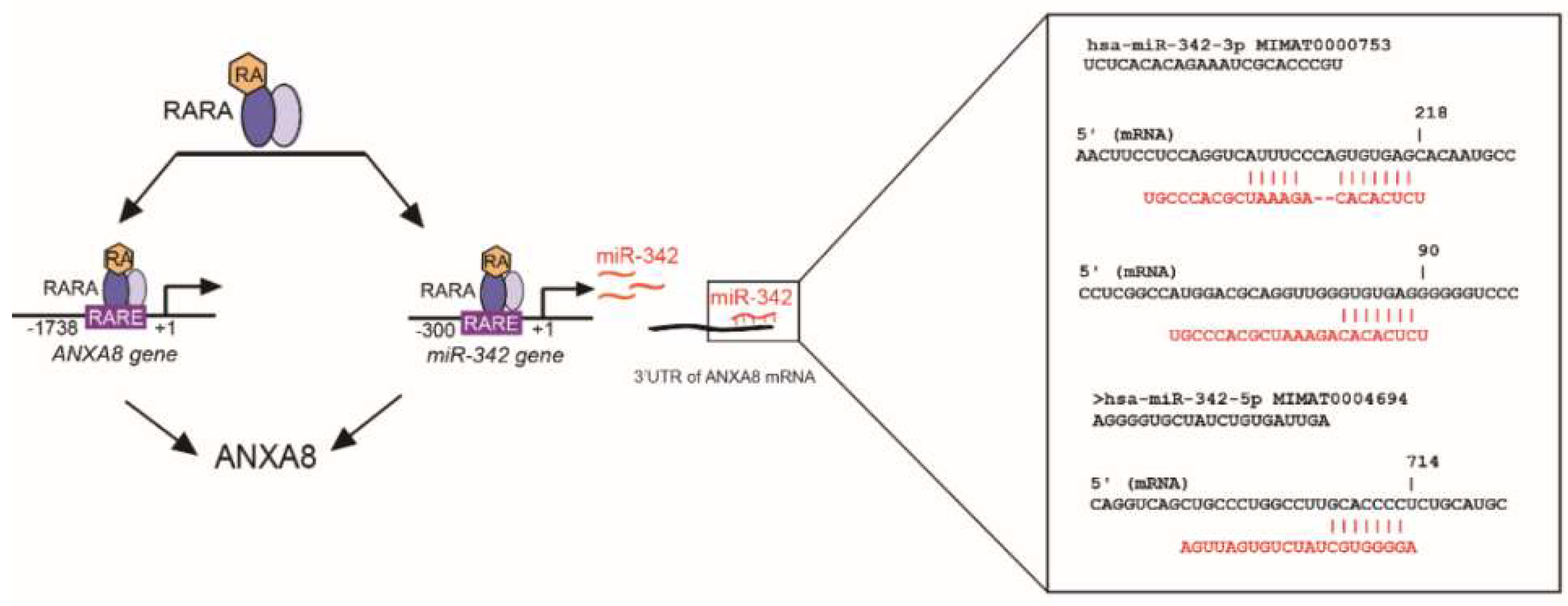
2.4. An ANXA8 Feedback Loop of Aberrant Mammary Morphogenesis
3. Discussion
4. Materials and Methods
4.1. Cells and Cell Culture
4.2. miRNA Prediction from Protein Changes
4.3. miRNA Pathway Enrichment Analysis
4.4. miRNA Database Mining
4.5. TCGA Analysis
4.6. In Silico Identification of RARE in miRNA-342 Promoter and Sites in the ANXA8 3′UTR mRNA
4.7. Western Blotting (WB)
4.8. Immunostaining of Normal and Aberrant 3D HME1 Acini and Confocal Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 2D | Two dimensional |
| 3D | Three dimensional |
| 3’ UTR | 3’ untranslated region |
| ANXA2 | Annexin 2 |
| ANXA8 | Annexin 8 |
| AKT | protein kinase B |
| DCIS | Ductal carcinoma in situ |
| ERA | Estrogen receptor alpha |
| GFP | Green Fluorescent Protein |
| HME1 | Human mammary epithelial 1 cell line |
| HME1-CtrlGFP | HME1 control cells carrying a stable RARE-GFP construct |
| HME1-RARA403 | HME1 cells carrying a dominant negative RARA403 mutant |
| HME1-MYC | HME1 cells with MYC ectopic expression |
| HME1-shERA | ERA knock down HME1 cells |
| HME1-shMTG16 | MTG16 knock down HME1 cells |
| HME1-shPER2 | PER2 knock down HME1 cells |
| HME1-ANXA8 | HME1 with ANXA8 ectopic expression |
| HME1-ANXA8GFP | HME1ANXA8 carrying a stable RARE-GFP construct |
| IPA | Ingenuity Pathway Analysis |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| MiRNA | microRNA |
| PI3KCA | Phosphatidylinositol-3Kinase catalytic subunit (p110) |
| P-AKT | phosphorylated AKT |
| PMID | PubMed unique research article identifier number |
| RA | All- Trans Retinoic acid |
| RARE | RA responsive element |
| RARA | RA receptor alpha |
| TCGA | The Cancer Genome Atlas |
References
- Arpino, G.; Laucirica, R.; Elledge, R.M. Premalignant and in situ breast disease: Biology and clinical implications. Ann. Intern. Med. 2005, 143, 446–457. [Google Scholar] [CrossRef] [PubMed]
- Meisner, A.L.; Fekrazad, M.H.; Royce, M.E. Breast disease: Benign and malignant. Med. Clin. North Am. 2008, 92, 1115–1141. [Google Scholar] [CrossRef] [PubMed]
- Bombonati, A.; Sgroi, D.C. The molecular pathology of breast cancer progression. J. Pathol. 2011, 223, 307–317. [Google Scholar] [CrossRef] [PubMed]
- Friel, A.M.; Corcoran, C.; Crown, J.; O’Driscoll, L. Relevance of circulating tumor cells, extracellular nucleic acids, and exosomes in breast cancer. Breast Cancer Res. Treat. 2010, 123, 613–625. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.L.; Marcato, P. Epigenetic modifications as biomarkers of tumor development, therapy response, and recurrence across the cancer care continuum. Cancers 2018, 10, 101. [Google Scholar] [CrossRef] [PubMed]
