Proteostasis in the Endoplasmic Reticulum: Road to Cure


{kind=link}
{kind=link}
Abstract
1. Introduction
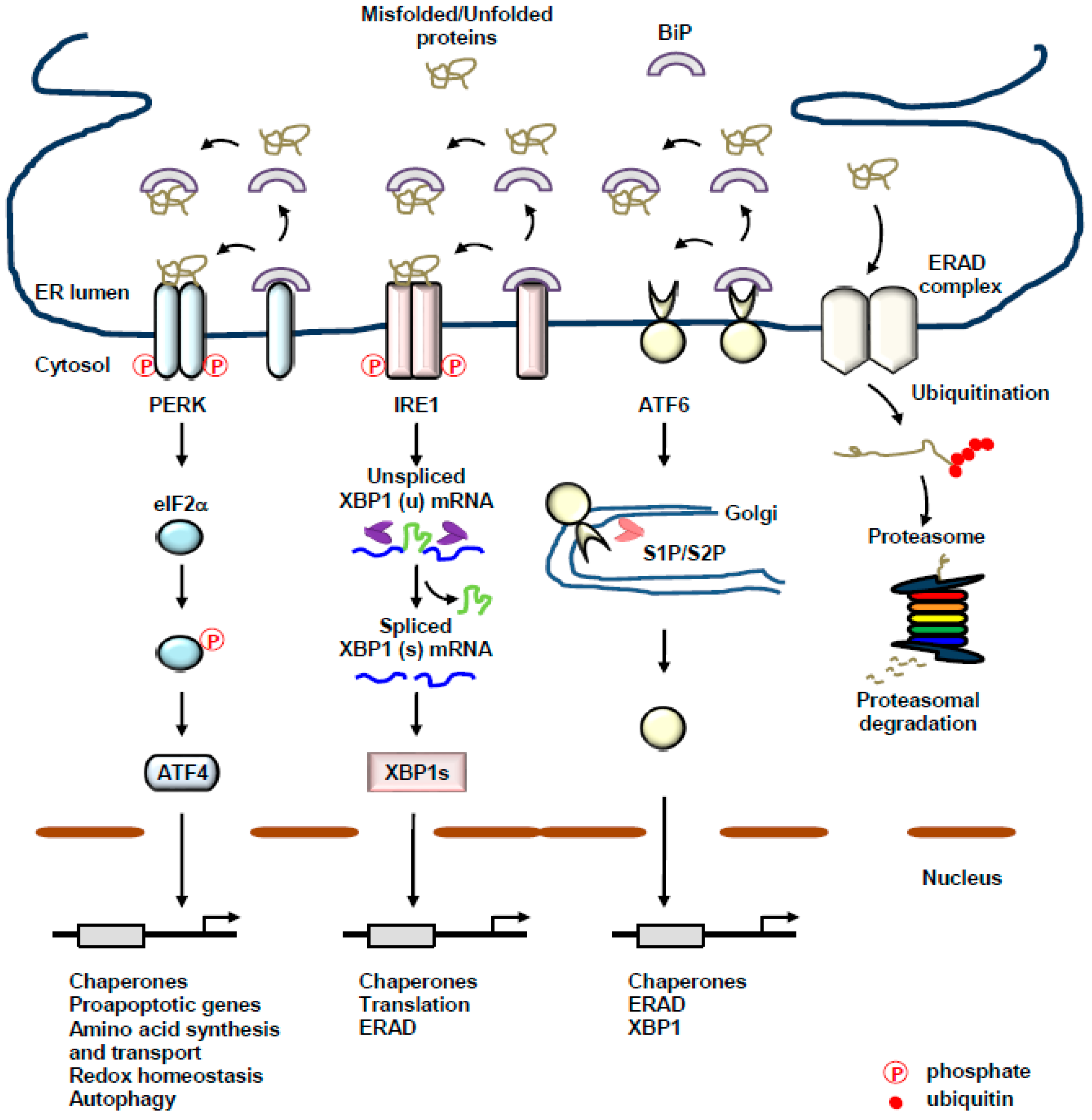
2. The UPR of the ER: A Complex Interplay between Three Transmembrane ER-Resident Stress Sensors
2.1. PERK
2.2. IRE1
2.3. ATF6
3. ER-Mitochondria Interconnection and UPR
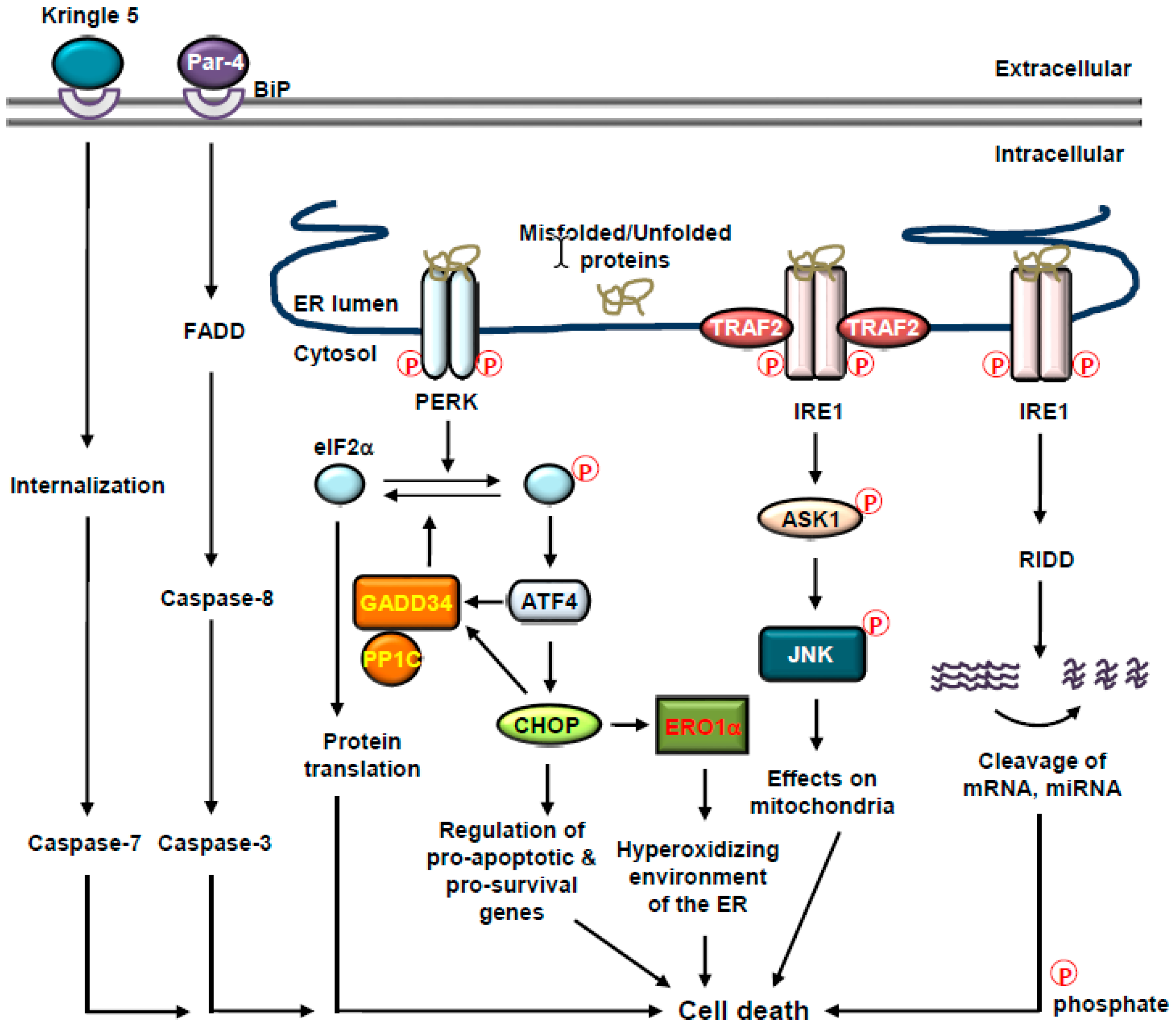
4. Cell Fate Decisions and UPR
4.1. Cell Fate Decisions and BiP
4.2. Cell Fate Decisions and PERK
4.3. Cell Fate Decisions and IRE1
4.4. Cell Fate Decisions and ER-Associated Caspases
5. UPR and Cancer
5.1. UPR and Tumorigenesis
5.1.1. Tumorigenesis and BiP
5.1.2. Tumorigenesis and IRE1
5.1.3. Tumorigenesis and PERK
5.1.4. Tumorigenesis and ATF6
5.2. UPR and Metastasis
5.3. UPR and Cancer Immunogenicity
6. Targeting the UPR in Cancer
6.1. Modulation of PERK
6.2. Modulation of IRE1
6.3. Modulation of ATF6
6.4. Modulation of ERAD
6.5. Modulation of Chaperones
6.6. ER Stress and Immunogenic Cell Death
7. Conclusions and Future Perspectives
Funding
Conflicts of Interest
References
- Alberts, B.; Johnson, A.; Lewis, J.; Raff, M.; Roberts, K.; Walter, P. The endoplasmic reticulum. In Molecular Biology of the Cell, 4th ed.; Garland Science: New York, NY, USA, 2002. [Google Scholar]
- Görlach, A.; Klappa, P.; Kietzmann, D.T. The endoplasmic reticulum: Folding, calcium homeostasis, signaling, and redox control. Antioxid. Redox Signal. 2006, 8, 1391–1418. [Google Scholar] [CrossRef] [PubMed]
- Berridge, M.J.; Lipp, P.; Bootman, M.D. The versatility and universality of calcium signalling. Nat. Rev. Mol. Cell Biol. 2000, 1, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Oakes, S.A.; Papa, F.R. The role of endoplasmic reticulum stress in human pathology. Annu. Rev. Pathol. Mech. Dis. 2015, 10, 173–194. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Kaufman, R.J. The impact of the endoplasmic reticulum protein-folding environment on cancer development. Nat. Rev. Cancer 2014, 14, 581–597. [Google Scholar] [CrossRef]
- Schröder, M.; Kaufman, R.J. The mammalian unfolded protein response. Annu. Rev. Biochem. 2005, 74, 739–789. [Google Scholar] [CrossRef]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef]
- Chiti, F.; Dobson, C.M. Protein misfolding, functional amyloid, and human disease. Annu. Rev. Biochem. 2006, 75, 333–366. [Google Scholar] [CrossRef]
- Wang, M.; Kaufman, R.J. Protein misfolding in the endoplasmic reticulum as a conduit to human disease. Nature 2016, 529, 326–335. [Google Scholar] [CrossRef] [PubMed]
- Schröder, M. Endoplasmic reticulum stress responses. Cell. Mol. Life Sci. 2008, 65, 862–894. [Google Scholar] [CrossRef] [PubMed]
- Walter, P.; Ron, D. The unfolded protein response: From stress pathway to homeostatic regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef] [PubMed]
- Moon, H.W.; Han, H.G.; Jeon, Y.J. Protein quality control in the endoplasmic reticulum and cancer. Int. J. Mol. Sci. 2018, 19, 3020. [Google Scholar] [CrossRef] [PubMed]
- Yoo, Y.S.; Han, H.G.; Jeon, Y.J. Unfolded protein response of the endoplasmic reticulum in tumor progression and immunogenicity. Oxidative Med. Cell. Longev. 2017, 2017, 2969271. [Google Scholar] [CrossRef] [PubMed]
- Bertolotti, A.; Wang, X.; Novoa, I.; Jungreis, R.; Schlessinger, K.; Cho, J.H.; West, A.B.; Ron, D. Increased sensitivity to dextran sodium sulfate colitis in ire1β-deficient mice. J. Clin. Investig. 2001, 107, 585–593. [Google Scholar] [CrossRef] [PubMed]
- Otero, J.H.; Lizák, B.; Hendershot, L.M. Life and death of a bip substrate. Semin. Cell Dev. Biol. 2010, 21, 472–478. [Google Scholar] [CrossRef] [PubMed]
- Bertolotti, A.; Zhang, Y.; Hendershot, L.M.; Harding, H.P.; Ron, D. Dynamic interaction of Bip and ER stress transducers in the unfolded-protein response. Nat. Cell Biol. 2000, 2, 326–332. [Google Scholar] [CrossRef]
