Transcriptomic Analyses Revealed Systemic Alterations in Gene Expression in Circulation and Tumor Microenvironment of Colorectal Cancer Patients

 , and
, and
Abstract
1. Introduction
2. Results
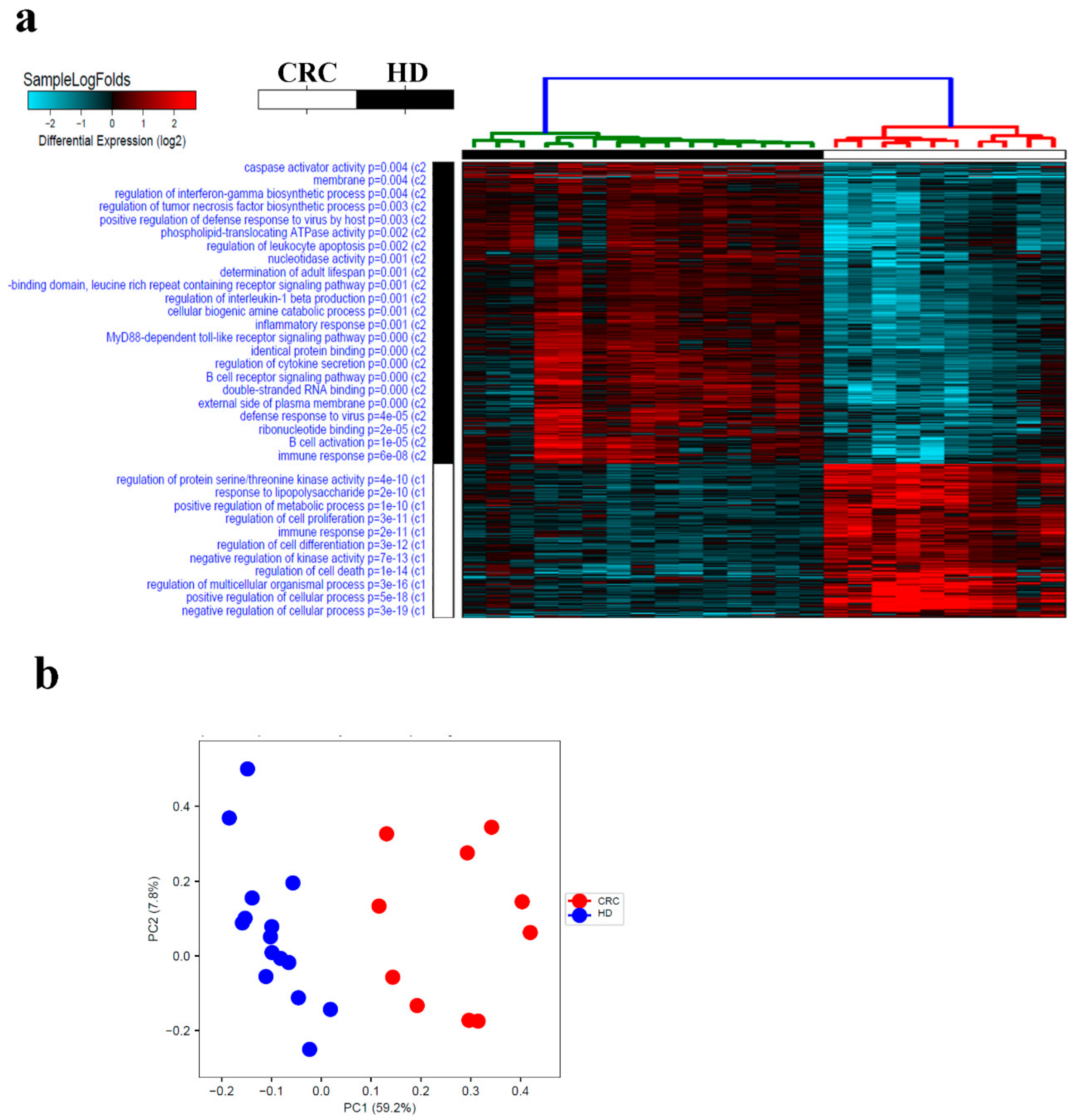
2.1. Transcriptome Analysis of PBMCs From CRC Patients Revealed Systemic Changes in Gene Expression
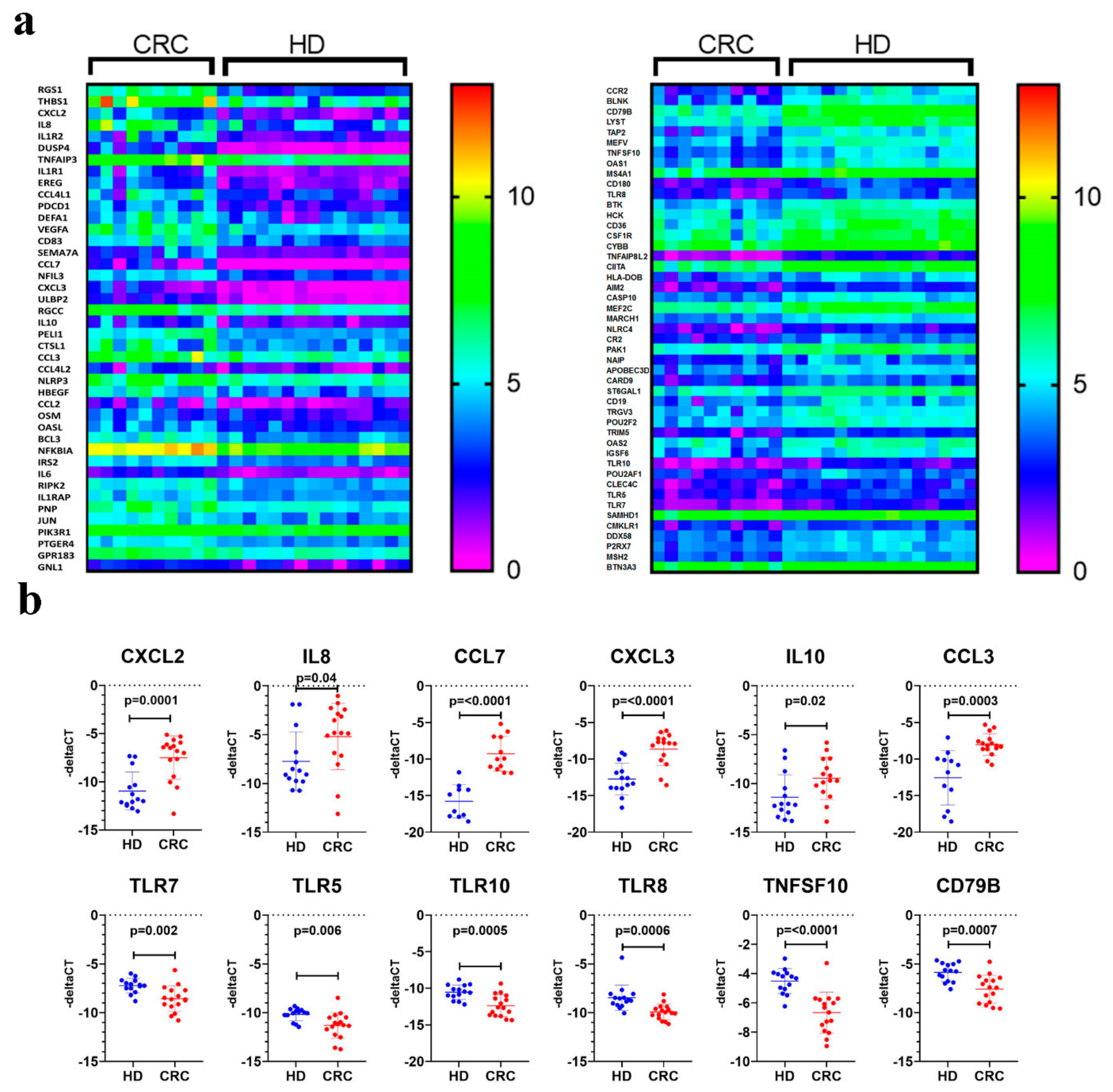
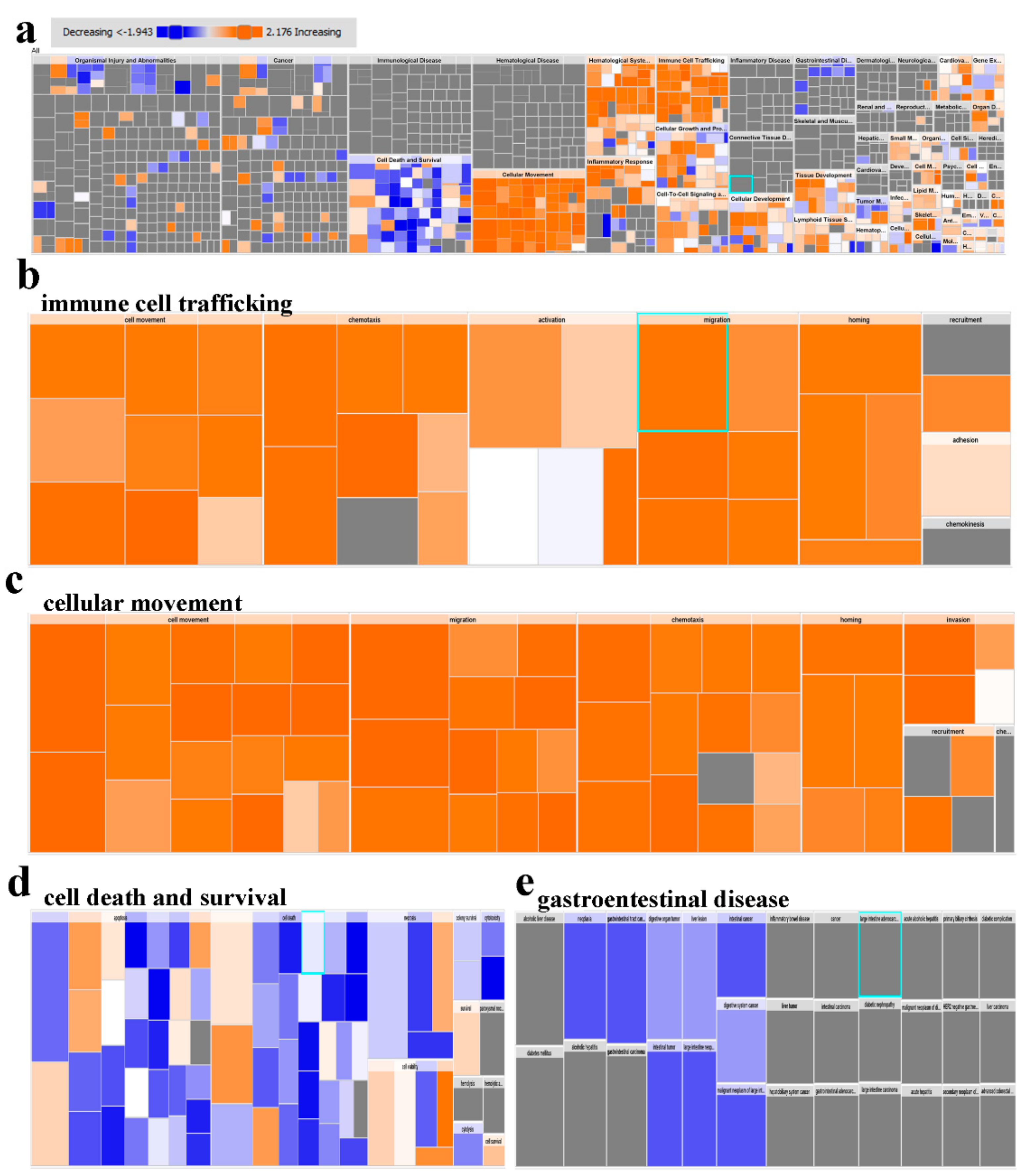
2.2. Downstream Effector Analysis of Differentially Expressed Genes in PBMCs from CRC and Healthy Subjects
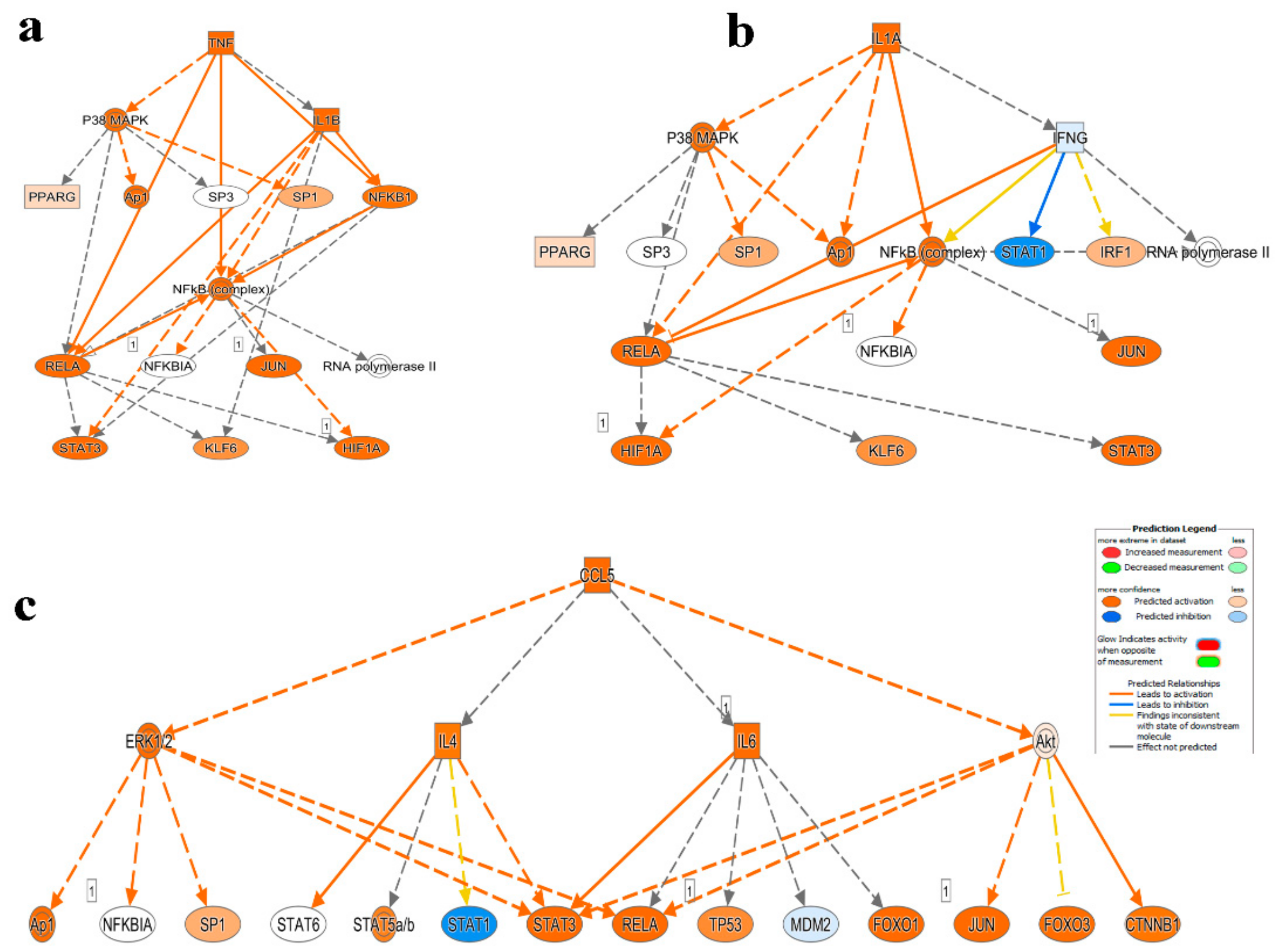
2.3. Mechanistic Network Analysis Predicts Multiple Altered Signaling Networks in PBMCs from CRC Compared to Healthy Subjects
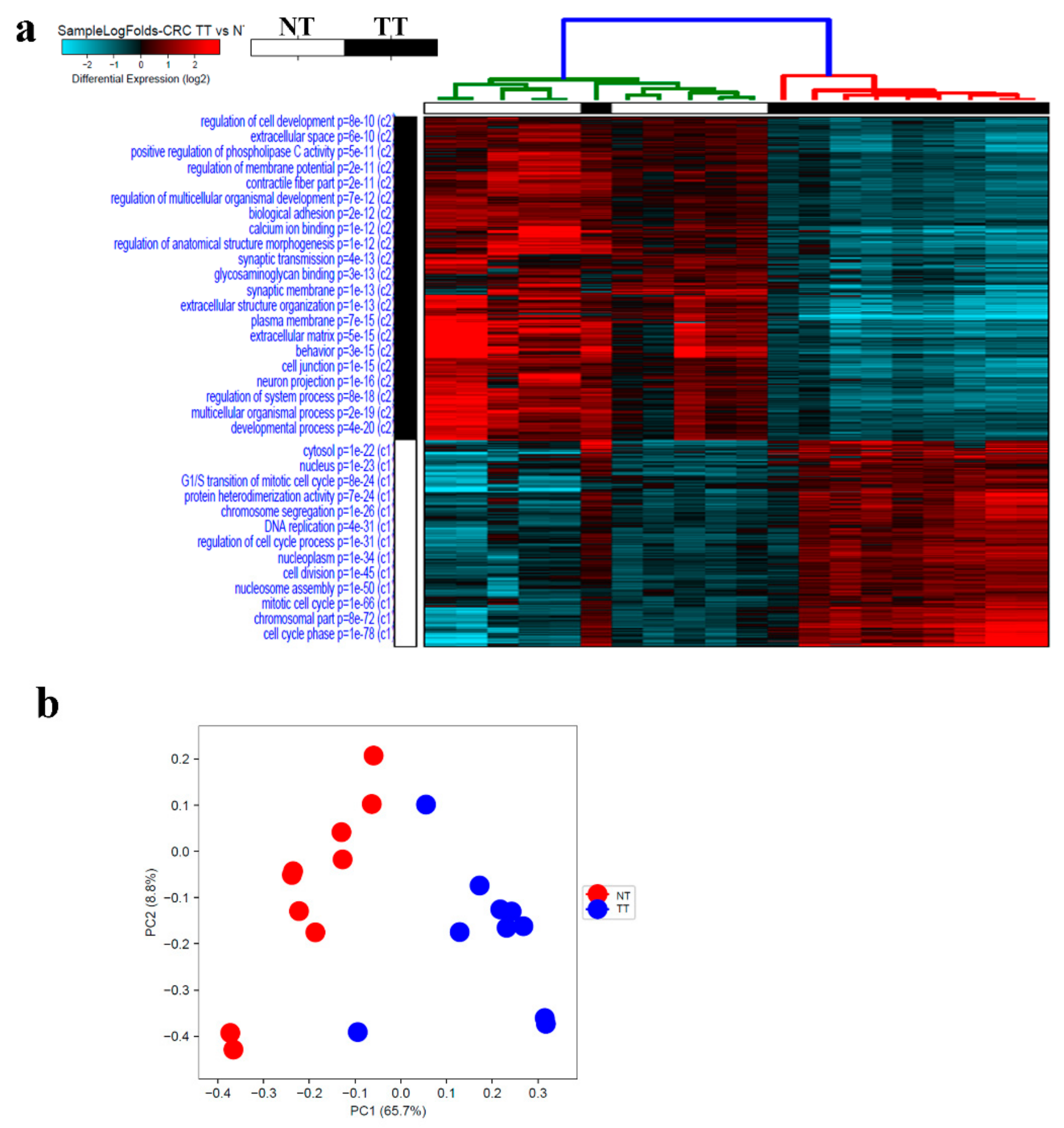
2.4. Alterations in CRC Tumor Tissue Compared to Adjacent Normal Tissue
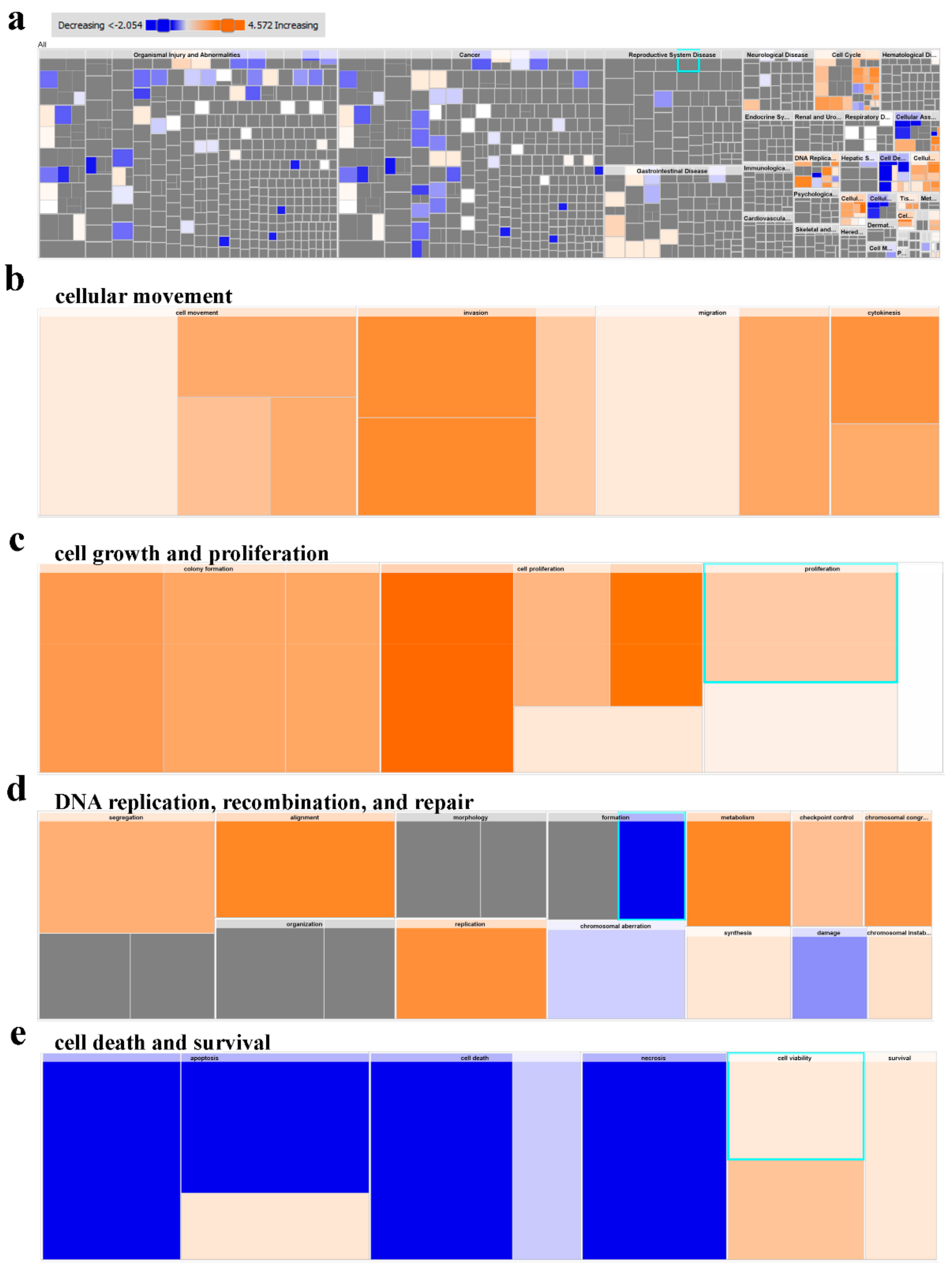
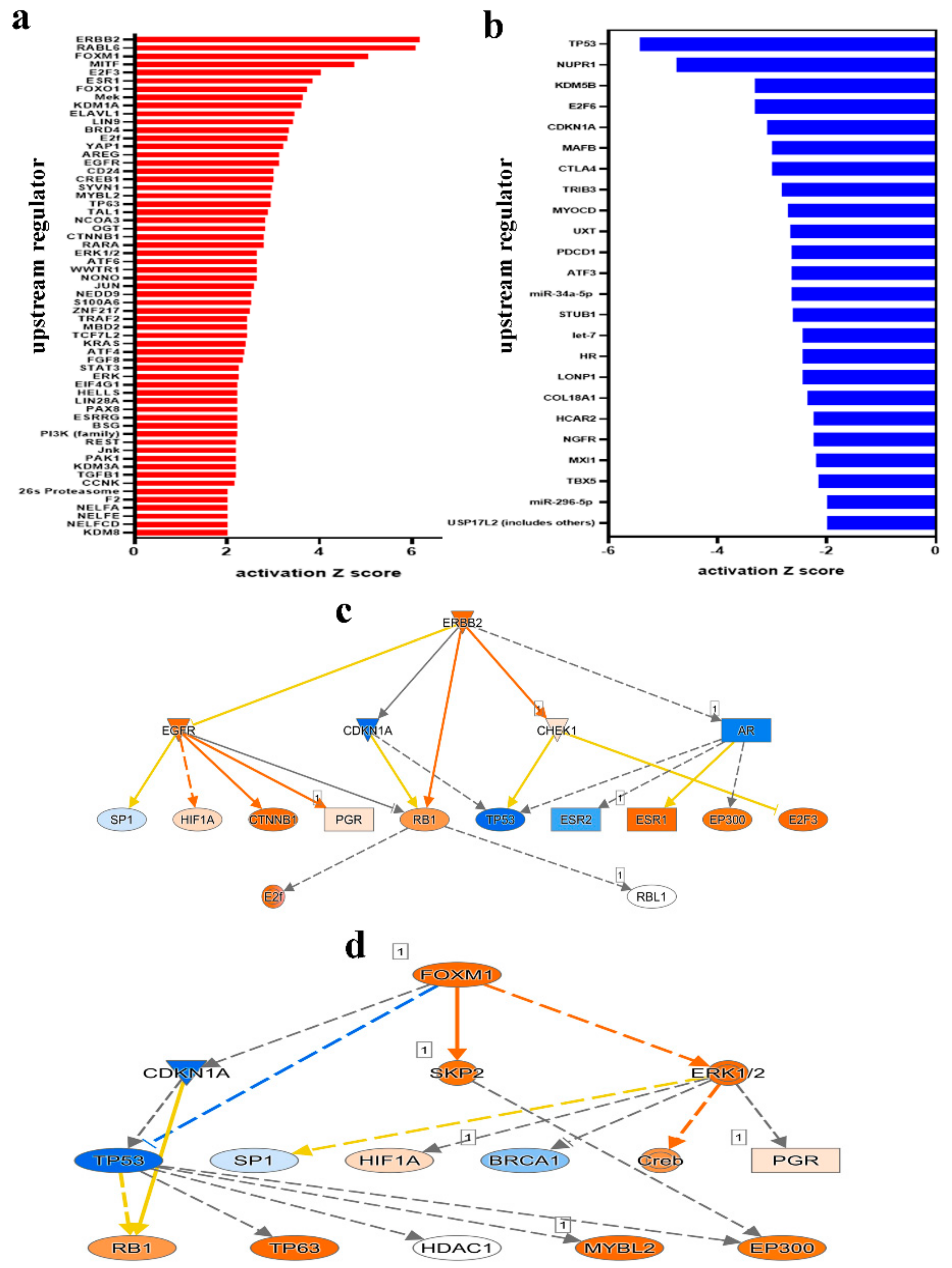
2.5. Multiple Altered Functional Categories and Signaling Networks in CRC
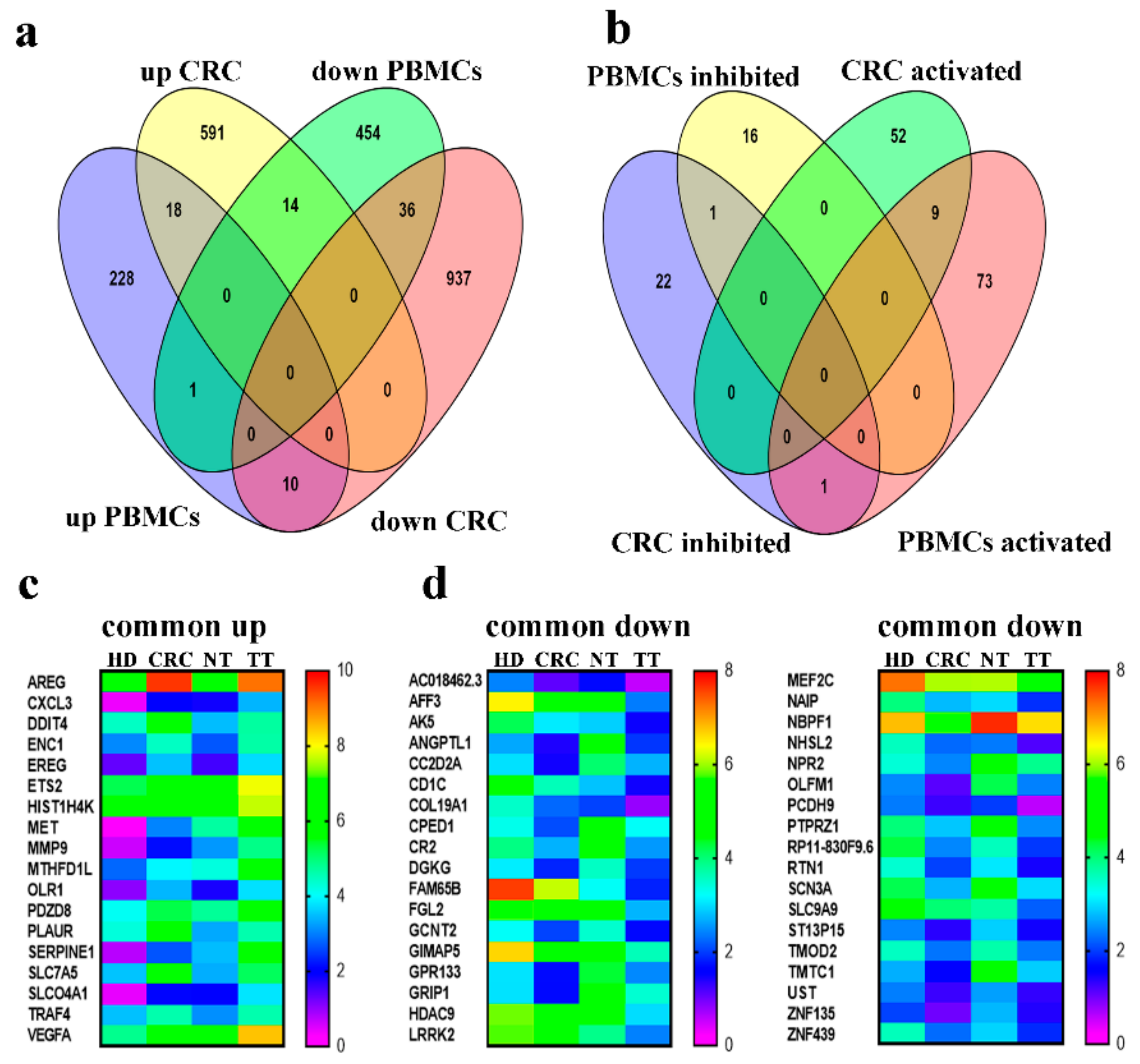
2.6. Commonality and Differences in Gene Expression Between PBMCs and CRC Tumor Tissue
3. Discussion
4. Materials and Methods
4.1. Ethics and Sample Collection
4.2. PBMC Isolation and RNA Extraction
4.3. Tumor Tissue and RNA Extraction
4.4. RNA Concentration and Quality Assessment
4.5. Library Preparation and RNA sequencing
4.6. RNA-Seq and Ingenuity Pathway Analyses
4.7. Quantitative Reverse Transcription PCR (qRT-PCR)
4.8. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Society, A.C. Key Statistics for Colorectal Cancer. Available online: https://www.cancer.org/cancer/colon-rectal-cancer/about/key-statistics.html#references (accessed on 1 February 2019).
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA A Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef]
- Mishra, J.; Drummond, J.; Quazi, S.H.; Karanki, S.S.; Shaw, J.J.; Chen, B.; Kumar, N. Prospective of colon cancer treatments and scope for combinatorial approach to enhanced cancer cell apoptosis. Crit. Rev. Oncol. Hematol. 2013, 86, 232–250. [Google Scholar] [CrossRef]
- Kelty, E.; Ward, S.V.; Cadby, G.; McCarthy, N.S.; O’Leary, P.; Moses, E.K.; Ee, H.C.; Preen, D.B. Familial and non-familial risk factors associated with incidence of colorectal cancer in young and middle-aged persons in Western Australia. Cancer Epidemiol. 2019, 62, 101591. [Google Scholar] [CrossRef]
- Jasperson, K.W.; Tuohy, T.M.; Neklason, D.W.; Burt, R.W. Hereditary and familial colon cancer. Gastroenterology 2010, 138, 2044–2058. [Google Scholar] [CrossRef]