- Petersen, O.W.; Ronnov-Jessen, L.; Howlett, A.R.; Bissell, M.J. Interaction with basement membrane serves to rapidly distinguish growth and differentiation pattern of normal and malignant human breast epithelial cells. Proc. Natl. Acad. Sci. USA 1992, 89, 9064–9068. [Google Scholar] [CrossRef]
- Bistulfi, G.; Pozzi, S.; Ren, M.; Rossetti, S.; Sacchi, N. A repressive epigenetic domino effect confers susceptibility to breast epithelial cell transformation: Implications for predicting breast cancer risk. Cancer Res. 2006, 66, 10308–10314. [Google Scholar] [CrossRef] [PubMed]
- Debnath, J.; Brugge, J.S. Modelling glandular epithelial cancers in three-dimensional cultures. Nat. Rev. Cancer 2005, 5, 675–688. [Google Scholar] [CrossRef]
- Corlazzoli, F.; Rossetti, S.; Bistulfi, G.; Ren, M.; Sacchi, N. Derangement of a factor upstream of RARalpha triggers the repression of a pleiotropic epigenetic network. PloS ONE 2009, 4, e4305. [Google Scholar] [CrossRef]
- Montesano, R.; Soulie, P. Retinoids induce lumen morphogenesis in mammary epithelial cells. J. Cell Sci. 2002, 115, 4419–4431. [Google Scholar] [CrossRef]
- Wang, Y.A.; Shen, K.; Wang, Y.; Brooks, S.C. Retinoic acid signaling is required for proper morphogenesis of mammary gland. Dev. Dyn. Off. Publ. Am. Assoc. Anat. 2005, 234, 892–899. [Google Scholar] [CrossRef]
- Seewaldt, V.L.; Caldwell, L.E.; Johnson, B.S.; Swisshelm, K.; Collins, S.J.; Tsai, S. Inhibition of retinoic acid receptor function in normal human mammary epithelial cells results in increased cellular proliferation and inhibits the formation of a polarized epithelium in vitro. Exp. Cell Res. 1997, 236, 16–28. [Google Scholar] [CrossRef]
- Rossetti, S.; Ren, M.; Visconti, N.; Corlazzoli, F.; Gagliostro, V.; Somenzi, G.; Yao, J.; Sun, Y.; Sacchi, N. Tracing anti-cancer and cancer-promoting actions of all-trans retinoic acid in breast cancer to a RARalpha epigenetic mechanism of mammary epithelial cell fate. Oncotarget 2016, 7, 87064–87080. [Google Scholar] [CrossRef] [PubMed]
- Ren, M.; Pozzi, S.; Bistulfi, G.; Somenzi, G.; Rossetti, S.; Sacchi, N. Impaired retinoic acid (RA) signal leads to RARbeta2 epigenetic silencing and RA resistance. Mol. Cell Biol. 2005, 25, 10591–10603. [Google Scholar] [CrossRef] [PubMed]
- Rossetti, S.; Corlazzoli, F.; Gregorski, A.; Azmi, N.H.; Sacchi, N. Identification of an estrogen-regulated circadian mechanism necessary for breast acinar morphogenesis. Cell Cycle 2012, 11, 3691–3700. [Google Scholar] [CrossRef]
- Laganiere, J.; Deblois, G.; Giguere, V. Functional genomics identifies a mechanism for estrogen activation of the retinoic acid receptor alpha1 gene in breast cancer cells. Mol. Endocrinol. 2005, 19, 1584–1592. [Google Scholar] [CrossRef] [PubMed]