- Pincus, D.; Chevalier, M.W.; Aragón, T.; Van Anken, E.; Vidal, S.E.; El-Samad, H.; Walter, P. Bip binding to the ER-stress sensor ire1 tunes the homeostatic behavior of the unfolded protein response. PLoS Biol. 2010, 8, e1000415. [Google Scholar] [CrossRef]
- Groenendyk, J.; Peng, Z.; Dudek, E.; Fan, X.; Mizianty, M.J.; Dufey, E.; Urra, H.; Sepulveda, D.; Rojas-Rivera, D.; Lim, Y. Interplay between the oxidoreductase pdia6 and microrna-322 controls the response to disrupted endoplasmic reticulum calcium homeostasis. Sci. Signal. 2014, 7, 54. [Google Scholar] [CrossRef]
- Eletto, D.; Eletto, D.; Dersh, D.; Gidalevitz, T.; Argon, Y. Protein disulfide isomerase a6 controls the decay of ire1α signaling via disulfide-dependent association. Mol. Cell 2014, 53, 562–576. [Google Scholar] [CrossRef]
- Higa, A.; Taouji, S.; Lhomond, S.; Jensen, D.; Fernandez-Zapico, M.E.; Simpson, J.C.; Pasquet, J.M.; Schekman, R.; Chevet, E. Endoplasmic reticulum stress-activated transcription factor atf6α requires the disulfide isomerase pdia5 to modulate chemoresistance. Mol. Cell. Biol. 2014, 34, 1839–1849. [Google Scholar] [CrossRef]
- Gardner, B.M.; Walter, P. Unfolded proteins are ire1-activating ligands that directly induce the unfolded protein response. Science 2011, 333, 1891–1894. [Google Scholar] [CrossRef]
- Korennykh, A.V.; Egea, P.F.; Korostelev, A.A.; Finer-Moore, J.; Zhang, C.; Shokat, K.M.; Stroud, R.M.; Walter, P. The unfolded protein response signals through high-order assembly of ire1. Nature 2009, 457, 687–693. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Korennykh, A.V.; Behrman, S.L.; Walter, P. Mammalian endoplasmic reticulum stress sensor ire1 signals by dynamic clustering. Proc. Natl. Acad. Sci. USA 2010, 107, 16113–16118. [Google Scholar] [CrossRef] [PubMed]
- Harding, H.P.; Zhang, Y.; Ron, D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 1999, 397, 271–274. [Google Scholar] [CrossRef] [PubMed]
- Harding, H.P.; Zhang, Y.; Zeng, H.; Novoa, I.; Lu, P.D.; Calfon, M.; Sadri, N.; Yun, C.; Popko, B.; Paules, R. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol. Cell 2003, 11, 619–633. [Google Scholar] [CrossRef]
- Vattem, K.M.; Wek, R.C. Reinitiation involving upstream ORFs regulates ATF4 mRNA translation in mammalian cells. Proc. Natl. Acad. Sci. USA 2004, 101, 11269–11274. [Google Scholar] [CrossRef]
- Marciniak, S.J.; Yun, C.Y.; Oyadomari, S.; Novoa, I.; Zhang, Y.; Jungreis, R.; Nagata, K.; Harding, H.P.; Ron, D. Chop induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev. 2004, 18, 3066–3077. [Google Scholar] [CrossRef]
- Cullinan, S.B.; Diehl, J.A. Coordination of ER and oxidative stress signaling: The PERK/Nrf2 signaling pathway. Int. J. Biochem. Cell Biol. 2006, 38, 317–332. [Google Scholar] [CrossRef]
- Tirasophon, W.; Welihinda, A.A.; Kaufman, R.J. A stress response pathway from the endoplasmic reticulum to the nucleus requires a novel bifunctional protein kinase/endoribonuclease (ire1p) in mammalian cells. Genes Dev. 1998, 12, 1812–1824. [Google Scholar] [CrossRef]
- Sepulveda, D.; Rojas-Rivera, D.; Rodriguez, D.A.; Groenendyk, J.; Köhler, A.; Lebeaupin, C.; Ito, S.; Urra, H.; Carreras-Sureda, A.; Hazari, Y. Interactome screening identifies the ER luminal chaperone hsp47 as a regulator of the unfolded protein response transducer IRE1α. Mol. Cell 2018, 69, 238–252. [Google Scholar] [CrossRef]
- Lamriben, L.; Hebert, D.N. Activating and repressing IRE1α: The Hsp47 and Bip Tug of War. Mol. Cell 2018, 69, 159–160. [Google Scholar] [CrossRef]
- Yoshida, H.; Matsui, T.; Yamamoto, A.; Okada, T.; Mori, K. Xbp1 mrna is induced by atf6 and spliced by ire1 in response to ER stress to produce a highly active transcription factor. Cell 2001, 107, 881–891. [Google Scholar] [CrossRef]
- Liu, Y.; Adachi, M.; Zhao, S.; Hareyama, M.; Koong, A.; Luo, D.; Rando, T.; Imai, K.; Shinomura, Y. Preventing oxidative stress: A new role for XBP1. Cell Death Differ. 2009, 16, 847–857. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.H.; Iwakoshi, N.N.; Glimcher, L.H. Xbp-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol. Cell. Biol. 2003, 23, 7448–7459. [Google Scholar] [CrossRef] [PubMed]