- Lynch, H.T.; de la Chapelle, A. Genetic susceptibility to non-polyposis colorectal cancer. J. Med. Genet. 1999, 36, 801–818. [Google Scholar]
- Zare-Bandamiri, M.; Fararouei, M.; Zohourinia, S.; Daneshi, N.; Dianatinasab, M. Risk Factors Predicting Colorectal Cancer Recurrence Following Initial Treatment: A 5-year Cohort Study. Asian Pac. J. Cancer Prev. 2017, 18, 2465–2470. [Google Scholar] [CrossRef]
- Gupta, R.; Bhatt, L.K.; Johnston, T.P.; Prabhavalkar, K.S. Colon cancer stem cells: Potential target for the treatment of colorectal cancer. Cancer Biol. Ther. 2019, 20, 1068–1082. [Google Scholar] [CrossRef]
- Tao, L.; Zhu, Y.; Gu, Y.; Zheng, J.; Yang, J. ARHGAP25: A negative regulator of colorectal cancer (CRC) metastasis via the Wnt/beta-catenin pathway. Eur. J. Pharmacol. 2019, 858, 172476. [Google Scholar] [CrossRef]
- Tsao, T.; Beretov, J.; Ni, J.; Bai, X.; Bucci, J.; Graham, P.; Li, Y. Cancer stem cells in prostate cancer radioresistance. Cancer Lett. 2019, 28, 94–104. [Google Scholar] [CrossRef]
- Ciarloni, L.; Hosseinian, S.; Monnier-Benoit, S.; Imaizumi, N.; Dorta, G.; Ruegg, C.; Group, D.-C.-S. Discovery of a 29-gene panel in peripheral blood mononuclear cells for the detection of colorectal cancer and adenomas using high throughput real-time PCR. PLoS ONE 2015, 10, e0123904. [Google Scholar] [CrossRef]
- Le, D.T.; Kim, T.W.; Van Cutsem, E.; Geva, R.; Jager, D.; Hara, H.; Burge, M.; O’Neil, B.; Kavan, P.; Yoshino, T.; et al. Phase II Open-Label Study of Pembrolizumab in Treatment-Refractory, Microsatellite Instability-High/Mismatch Repair-Deficient Metastatic Colorectal Cancer: KEYNOTE-164. J. Clin. Oncol. 2019. [Google Scholar] [CrossRef]
- Zhang, L.; Yu, X.; Zheng, L.; Zhang, Y.; Li, Y.; Fang, Q.; Gao, R.; Kang, B.; Zhang, Q.; Huang, J.Y.; et al. Lineage tracking reveals dynamic relationships of T cells in colorectal cancer. Nature 2018, 564, 268–272. [Google Scholar] [CrossRef]
- Zhang, Y.; Zheng, L.; Zhang, L.; Hu, X.; Ren, X.; Zhang, Z. Deep single-cell RNA sequencing data of individual T cells from treatment-naive colorectal cancer patients. Sci. Data 2019, 6, 131. [Google Scholar] [CrossRef]
- Toor, S.M.; Syed Khaja, A.S.; El Salhat, H.; Bekdache, O.; Kanbar, J.; Jaloudi, M.; Elkord, E. Increased Levels of Circulating and Tumor-Infiltrating Granulocytic Myeloid Cells in Colorectal Cancer Patients. Front. Immunol. 2016, 7, 560. [Google Scholar] [CrossRef]
- Aljebreen, A.M. Clinico-pathological patterns of colorectal cancer in Saudi Arabia: Younger with an advanced stage presentation. Saudi J. Gastroenterol. 2007, 13, 84–87. [Google Scholar] [CrossRef]