- Ross-Innes, C.S.; Stark, R.; Holmes, K.A.; Schmidt, D.; Spyrou, C.; Russell, R.; Massie, C.E.; Vowler, S.L.; Eldridge, M.; Carroll, J.S. Cooperative interaction between retinoic acid receptor-alpha and estrogen receptor in breast cancer. Genes Dev. 2010, 24, 171–182. [Google Scholar] [CrossRef]
- Xiang, S.; Coffelt, S.B.; Mao, L.; Yuan, L.; Cheng, Q.; Hill, S.M. Period-2: A tumor suppressor gene in breast cancer. J.Circadian Rhythm. 2008, 6, 4. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.H.; Farkas, M.E. Altered circadian rhythms and breast cancer: From the human to the molecular level. Front. Endocrinol. 2018, 9, 219. [Google Scholar] [CrossRef]
- Rossetti, S.; Hoogeveen, A.T.; Esposito, J.; Sacchi, N. Loss of MTG16a (CBFA2T3), a novel rDNA repressor, leads to increased ribogenesis and disruption of breast acinar morphogenesis. J. Cell Mol. Med. 2010, 14, 1358–1370. [Google Scholar] [CrossRef]
- Kochetkova, M.; McKenzie, O.L.; Bais, A.J.; Martin, J.M.; Secker, G.A.; Seshadri, R.; Powell, J.A.; Hinze, S.J.; Gardner, A.E.; Spendlove, H.E.; et al. CBFA2T3 (MTG16) is a putative breast tumor suppressor gene from the breast cancer loss of heterozygosity region at 16q24.3. Cancer Res. 2002, 62, 4599–4604. [Google Scholar] [PubMed]
- Rossetti, S.; Wierzbicki, A.J.; Sacchi, N. Mammary epithelial morphogenesis and early breast cancer. Evidence of involvement of basal components of the RNA Polymerase I transcription machinery. Cell Cycle 2016, 15, 2515–2526. [Google Scholar] [CrossRef] [PubMed]
- Uribesalgo, I.; Buschbeck, M.; Gutierrez, A.; Teichmann, S.; Demajo, S.; Kuebler, B.; Nomdedeu, J.F.; Martin-Caballero, J.; Roma, G.; Benitah, S.A.; et al. E-box-independent regulation of transcription and differentiation by MYC. Nat. Cell Biol. 2011, 13, 1443–1449. [Google Scholar] [CrossRef] [PubMed]
- Rossetti, S.; Bshara, W.; Reiners, J.A.; Corlazzoli, F.; Miller, A.; Sacchi, N. Harnessing 3D models of mammary epithelial morphogenesis: An off the beaten path approach to identify candidate biomarkers of early stage breast cancer. Cancer Lett. 2016, 380, 375–383. [Google Scholar] [CrossRef]
- Chang, K.S.; Wang, G.; Freireich, E.J.; Daly, M.; Naylor, S.L.; Trujillo, J.M.; Stass, S.A. Specific expression of the annexin VIII gene in acute promyelocytic leukemia. Blood 1992, 79, 1802–1810. [Google Scholar] [PubMed]
- Careccia, S.; Mainardi, S.; Pelosi, A.; Gurtner, A.; Diverio, D.; Riccioni, R.; Testa, U.; Pelosi, E.; Piaggio, G.; Sacchi, A.; et al. A restricted signature of miRNAs distinguishes APL blasts from normal promyelocytes. Oncogene 2009, 28, 4034–4040. [Google Scholar] [CrossRef] [PubMed]
- Lewis, B.P.; Shih, I.H.; Jones-Rhoades, M.W.; Bartel, D.P.; Burge, C.B. Prediction of mammalian microRNA targets. Cell 2003, 115, 787–798. [Google Scholar] [CrossRef]