- Haze, K.; Yoshida, H.; Yanagi, H.; Yura, T.; Mori, K. Mammalian transcription factor atf6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol. Biol. Cell 1999, 10, 3787–3799. [Google Scholar] [CrossRef]
- Lee, K.; Tirasophon, W.; Shen, X.; Michalak, M.; Prywes, R.; Okada, T.; Yoshida, H.; Mori, K.; Kaufman, R.J. IRE1-mediated unconventional mRNA splicing and S2P-mediated ATF6 cleavage merge to regulate XBP1 in signaling the unfolded protein response. Genes Dev. 2002, 16, 452–466. [Google Scholar] [CrossRef] [PubMed]
- Diedrich, K.; Fauser, B.C.J.M.; Devroey, P.; Griesinger, G. The role of the endometrium and embryo in human implantation. Hum. Reprod. Update 2007, 13, 365–377. [Google Scholar] [CrossRef]
- Bommiasamy, H.; Back, S.H.; Fagone, P.; Lee, K.; Meshinchi, S.; Vink, E.; Sriburi, R.; Frank, M.; Jackowski, S.; Kaufman, R.J. Atf6α induces xbp1-independent expansion of the endoplasmic reticulum. J. Cell Sci. 2009, 122, 1626–1636. [Google Scholar] [CrossRef]
- Rutkowski, D.T.; Hegde, R.S. Regulation of basal cellular physiology by the homeostatic unfolded protein response. J. Cell Biol. 2010, 189, 783–794. [Google Scholar] [CrossRef]
- Bernhard, W.; Rouiller, C. Close topographical relationship between mitochondria and ergastoplasm of liver cells in a definite phase of cellular activity. J. Cell Biol. 1956, 2, 73–78. [Google Scholar] [CrossRef]
- Sassano, M.L.; van Vliet, A.R.; Agostinis, P. Mitochondria-associated membranes as networking platforms and regulators of cancer cell fate. Front. Oncol. 2017, 7, 174. [Google Scholar] [CrossRef]
- Doghman-Bouguerra, M.; Lalli, E. Er-mitochondria interactions: Both strength and weakness within cancer cells. Biochim. Biophys. Acta BBA Mol. Cell Res. 2019, 1866, 650–662. [Google Scholar] [CrossRef]
- Cárdenas, C.; Miller, R.A.; Smith, I.; Bui, T.; Molgó, J.; Müller, M.; Vais, H.; Cheung, K.H.; Yang, J.; Parker, I. Essential regulation of cell bioenergetics by constitutive InsP3 receptor Ca2+ transfer to mitochondria. Cell 2010, 142, 270–283. [Google Scholar] [CrossRef]
- van Vliet, A.R.; Verfaillie, T.; Agostinis, P. New functions of mitochondria associated membranes in cellular signaling. Biochim. Biophys. Acta BBA Mol. Cell Res. 2014, 1843, 2253–2262. [Google Scholar] [CrossRef] [PubMed]
- van Vliet, A.R.; Agostinis, P. When under pressure, get closer: Perking up membrane contact sites during ER stress. Biochem. Soc. Trans. 2016, 44, 499–504. [Google Scholar] [CrossRef] [PubMed]
- Carreras-Sureda, A.; Pihán, P.; Hetz, C. The unfolded protein response: At the intersection between endoplasmic reticulum function and mitochondrial bioenergetics. Front. Oncol. 2017, 7, 55. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez, T.; Simmen, T. Endoplasmic reticulum chaperones tweak the mitochondrial calcium rheostat to control metabolism and cell death. Cell Calcium 2018, 70, 64–75. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, T.; Su, T.P. Sigma-1 receptor chaperones at the ER-mitochondrion interface regulate Ca2+ signaling and cell survival. Cell 2007, 131, 596–610. [Google Scholar] [CrossRef] [PubMed]
- Verfaillie, T.; Rubio, N.; Garg, A.; Bultynck, G.; Rizzuto, R.; Decuypere, J.; Piette, J.; Linehan, C.; Gupta, S.; Samali, A. Perk is required at the ER-mitochondrial contact sites to convey apoptosis after ROS-based ER stress. Cell Death Differ. 2012, 19, 1880–1891. [Google Scholar] [CrossRef]
- Chami, M.; Oulès, B.; Szabadkai, G.; Tacine, R.; Rizzuto, R.; Paterlini-Bréchot, P. Role of serca1 truncated isoform in the proapoptotic calcium transfer from ER to mitochondria during ER stress. Mol. Cell 2008, 32, 641–651. [Google Scholar] [CrossRef]