- Arafa, M.A.; Farhat, K. Colorectal Cancer in the Arab World—Screening Practices and Future Prospects. Asian Pac. J. Cancer Prev. 2015, 16, 7425–7430. [Google Scholar]
- Nichita, C.; Ciarloni, L.; Monnier-Benoit, S.; Hosseinian, S.; Dorta, G.; Ruegg, C. A novel gene expression signature in peripheral blood mononuclear cells for early detection of colorectal cancer. Aliment. Pharmacol. Ther. 2014, 39, 507–517. [Google Scholar] [CrossRef]
- Zhang, G.L.; Pan, L.L.; Huang, T.; Wang, J.H. The transcriptome difference between colorectal tumor and normal tissues revealed by single-cell sequencing. J. Cancer 2019, 10, 5883–5890. [Google Scholar] [CrossRef]
- Kalmar, A.; Nagy, Z.B.; Galamb, O.; Csabai, I.; Bodor, A.; Wichmann, B.; Valcz, G.; Bartak, B.K.; Tulassay, Z.; Igaz, P.; et al. Genome-wide expression profiling in colorectal cancer focusing on lncRNAs in the adenoma-carcinoma transition. BMC Cancer 2019, 19, 1059. [Google Scholar] [CrossRef]
- Yang, Y.; Zhao, Y.; Zhang, W.; Bai, Y. Whole transcriptome sequencing identifies crucial genes associated with colon cancer and elucidation of their possible mechanisms of action. Onco Targets Ther. 2019, 12, 2737–2747. [Google Scholar] [CrossRef]
- Bien, S.A.; Su, Y.R.; Conti, D.V.; Harrison, T.A.; Qu, C.; Guo, X.; Lu, Y.; Albanes, D.; Auer, P.L.; Banbury, B.L.; et al. Genetic variant predictors of gene expression provide new insight into risk of colorectal cancer. Hum. Genet. 2019, 138, 307–326. [Google Scholar] [CrossRef]
- Chen, M.C.; Baskaran, R.; Lee, N.H.; Hsu, H.H.; Ho, T.J.; Tu, C.C.; Lin, Y.M.; Viswanadha, V.P.; Kuo, W.W.; Huang, C.Y. CXCL2/CXCR2 axis induces cancer stem cell characteristics in CPT-11-resistant LoVo colon cancer cells via Galphai-2 and Galphaq/11. J. Cell. Physiol. 2019, 234, 11822–11834. [Google Scholar] [CrossRef]
- Yu, Y.; Zhang, X.; Tian, H.; Zhang, Z.; Tian, Y. Knockdown of long non-coding RNA HOTAIR increases cisplatin sensitivity in ovarian cancer by inhibiting cisplatin-induced autophagy. J. BUON 2018, 23, 1396–1401. [Google Scholar]
- McLean, M.H.; Murray, G.I.; Stewart, K.N.; Norrie, G.; Mayer, C.; Hold, G.L.; Thomson, J.; Fyfe, N.; Hope, M.; Mowat, N.A.; et al. The inflammatory microenvironment in colorectal neoplasia. PLoS ONE 2011, 6, e15366. [Google Scholar] [CrossRef]
- Sasaki, S.; Baba, T.; Shinagawa, K.; Matsushima, K.; Mukaida, N. Crucial involvement of the CCL3-CCR5 axis-mediated fibroblast accumulation in colitis-associated carcinogenesis in mice. Int. J. Cancer 2014, 135, 1297–1306. [Google Scholar] [CrossRef]