- Vlachos, I.S.; Kostoulas, N.; Vergoulis, T.; Georgakilas, G.; Reczko, M.; Maragkakis, M.; Paraskevopoulou, M.D.; Prionidis, K.; Dalamagas, T.; Hatzigeorgiou, A.G. DIANA miRPath v.2.0: Investigating the combinatorial effect of microRNAs in pathways. Nucleic Acids Res. 2012, 40, W498–W504. [Google Scholar] [CrossRef]
- Chen, W.X.; Hu, Q.; Qiu, M.T.; Zhong, S.L.; Xu, J.J.; Tang, J.H.; Zhao, J.H. miR-221/222: Promising biomarkers for breast cancer. Tumor Biol. 2013, 34, 1361–1370. [Google Scholar] [CrossRef]
- Li, P.; Sheng, C.; Huang, L.; Zhang, H.; Huang, L.; Cheng, Z.; Zhu, Q. MiR-183/-96/-182 cluster is up-regulated in most breast cancers and increases cell proliferation and migration. Breast Cancer Res. BCR 2014, 16, 473. [Google Scholar] [CrossRef]
- Zhang, J.; Kong, X.; Li, J.; Luo, Q.; Li, X.; Shen, L.; Chen, L.; Fang, L. MiR-96 promotes tumor proliferation and invasion by targeting RECK in breast cancer. Oncol. Rep. 2014, 31, 1357–1363. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.; Zhu, J.; Su, S.; Wu, W.; Liu, Q.; Su, F.; Yu, F. MiR-27 as a prognostic marker for breast cancer progression and patient survival. PloS ONE 2012, 7, e51702. [Google Scholar] [CrossRef] [PubMed]
- Van Schooneveld, E.; Wouters, M.C.; Van der Auwera, I.; Peeters, D.J.; Wildiers, H.; Van Dam, P.A.; Vergote, I.; Vermeulen, P.B.; Dirix, L.Y.; Van Laere, S.J. Expression profiling of cancerous and normal breast tissues identifies microRNAs that are differentially expressed in serum from patients with (metastatic) breast cancer and healthy volunteers. Breast Cancer Res. BCR 2012, 14, R34. [Google Scholar] [CrossRef]
- Sakr, R.A.; Weigelt, B.; Chandarlapaty, S.; Andrade, V.P.; Guerini-Rocco, E.; Giri, D.; Ng, C.K.; Cowell, C.F.; Rosen, N.; Reis-Filho, J.S.; et al. PI3K pathway activation in high-grade ductal carcinoma in situ-implications for progression to invasive breast carcinoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2014, 20, 2326–2337. [Google Scholar] [CrossRef]
- Romero-Cordoba, S.L.; Rodriguez-Cuevas, S.; Bautista-Pina, V.; Maffuz-Aziz, A.; D’Ippolito, E.; Cosentino, G.; Baroni, S.; Iorio, M.V.; Hidalgo-Miranda, A. Loss of function of miR-342-3p results in MCT1 over-expression and contributes to oncogenic metabolic reprogramming in triple negative breast cancer. Sci. Rep. 2018, 8, 12252. [Google Scholar] [CrossRef] [PubMed]
- Young, J.; Kawaguchi, T.; Yan, L.; Qi, Q.; Liu, S.; Takabe, K. Tamoxifen sensitivity-related microRNA-342 is a useful biomarker for breast cancer survival. Oncotarget 2017, 8, 99978–99989. [Google Scholar] [CrossRef]
- Staquicini, D.I.; Rangel, R.; Guzman-Rojas, L.; Staquicini, F.I.; Dobroff, A.S.; Tarleton, C.A.; Ozbun, M.A.; Kolonin, M.G.; Gelovani, J.G.; Marchio, S.; et al. Intracellular targeting of annexin A2 inhibits tumor cell adhesion, migration, and in vivo grafting. Sci. Rep. 2017, 7, 4243. [Google Scholar] [CrossRef]