- Calì, T.; Ottolini, D.; Negro, A.; Brini, M. Enhanced parkin levels favor ER-mitochondria crosstalk and guarantee Ca2+ transfer to sustain cell bioenergetics. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2013, 1832, 495–508. [Google Scholar] [CrossRef]
- Urra, H.; Dufey, E.; Lisbona, F.; Rojas-Rivera, D.; Hetz, C. When ER stress reaches a dead end. Biochim. Biophys. Acta BBA Mol. Cell Res. 2013, 1833, 3507–3517. [Google Scholar] [CrossRef] [PubMed]
- Misra, U.K.; Deedwania, R.; Pizzo, S.V. Activation and cross-talk between Akt, Nf-κB, and unfolded protein response signaling in 1-LN prostate cancer cells consequent to ligation of cell surface-associated GRP78. J. Biol. Chem. 2006, 281, 13694–13707. [Google Scholar] [CrossRef] [PubMed]
- Burikhanov, R.; Zhao, Y.; Goswami, A.; Qiu, S.; Schwarze, S.R.; Rangnekar, V.M. The tumor suppressor Par-4 activates an extrinsic pathway for apoptosis. Cell 2009, 138, 377–388. [Google Scholar] [CrossRef] [PubMed]
- Davidson, D.J.; Haskell, C.; Majest, S.; Kherzai, A.; Egan, D.A.; Walter, K.A.; Schneider, A.; Gubbins, E.F.; Solomon, L.; Chen, Z. Kringle 5 of human plasminogen induces apoptosis of endothelial and tumor cells through surface-expressed glucose-regulated protein 78. Cancer Res. 2005, 65, 4663–4672. [Google Scholar] [CrossRef]
- Itoh, K.; Chiba, T.; Takahashi, S.; Ishii, T.; Igarashi, K.; Katoh, Y.; Oyake, T.; Hayashi, N.; Satoh, K.; Hatayama, I. An Nrf2/small Maf heterodimer mediates the induction of phase ii detoxifying enzyme genes through antioxidant response elements. Biochem. Biophys. Res. Commun. 1997, 236, 313–322. [Google Scholar] [CrossRef]
- Kensler, T.W.; Wakabayashi, N.; Biswal, S. Cell survival responses to environmental stresses via the keap1-Nrf2-are pathway. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 89–116. [Google Scholar] [CrossRef]
- Niture, S.K.; Jaiswal, A.K. Nrf2 protein up-regulates antiapoptotic protein Bcl-2 and prevents cellular apoptosis. J. Biol. Chem. 2012, 287, 9873–9886. [Google Scholar] [CrossRef]
- Niture, S.K.; Jaiswal, A.K. Nrf2-induced antiapoptotic Bcl-xL protein enhances cell survival and drug resistance. Free Radic. Biol. Med. 2013, 57, 119–131. [Google Scholar] [CrossRef]
- Chitnis, N.S.; Pytel, D.; Bobrovnikova-Marjon, E.; Pant, D.; Zheng, H.; Maas, N.L.; Frederick, B.; Kushner, J.A.; Chodosh, L.A.; Koumenis, C. Mir-211 is a prosurvival microrna that regulates chop expression in a perk-dependent manner. Mol. Cell 2012, 48, 353–364. [Google Scholar] [CrossRef]
- Lin, J.H.; Li, H.; Zhang, Y.; Ron, D.; Walter, P. Divergent effects of perk and ire1 signaling on cell viability. PLoS ONE 2009, 4, e4170. [Google Scholar] [CrossRef]
- Song, B.; Scheuner, D.; Ron, D.; Pennathur, S.; Kaufman, R.J. Chop deletion reduces oxidative stress, improves β cell function, and promotes cell survival in multiple mouse models of diabetes. J. Clin. Investig. 2008, 118, 3378–3389. [Google Scholar] [CrossRef] [PubMed]
- Woo, C.W.; Kutzler, L.; Kimball, S.R.; Tabas, I. Toll-like receptor activation suppresses ER stress factor CHOP and translation inhibition through activation of eiF2B. Nat. Cell Biol. 2012, 14, 192–200. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Back, S.H.; Hur, J.; Lin, Y.H.; Gildersleeve, R.; Shan, J.; Yuan, C.L.; Krokowski, D.; Wang, S.; Hatzoglou, M. Er-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat. Cell Biol. 2013, 15, 481–490. [Google Scholar] [CrossRef]
- Brush, M.H.; Weiser, D.C.; Shenolikar, S. Growth arrest and DNA damage-inducible protein GADD34 targets protein phosphatase 1α to the endoplasmic reticulum and promotes dephosphorylation of the α subunit of eukaryotic translation initiation factor 2. Mol. Cell. Biol. 2003, 23, 1292–1303. [Google Scholar] [CrossRef]
- Kaufman, R.J.; Malhotra, J.D. Calcium trafficking integrates endoplasmic reticulum function with mitochondrial bioenergetics. Biochim. Biophys. Acta BBA Mol. Cell Res. 2014, 1843, 2233–2239. [Google Scholar] [CrossRef]