- Servais, C.; Erez, N. From sentinel cells to inflammatory culprits: Cancer-associated fibroblasts in tumour-related inflammation. J. Pathol. 2013, 229, 198–207. [Google Scholar] [CrossRef]
- Cho, Y.B.; Lee, W.Y.; Choi, S.J.; Kim, J.; Hong, H.K.; Kim, S.H.; Choi, Y.L.; Kim, H.C.; Yun, S.H.; Chun, H.K.; et al. CC chemokine ligand 7 expression in liver metastasis of colorectal cancer. Oncol. Rep 2012, 28, 689–694. [Google Scholar] [CrossRef]
- Liu, G.; Zhang, L.; Zhao, Y. Modulation of immune responses through direct activation of Toll-like receptors to T cells. Clin. Exp. Immunol. 2010, 160, 168–175. [Google Scholar] [CrossRef]
- Braunstein, M.J.; Kucharczyk, J.; Adams, S. Targeting Toll-Like Receptors for Cancer Therapy. Target. Oncol. 2018, 13, 583–598. [Google Scholar] [CrossRef]
- Kawai, T.; Akira, S. Signaling to NF-kappaB by Toll-like receptors. Trends Mol. Med. 2007, 13, 460–469. [Google Scholar] [CrossRef]
- Wang, W.; Li, F.; Mao, Y.; Zhou, H.; Sun, J.; Li, R.; Liu, C.; Chen, W.; Hua, D.; Zhang, X. A miR-570 binding site polymorphism in the B7-H1 gene is associated with the risk of gastric adenocarcinoma. Hum. Genet. 2013, 132, 641–648. [Google Scholar] [CrossRef] [PubMed]
- Pope, B.L.; Sigindere, J.; Chourmouzis, E.; MacIntyre, P.; Goodman, M.G. 7-Allyl-8-oxoguanosine (loxoribine) inhibits the metastasis of B16 melanoma cells and has adjuvant activity in mice immunized with a B16 tumor vaccine. Cancer Immunol. Immunother. 1994, 38, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Walter, A.; Schafer, M.; Cecconi, V.; Matter, C.; Urosevic-Maiwald, M.; Belloni, B.; Schonewolf, N.; Dummer, R.; Bloch, W.; Werner, S.; et al. Aldara activates TLR7-independent immune defence. Nat. Commun. 2013, 4, 1560. [Google Scholar] [CrossRef] [PubMed]
- Chi, H.; Li, C.; Zhao, F.S.; Zhang, L.; Ng, T.B.; Jin, G.; Sha, O. Anti-tumor Activity of Toll-Like Receptor 7 Agonists. Front. Pharmacol. 2017, 8, 304. [Google Scholar] [CrossRef]
- Xia, Y.; Shen, S.; Verma, I.M. NF-kappaB, an active player in human cancers. Cancer Immunol. Res. 2014, 2, 823–830. [Google Scholar] [CrossRef]
- Turner, M.D.; Nedjai, B.; Hurst, T.; Pennington, D.J. Cytokines and chemokines: At the crossroads of cell signalling and inflammatory disease. Biochim. Biophys. Acta. 2014, 1843, 2563–2582. [Google Scholar] [CrossRef]
- Okazaki, S.; Stintzing, S.; Sunakawa, Y.; Cao, S.; Zhang, W.; Yang, D.; Ning, Y.; Matsusaka, S.; Berger, M.D.; Miyamoto, Y.; et al. Predictive value of TLR7 polymorphism for cetuximab-based chemotherapy in patients with metastatic colorectal cancer. Int. J. Cancer 2017, 141, 1222–1230. [Google Scholar] [CrossRef]