- Lewis, B.P.; Burge, C.B.; Bartel, D.P. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 2005, 120, 15–20. [Google Scholar] [CrossRef]
- Lu, T.-P.; Lee, C.-Y.; Tsai, M.-H.; Chiu, Y.-C.; Hsiao, C.K.; Lai, L.-C.; Chuang, E.Y. MiRSystem: An integrated system for characterizing enriched functions and pathways of microRNA targets. PloS ONE 2012, 7, e42390. [Google Scholar] [CrossRef]
- Ai, J.; Huang, H.; Lv, X.; Tang, Z.; Chen, M.; Chen, T.; Duan, W.; Sun, H.; Li, Q.; Tan, R.; et al. FLNA and PGK1 are two potential markers for progression in hepatocellular carcinoma. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2011, 27, 207–216. [Google Scholar] [CrossRef]
- Wang, D.; Gu, J.; Wang, T.; Ding, Z. OncomiRDB: A database for the experimentally verified oncogenic and tumor-suppressive microRNAs. Bioinformatics 2014, 30, 2237–2238. [Google Scholar] [CrossRef] [PubMed]
- Xie, B.; Ding, Q.; Han, H.; Wu, D. miRCancer: A microRNA-cancer association database constructed by text mining on literature. Bioinformatics 2013, 29, 638–644. [Google Scholar] [CrossRef]
- Jiang, Q.; Wang, Y.; Hao, Y.; Juan, L.; Teng, M.; Zhang, X.; Li, M.; Wang, G.; Liu, Y. MiR2Disease: A manually curated database for microRNA deregulation in human disease. Nucleic Acids Res. 2009, 37, D98–D104. [Google Scholar] [CrossRef] [PubMed]
- Ciriello, G.; Cerami, E.; Sander, C.; Schultz, N. Mutual exclusivity analysis identifies oncogenic network modules. Genome Res. 2012, 22, 398–406. [Google Scholar] [CrossRef] [PubMed]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef]
- Vejnar, C.E.; Zdobnov, E.M. MiRmap: Comprehensive prediction of microRNA target repression strength. Nucleic Acids Res. 2012, 40, 11673–11683. [Google Scholar] [CrossRef]










© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rossetti, S.; Sacchi, N. 3D Mammary Epithelial Cell Models: A Goldmine of DCIS Biomarkers and Morphogenetic Mechanisms. Cancers 2019, 11, 130. https://doi.org/10.3390/cancers11020130
Rossetti S, Sacchi N. 3D Mammary Epithelial Cell Models: A Goldmine of DCIS Biomarkers and Morphogenetic Mechanisms. Cancers. 2019; 11(2):130. https://doi.org/10.3390/cancers11020130
Chicago/Turabian StyleRossetti, Stefano, and Nicoletta Sacchi. 2019. "3D Mammary Epithelial Cell Models: A Goldmine of DCIS Biomarkers and Morphogenetic Mechanisms" Cancers 11, no. 2: 130. https://doi.org/10.3390/cancers11020130
APA StyleRossetti, S., & Sacchi, N. (2019). 3D Mammary Epithelial Cell Models: A Goldmine of DCIS Biomarkers and Morphogenetic Mechanisms. Cancers, 11(2), 130. https://doi.org/10.3390/cancers11020130