- McCullough, K.D.; Martindale, J.L.; Klotz, L.O.; Aw, T.Y.; Holbrook, N.J. GADD153 sensitizes cells to endoplasmic reticulum stress by down-regulating bcl2 and perturbing the cellular redox state. Mol. Cell. Biol. 2001, 21, 1249–1259. [Google Scholar] [CrossRef]
- Emily, H.Y.C.; Wei, M.C.; Weiler, S.; Flavell, R.A.; Mak, T.W.; Lindsten, T.; Korsmeyer, S.J. BCL-2, BCL-XL sequester BH3 domain-only molecules preventing BAX-and BAK-mediated mitochondrial apoptosis. Mol. Cell 2001, 8, 705–711. [Google Scholar]
- Rodriguez, D.; Rojas-Rivera, D.; Hetz, C. Integrating stress signals at the endoplasmic reticulum: The BCL-2 protein family rheostat. Biochim. Biophys. Acta BBA Mol. Cell Res. 2011, 1813, 564–574. [Google Scholar] [CrossRef]
- Galehdar, Z.; Swan, P.; Fuerth, B.; Callaghan, S.M.; Park, D.S.; Cregan, S.P. Neuronal apoptosis induced by endoplasmic reticulum stress is regulated by ATF4–CHOP-mediated induction of the Bcl-2 homology 3-only member puma. J. Neurosci. 2010, 30, 16938–16948. [Google Scholar] [CrossRef]
- Puthalakath, H.; O’Reilly, L.A.; Gunn, P.; Lee, L.; Kelly, P.N.; Huntington, N.D.; Hughes, P.D.; Michalak, E.M.; McKimm-Breschkin, J.; Motoyama, N. ER stress triggers apoptosis by activating BH3-only protein Bim. Cell 2007, 129, 1337–1349. [Google Scholar] [CrossRef]
- Yamaguchi, H.; Wang, H.G. CHOP is involved in endoplasmic reticulum stress-induced apoptosis by enhancing DR5 expression in human carcinoma cells. J. Biol. Chem. 2004, 279, 45495–45502. [Google Scholar] [CrossRef]
- Du, K.; Herzig, S.; Kulkarni, R.N.; Montminy, M. Trb3: A tribbles homolog that inhibits AKT/PKB activation by insulin in liver. Science 2003, 300, 1574–1577. [Google Scholar] [CrossRef]
- Grivennikov, S.I.; Karin, M. Dangerous liaisons: Stat3 and NF-κB collaboration and crosstalk in cancer. Cytokine Growth Factor Rev. 2010, 21, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Woehlbier, U.; Hetz, C. Modulating stress responses by the uprosome: A matter of life and death. Trends Biochem. Sci. 2011, 36, 329–337. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C.; Glimcher, L.H. Fine-tuning of the unfolded protein response: Assembling the ire1α interactome. Mol. Cell 2009, 35, 551–561. [Google Scholar] [CrossRef] [PubMed]
- Lisbona, F.; Hetz, C. Turning off the Unfolded Protein Response: An Interplay between the Apoptosis Machinery and ER Stress Signaling; Taylor & Francis: Abingdon, UK, 2009. [Google Scholar]
- Hetz, C.; Bernasconi, P.; Fisher, J.; Lee, A.H.; Bassik, M.C.; Antonsson, B.; Brandt, G.S.; Iwakoshi, N.N.; Schinzel, A.; Glimcher, L.H. Proapoptotic BAX and BAK modulate the unfolded protein response by a direct interaction with IRE1α. Science 2006, 312, 572–576. [Google Scholar] [CrossRef]
- Luo, D.; He, Y.; Zhang, H.; Yu, L.; Chen, H.; Xu, Z.; Tang, S.; Urano, F.; Min, W. AIP1 is critical in transducing IRE1-mediated endoplasmic reticulum stress response. J. Biol. Chem. 2008, 283, 11905–11912. [Google Scholar] [CrossRef]
- Gu, F.; Nguyên, D.T.; Stuible, M.; Dubé, N.; Tremblay, M.L.; Chevet, E. Protein-tyrosine phosphatase 1B potentiates IRE1 signaling during endoplasmic reticulum stress. J. Biol. Chem. 2004, 279, 49689–49693. [Google Scholar] [CrossRef]
- Gupta, S.; Deepti, A.; Deegan, S.; Lisbona, F.; Hetz, C.; Samali, A. HSP72 protects cells from ER stress-induced apoptosis via enhancement of IRE1Α-XBP1 signaling through a physical interaction. PLoS Biol. 2010, 8, e1000410. [Google Scholar] [CrossRef]
- Urano, F.; Wang, X.; Bertolotti, A.; Zhang, Y.; Chung, P.; Harding, H.P.; Ron, D. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science 2000, 287, 664–666. [Google Scholar] [CrossRef]
- Nishitoh, H.; Matsuzawa, A.; Tobiume, K.; Saegusa, K.; Takeda, K.; Inoue, K.; Hori, S.; Kakizuka, A.; Ichijo, H. ASK1 is essential for endoplasmic reticulum stress-induced neuronal cell death triggered by expanded polyglutamine repeats. Genes Dev. 2002, 16, 1345–1355. [Google Scholar] [CrossRef]