- Rhee, S.H.; Im, E.; Pothoulakis, C. Toll-like receptor 5 engagement modulates tumor development and growth in a mouse xenograft model of human colon cancer. Gastroenterology 2008, 135, 518–528. [Google Scholar] [CrossRef]
- Klimosch, S.N.; Forsti, A.; Eckert, J.; Knezevic, J.; Bevier, M.; von Schonfels, W.; Heits, N.; Walter, J.; Hinz, S.; Lascorz, J.; et al. Functional TLR5 genetic variants affect human colorectal cancer survival. Cancer Res. 2013, 73, 7232–7242. [Google Scholar] [CrossRef]
- Waldner, M.J.; Foersch, S.; Neurath, M.F. Interleukin-6—A key regulator of colorectal cancer development. Int. J. Biol. Sci. 2012, 8, 1248–1253. [Google Scholar] [CrossRef]
- Grimm, M.; Kim, M.; Rosenwald, A.; Heemann, U.; Germer, C.T.; Waaga-Gasser, A.M.; Gasser, M. Toll-like receptor (TLR) 7 and TLR8 expression on CD133+ cells in colorectal cancer points to a specific role for inflammation-induced TLRs in tumourigenesis and tumour progression. Eur. J. Cancer 2010, 46, 2849–2857. [Google Scholar] [CrossRef] [PubMed]
- Kelly, M.G.; Alvero, A.B.; Chen, R.; Silasi, D.A.; Abrahams, V.M.; Chan, S.; Visintin, I.; Rutherford, T.; Mor, G. TLR-4 signaling promotes tumor growth and paclitaxel chemoresistance in ovarian cancer. Cancer Res. 2006, 66, 3859–3868. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Zhao, J.; Li, H.; He, K.L.; Chen, Y.; Chen, S.H.; Mayer, L.; Unkeless, J.C.; Xiong, H. Toll-like receptors on tumor cells facilitate evasion of immune surveillance. Cancer Res. 2005, 65, 9108. [Google Scholar] [CrossRef] [PubMed]
- Bohnhorst, J.; Rasmussen, T.; Moen, S.H.; Flottum, M.; Knudsen, L.; Borset, M.; Espevik, T.; Sundan, A. Toll-like receptors mediate proliferation and survival of multiple myeloma cells. Leukemia 2006, 20, 1138–1144. [Google Scholar] [CrossRef]
- Burstein, H.J. The distinctive nature of HER2-positive breast cancers. N. Engl. J. Med. 2005, 353, 1652–1654. [Google Scholar] [CrossRef]
- Ruschoff, J.; Hanna, W.; Bilous, M.; Hofmann, M.; Osamura, R.Y.; Penault-Llorca, F.; van de Vijver, M.; Viale, G. HER2 testing in gastric cancer: a practical approach. Mod. Path. 2012, 25, 637–650. [Google Scholar] [CrossRef]
- Chiosea, S.I.; Williams, L.; Griffith, C.C.; Thompson, L.D.; Weinreb, I.; Bauman, J.E.; Luvison, A.; Roy, S.; Seethala, R.R.; Nikiforova, M.N. Molecular characterization of apocrine salivary duct carcinoma. Am. J. Surg. Pathol. 2015, 39, 744–752. [Google Scholar] [CrossRef]
- Chung, Y.W.; Kim, S.; Hong, J.H.; Lee, J.K.; Lee, N.W.; Lee, Y.S.; Song, J.Y. Overexpression of HER2/HER3 and clinical feature of ovarian cancer. J. Gynecol. Oncol. 2019, 30, e75. [Google Scholar] [CrossRef]