- Yoneda, T.; Imaizumi, K.; Oono, K.; Yui, D.; Gomi, F.; Katayama, T.; Tohyama, M. Activation of caspase-12, an endoplastic reticulum (ER) resident caspase, through tumor necrosis factor receptor-associated factor 2-dependent mechanism in response to the ER stress. J. Biol. Chem. 2001, 276, 13935–13940. [Google Scholar] [CrossRef]
- Szegezdi, E.; Logue, S.E.; Gorman, A.M.; Samali, A. Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Rep. 2006, 7, 880–885. [Google Scholar] [CrossRef] [PubMed]
- Szegezdi, E.; MacDonald, D.C.; Ní Chonghaile, T.O.; Gupta, S.; Samali, A. BCL-2 family on guard at the ER. Am. J. Physiol. Cell Physiol. 2009, 296, C941–C953. [Google Scholar] [CrossRef] [PubMed]
- Klee, M.; Pallauf, K.; Alcalá, S.; Fleischer, A.; Pimentel-Muiños, F.X. Mitochondrial apoptosis induced by BH3-only molecules in the exclusive presence of endoplasmic reticular Bak. EMBO J. 2009, 28, 1757–1768. [Google Scholar] [CrossRef] [PubMed]
- Lisbona, F.; Rojas-Rivera, D.; Thielen, P.; Zamorano, S.; Todd, D.; Martinon, F.; Glavic, A.; Kress, C.; Lin, J.H.; Walter, P. Bax inhibitor-1 is a negative regulator of the ER stress sensor ire1α. Mol. Cell 2009, 33, 679–691. [Google Scholar] [CrossRef]
- Lee, G.H.; Kim, H.K.; Chae, S.W.; Kim, D.S.; Ha, K.C.; Cuddy, M.; Kress, C.; Reed, J.C.; Kim, H.R.; Chae, H.J. Bax inhibitor-1 regulates endoplasmic reticulum stress-associated reactive oxygen species and heme oxygenase-1 expression. J. Biol. Chem. 2007, 282, 21618–21628. [Google Scholar] [CrossRef]
- Bailly-Maitre, B.; Belgardt, B.F.; Jordan, S.D.; Coornaert, B.; von Freyend, M.J.; Kleinridders, A.; Mauer, J.; Cuddy, M.; Kress, C.L.; Willmes, D. Hepatic bax inhibitor-1 inhibits IRE1α and protects from obesity-associated insulin resistance and glucose intolerance. J. Biol. Chem. 2010, 285, 6198–6207. [Google Scholar] [CrossRef]
- Bailly-Maitre, B.; Fondevila, C.; Kaldas, F.; Droin, N.; Luciano, F.; Ricci, J.E.; Croxton, R.; Krajewska, M.; Zapata, J.M.; Kupiec-Weglinski, J.W. Cytoprotective gene bi-1 is required for intrinsic protection from endoplasmic reticulum stress and ischemia-reperfusion injury. Proc. Natl. Acad. Sci. USA 2006, 103, 2809–2814. [Google Scholar] [CrossRef]
- Rong, J.; Chen, L.; Toth, J.I.; Tcherpakov, M.; Petroski, M.D.; Reed, J.C. Bifunctional apoptosis regulator (bar), an endoplasmic reticulum (ER)-associated E3 ubiquitin ligase, modulates BI-1 protein stability and function in ER stress. J. Biol. Chem. 2011, 286, 1453–1463. [Google Scholar] [CrossRef]
- Hollien, J.; Weissman, J.S. Decay of endoplasmic reticulum-localized mRNAS during the unfolded protein response. Science 2006, 313, 104–107. [Google Scholar] [CrossRef] [PubMed]
- Han, D.; Lerner, A.G.; Walle, L.V.; Upton, J.P.; Xu, W.; Hagen, A.; Backes, B.J.; Oakes, S.A.; Papa, F.R. Ire1α kinase activation modes control alternate endoribonuclease outputs to determine divergent cell fates. Cell 2009, 138, 562–575. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, S.; Wakasa, Y.; Ozawa, K.; Takaiwa, F. Characterization of IRE 1 ribonuclease-mediated mRNA decay in plants using transient expression analyses in rice protoplasts. New Phytol. 2016, 210, 1259–1268. [Google Scholar] [CrossRef] [PubMed]
- Oikawa, D.; Tokuda, M.; Hosoda, A.; Iwawaki, T. Identification of a consensus element recognized and cleaved by IRE1α. Nucleic Acids Res. 2010, 38, 6265–6273. [Google Scholar] [CrossRef]
- Ghosh, R.; Wang, L.; Wang, E.S.; Perera, B.G.K.; Igbaria, A.; Morita, S.; Prado, K.; Thamsen, M.; Caswell, D.; Macias, H. Allosteric inhibition of the IRE1α RNase preserves cell viability and function during endoplasmic reticulum stress. Cell 2014, 158, 534–548. [Google Scholar] [CrossRef] [PubMed]
- Lerner, A.G.; Upton, J.P.; Praveen, P.; Ghosh, R.; Nakagawa, Y.; Igbaria, A.; Shen, S.; Nguyen, V.; Backes, B.J.; Heiman, M. IRE1α induces thioredoxin-interacting protein to activate the NLRP3 inflammasome and promote programmed cell death under irremediable ER stress. Cell Metab. 2012, 16, 250–264. [Google Scholar] [CrossRef]
- Upton, J.P.; Wang, L.; Han, D.; Wang, E.S.; Huskey, N.E.; Lim, L.; Truitt, M.; McManus, M.T.; Ruggero, D.; Goga, A. IRE1α cleaves select micrornas during ER stress to derepress translation of proapoptotic caspase-2. Science 2012, 338, 818–822. [Google Scholar] [CrossRef]
- Shiraishi, H.; Okamoto, H.; Yoshimura, A.; Yoshida, H. ER stress-induced apoptosis and caspase-12 activation occurs downstream of mitochondrial apoptosis involving apaf-1. J. Cell Sci. 2006, 119, 3958–3966. [Google Scholar] [CrossRef]
- Saleh, M.; Mathison, J.C.; Wolinski, M.K.; Bensinger, S.J.; Fitzgerald, P.; Droin, N.; Ulevitch, R.J.; Green, D.R.; Nicholson, D.W. Enhanced bacterial clearance and sepsis resistance in caspase-12-deficient mice. Nature 2006, 440, 1064–1068. [Google Scholar] [CrossRef]
- Nakagawa, T.; Zhu, H.; Morishima, N.; Li, E.; Xu, J.; Yankner, B.A.; Yuan, J. Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-β. Nature 2000, 403, 98–103. [Google Scholar] [CrossRef]
- Lamkanfi, M.; Kalai, M.; Vandenabeele, P. Caspase-12: An overview. Cell Death Differ. 2004, 11, 365–368. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; Zhang, Z.; Hitomi, E.; Lee, Y.C.; Mukherjee, A.B. Endoplasmic reticulum stress-induced caspase-4 activation mediates apoptosis and neurodegeneration in incl. Hum. Mol. Genet. 2006, 15, 1826–1834. [Google Scholar] [CrossRef] [PubMed]
- Hitomi, J.; Katayama, T.; Eguchi, Y.; Kudo, T.; Taniguchi, M.; Koyama, Y.; Manabe, T.; Yamagishi, S.; Bando, Y.; Imaizumi, K. Involvement of caspase-4 in endoplasmic reticulum stress-induced apoptosis and aβ-induced cell death. J. Cell Biol. 2004, 165, 347–356. [Google Scholar] [CrossRef] [PubMed]
- Breckenridge, D.G.; Stojanovic, M.; Marcellus, R.C.; Shore, G.C. Caspase cleavage product of BAP31 induces mitochondrial fission through endoplasmic reticulum calcium signals, enhancing cytochrome c release to the cytosol. J. Cell Biol. 2003, 160, 1115–1127. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Hendershot, L.M. The role of the unfolded protein response in tumour development: Friend or foe? Nat. Rev. Cancer 2004, 4, 966–977. [Google Scholar] [CrossRef]
- Urra, H.; Dufey, E.; Avril, T.; Chevet, E.; Hetz, C. Endoplasmic reticulum stress and the hallmarks of cancer. Trends Cancer 2016, 2, 252–262. [Google Scholar] [CrossRef]
- Vanacker, H.; Vetters, J.; Moudombi, L.; Caux, C.; Janssens, S.; Michallet, M.C. Emerging role of the unfolded protein response in tumor immunosurveillance. Trends Cancer 2017, 3, 491–505. [Google Scholar] [CrossRef]
- Volmer, R.; Ron, D. Lipid-dependent regulation of the unfolded protein response. Curr. Opin. Cell Biol. 2015, 33, 67–73. [Google Scholar] [CrossRef]
- Clarke, H.J.; Chambers, J.E.; Liniker, E.; Marciniak, S.J. Endoplasmic reticulum stress in malignancy. Cancer Cell 2014, 25, 563–573. [Google Scholar] [CrossRef]
- Andruska, N.; Zheng, X.; Yang, X.; Helferich, W.G.; Shapiro, D.J. Anticipatory estrogen activation of the unfolded protein response is linked to cell proliferation and poor survival in estrogen receptor α-positive breast cancer. Oncogene 2015, 34, 3760–3769. [Google Scholar] [CrossRef] [PubMed]
- Dong, D.; Ni, M.; Li, J.; Xiong, S.; Ye, W.; Virrey, J.J.; Mao, C.; Ye, R.; Wang, M.; Pen, L. Critical role of the stress chaperone GRP78/BiP in tumor proliferation, survival, and tumor angiogenesis in transgene-induced mammary tumor development. Cancer Res. 2008, 68, 498–505. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Wey, S.; Wang, M.; Ye, R.; Liao, C.P.; Roy-Burman, P.; Lee, A.S. Pten null prostate tumorigenesis and AKT activation are blocked by targeted knockout of ER chaperone GRP78/BiP in prostate epithelium. Proc. Natl. Acad. Sci. USA 2008, 105, 19444–19449. [Google Scholar] [CrossRef] [PubMed]