- Jeong, J.H.; Kim, J.; Hong, Y.S.; Kim, D.; Kim, J.E.; Kim, S.Y.; Kim, K.P.; Yoon, Y.K.; Kim, D.; Chun, S.M.; et al. HER2 Amplification and Cetuximab Efficacy in Patients With Metastatic Colorectal Cancer Harboring Wild-type RAS and BRAF. Clin. Colorectal Cancer 2017, 16, e147–e152. [Google Scholar] [CrossRef]
- Siravegna, G.; Sartore-Bianchi, A.; Nagy, R.J.; Raghav, K.; Odegaard, J.I.; Lanman, R.B.; Trusolino, L.; Marsoni, S.; Siena, S.; Bardelli, A. Plasma HER2 (ERBB2) Copy Number Predicts Response to HER2-targeted Therapy in Metastatic Colorectal Cancer. Clin. Cancer Res. 2019, 25, 3046–3053. [Google Scholar] [CrossRef]
- Wierstra, I.; Alves, J. FOXM1, a typical proliferation-associated transcription factor. Biol. Chem. 2007, 388, 1257–1274. [Google Scholar] [CrossRef] [PubMed]
- Vishnubalaji, R.; Hamam, R.; Yue, S.; Al-Obeed, O.; Kassem, M.; Liu, F.F.; Aldahmash, A.; Alajez, N.M. MicroRNA-320 suppresses colorectal cancer by targeting SOX4, FOXM1 and FOXQ1. Oncotarget 2016, 7, 35789–35802. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Yang, R.; Yang, L.; Wang, D.; Zhou, X.; Sun, Y. circANKS1B regulates FOXM1 expression and promotes cell migration and invasion by functioning as a sponge of the miR-149 in colorectal cancer. Onco Targets Ther. 2019, 12, 4065–4073. [Google Scholar] [CrossRef] [PubMed]
- Fei, B.Y.; He, X.; Ma, J.; Zhang, M.; Chai, R. FoxM1 is associated with metastasis in colorectal cancer through induction of the epithelial-mesenchymal transition. Oncol. Lett. 2017, 14, 6553–6561. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Yang, K.; Jiang, B.; Lu, Y.; Shu, Q.; Zhai, P.; Zhi, Q.; Li, Q. FOXM1 promotes the growth and metastasis of colorectal cancer via activation of beta-catenin signaling pathway. Cancer Manag. Res. 2019, 11, 3779–3790. [Google Scholar] [CrossRef] [PubMed]
- Elango, R.; Vishnubalaji, R.; Manikandan, M.; Binhamdan, S.I.; Siyal, A.A.; Alshawakir, Y.A.; Alfayez, M.; Aldahmash, A.; Alajez, N.M. Concurrent targeting of BMI1 and CDK4/6 abrogates tumor growth in vitro and in vivo. Sci. Rep. 2019, 9, 13696. [Google Scholar] [CrossRef]
- Donehower, L.A.; Harvey, M.; Slagle, B.L.; McArthur, M.J.; Montgomery, C.A., Jr.; Butel, J.S.; Bradley, A. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature 1992, 356, 215–221. [Google Scholar] [CrossRef]
- Vousden, K.H.; Lu, X. Live or let die: The cell’s response to p53. Nat. Rev. Cancer 2002, 2, 594–604. [Google Scholar] [CrossRef]
- Velculescu, V.E.; El-Deiry, W.S. Biological and clinical importance of the p53 tumor suppressor gene. Clin. Chem. 1996, 42, 858–868. [Google Scholar]
- Busser, B.; Sancey, L.; Brambilla, E.; Coll, J.-L.; Hurbin, A. The multiple roles of amphiregulin in human cancer. Biochim/Biophys. Acta 2011, 1816, 119–131. [Google Scholar] [CrossRef]
- Lin, Y.M.; Lu, C.C.; Hsiang, Y.P.; Pi, S.C.; Chen, C.I.; Cheng, K.C.; Pan, H.L.; Chien, P.H.; Chen, Y.J. c-Met inhibition is required for the celecoxib-attenuated stemness property of human colorectal cancer cells. J. Cell. Physiol. 2019, 234, 10336–10344. [Google Scholar] [CrossRef] [PubMed]
- Pentheroudakis, G.; Mavroeidis, L.; Papadopoulou, K.; Koliou, G.-A.; Bamia, C.; Chatzopoulos, K.; Samantas, E.; Mauri, D.; Efstratiou, I.; Pectasides, D. Angiogenic and Antiangiogenic VEGFA Splice Variants in Colorectal Cancer: Prospective Retrospective Cohort Study in Patients Treated With Irinotecan-Based Chemotherapy and Bevacizumab. Clin. Colorectal Cancer 2019, 18, 370–384. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; Zhang, W.; Ma, J.; Dai, Y.; Zhang, Q.; Liu, Q.; Yang, B.; Li, G. MicroRNA-139-5p regulates chronic inflammation by suppressing nuclear factor-κB activity to inhibit cell proliferation and invasion in colorectal cancer. Exp. Ther. Med. 2019, 18, 4049–4057. [Google Scholar] [CrossRef] [PubMed]
- Onder, T.T.; Gupta, P.B.; Mani, S.A.; Yang, J.; Lander, E.S.; Weinberg, R.A. Loss of E-cadherin promotes metastasis via multiple downstream transcriptional pathways. Cancer Res. 2008, 68, 3645–3654. [Google Scholar] [CrossRef] [PubMed]
- Shahine, A.; Reinink, P.; Reijneveld, J.F.; Gras, S.; Holzheimer, M.; Cheng, T.-Y.; Minnaard, A.J.; Altman, J.D.; Lenz, S.; Prandi, J. A T-cell receptor escape channel allows broad T-cell response to CD1b and membrane phospholipids. Nat. Commun. 2019, 10, 56. [Google Scholar] [CrossRef] [PubMed]
- Schanze, D.; Kayserili, H.; Satkın, B.N.; Altunoglu, U.; Zenker, M. Fraser syndrome due to mutations in GRIP1—clinical phenotype in two families and expansion of the mutation spectrum. Am. J. Med. Genet. A 2014, 164, 837–840. [Google Scholar] [CrossRef]
- Bray, N.L.; Pimentel, H.; Melsted, P.; Pachter, L. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 2016, 34, 525–527. [Google Scholar] [CrossRef]
- Olsson, A.; Venkatasubramanian, M.; Chaudhri, V.K.; Aronow, B.J.; Salomonis, N.; Singh, H.; Grimes, H.L. Single-cell analysis of mixed-lineage states leading to a binary cell fate choice. Nature 2016, 537, 698–702. [Google Scholar] [CrossRef]
- Vishnubalaji, R.; Shaath, H.; Elkord, E.; Alajez, N.M. Long non-coding RNA (lncRNA) transcriptional landscape in breast cancer identifies LINC01614 as non-favorable prognostic biomarker regulated by TGFbeta and focal adhesion kinase (FAK) signaling. Cell Death Discov. 2019, 5, 109. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HD | CRC Patients | |
|---|---|---|
| Number | 15 | 10 |
| Age (Median) | 30 (21–47) * | 41 (36–62) * |
| Gender (Male, Female) | 7:8 | 6:4 |
| TNM stage | ||
| I | 0 | |
| II | 3 | |
| III | 7 | |
| IV | 0 | |
| Histological grade | 7 | |
| Poorly differentiated | ||
| Well/Moderately differentiated | 3 |
| Gene | Forward Primer (5′-3′) | Reverse Primer (5′-3′) | Product Size |
|---|---|---|---|
| CXCL2 | GAAAGCTTGTCTCAACCCCG | TGGTCAGTTGGATTTGCCATTTT | 82 |
| IL8 | GGAGAAGTTTTTGAAGAGGGCTG | TTTGCTTGAAGTTTCACTGGCA | 95 |
| CCL7 | TTGCTCAGCCAGTTGGGATTA | GCTCTCCAGCCTCTGCTTAG | 86 |
| CXCL3 | AGATACTGAACAAGGGGAGCAC | TCCTTTCCAGCTGTCCCTAGA | 150 |
| IL10 | TACGGCGCTGTCATCGATTT | TAGAGTCGCCACCCTGATGT | 191 |
| CCL3 | GCTCTCTGCAACCAGTTCTCT | TCGCTTGGTTAGGAAGATGACA | 159 |
| TLR7 | TCAAGAAAGTTGATGCTATTGGGC | ACCATCTAGCCCCAAGGAGT | 195 |
| TLR5 | GTCACCAAACCAGGGATGCT | GGGCAAAGTCAATTGCCAGG | 100 |
| TLR10 | GCCAATTCTGACCGTGTCAAC | CTGCTGAATTCCCACGGCTT | 79 |
| TLR8 | GAAACATGGTTCTCTTGACACTTCA | GGCTTCCTTCAAGGTCTCCT | 75 |
| TNFSF10 | TGAAGCAGATGCAGGACAAGT | TGGTTTCCTCAGAGGTTCTCAAAA | 174 |
| CD79B | CGGAATCCCAAAGGTAGTGCT | TCCAGAGCCAGCTCACATTG | 130 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shaath, H.; Toor, S.; Nair, V.S.; Elkord, E.; Alajez, N.M. Transcriptomic Analyses Revealed Systemic Alterations in Gene Expression in Circulation and Tumor Microenvironment of Colorectal Cancer Patients. Cancers 2019, 11, 1994. https://doi.org/10.3390/cancers11121994
Shaath H, Toor S, Nair VS, Elkord E, Alajez NM. Transcriptomic Analyses Revealed Systemic Alterations in Gene Expression in Circulation and Tumor Microenvironment of Colorectal Cancer Patients. Cancers. 2019; 11(12):1994. https://doi.org/10.3390/cancers11121994
Chicago/Turabian StyleShaath, Hibah, Salman Toor, Varun Sasidharan Nair, Eyad Elkord, and Nehad M. Alajez. 2019. "Transcriptomic Analyses Revealed Systemic Alterations in Gene Expression in Circulation and Tumor Microenvironment of Colorectal Cancer Patients" Cancers 11, no. 12: 1994. https://doi.org/10.3390/cancers11121994
APA StyleShaath, H., Toor, S., Nair, V. S., Elkord, E., & Alajez, N. M. (2019). Transcriptomic Analyses Revealed Systemic Alterations in Gene Expression in Circulation and Tumor Microenvironment of Colorectal Cancer Patients. Cancers, 11(12), 1994. https://doi.org/10.3390/cancers11121994