- Luo, B.; Lee, A.S. The critical roles of endoplasmic reticulum chaperones and unfolded protein response in tumorigenesis and anticancer therapies. Oncogene 2013, 32, 805–818. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.S. GRP78 induction in cancer: Therapeutic and prognostic implications. Cancer Res. 2007, 67, 3496–3499. [Google Scholar] [CrossRef] [PubMed]
- Verfaillie, T.; Garg, A.D.; Agostinis, P. Targeting ER stress induced apoptosis and inflammation in cancer. Cancer Lett. 2013, 332, 249–264. [Google Scholar] [CrossRef]
- Lee, A.S. Glucose-regulated proteins in cancer: Molecular mechanisms and therapeutic potential. Nat. Rev. Cancer 2014, 14, 263–276. [Google Scholar] [CrossRef]
- Jamora, C.; Dennert, G.; Lee, A.S. Inhibition of tumor progression by suppression of stress protein GRP78/BiP induction in fibrosarcoma B/C10ME. Proc. Natl. Acad. Sci. USA 1996, 93, 7690–7694. [Google Scholar] [CrossRef]
- Wang, G.; Yang, Z.Q.; Zhang, K. Endoplasmic reticulum stress response in cancer: Molecular mechanism and therapeutic potential. Am. J. Transl. Res. 2010, 2, 65–74. [Google Scholar]
- Mintz, P.J.; Kim, J.; Do, K.A.; Wang, X.; Zinner, R.G.; Cristofanilli, M.; Arap, M.A.; Hong, W.K.; Troncoso, P.; Logothetis, C.J. Fingerprinting the circulating repertoire of antibodies from cancer patients. Nat. Biotechnol. 2003, 21, 57–63. [Google Scholar] [CrossRef]
- Denoyelle, C.; Abou-Rjaily, G.; Bezrookove, V.; Verhaegen, M.; Johnson, T.M.; Fullen, D.R.; Pointer, J.N.; Gruber, S.B.; Su, L.D.; Nikiforov, M.A. Anti-oncogenic role of the endoplasmic reticulum differentially activated by mutations in the mapk pathway. Nat. Cell Biol. 2006, 8, 1053–1063. [Google Scholar] [CrossRef] [PubMed]
- Uramoto, H.; Sugio, K.; Oyama, T.; Nakata, S.; Ono, K.; Yoshimastu, T.; Morita, M.; Yasumoto, K. Expression of endoplasmic reticulum molecular chaperone grp78 in human lung cancer and its clinical significance. Lung Cancer 2005, 49, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Hsu, W.M.; Hsieh, F.J.; Jeng, Y.M.; Kuo, M.L.; Tsao, P.N.; Lee, H.; Lin, M.T.; Lai, H.S.; Chen, C.N.; Lai, D.M. GRP78 expression correlates with histologic differentiation and favorable prognosis in neuroblastic tumors. Int. J. Cancer 2005, 113, 920–927. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.; Liu, K.; Anderson, J.; Patrene, K.; Lentzsch, S.; Roodman, G.D.; Ouyang, H. Expression of XBP1s in bone marrow stromal cells is critical for myeloma cell growth and osteoclast formation. Blood 2012, 119, 4205–4214. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, T.; Onda, M.; Nagai, H.; Nagahata, T.; Ogawa, K.; Emi, M. Upregulation and overexpression of human X-box binding protein 1 (hXBP-1) gene in primary breast cancers. Breast Cancer 2003, 10, 301–306. [Google Scholar] [CrossRef] [PubMed]
- Shuda, M.; Kondoh, N.; Imazeki, N.; Tanaka, K.; Okada, T.; Mori, K.; Hada, A.; Arai, M.; Wakatsuki, T.; Matsubara, O. Activation of the atf6, XBP1 and GRP78 genes in human hepatocellular carcinoma: A possible involvement of the ER stress pathway in hepatocarcinogenesis. J. Hepatol. 2003, 38, 605–614. [Google Scholar] [CrossRef]
- Sun, H.; Lin, D.C.; Guo, X.; Masouleh, B.K.; Gery, S.; Cao, Q.; Alkan, S.; Ikezoe, T.; Akiba, C.; Paquette, R. Inhibition of IRE1α-driven pro-survival pathways is a promising therapeutic application in acute myeloid leukemia. Oncotarget 2016, 7, 18736–18749. [Google Scholar]
- Chen, X.; Iliopoulos, D.; Zhang, Q.; Tang, Q.; Greenblatt, M.B.; Hatziapostolou, M.; Lim, E.; Tam, W.L.; Ni, M.; Chen, Y. XBP1 promotes triple-negative breast cancer by controlling the HIF1α pathway. Nature 2014, 508, 103–107. [Google Scholar] [CrossRef]
- McGrath, E.P.; Logue, S.E.; Mnich, K.; Deegan, S.; Jäger, R.; Gorman, A.M.; Samali, A. The unfolded protein response in breast cancer. Cancers 2018, 10, 344. [Google Scholar] [CrossRef]
- Zhao, N.; Cao, J.; Xu, L.; Tang, Q.; Dobrolecki, L.E.; Lv, X.; Talukdar, M.; Lu, Y.; Wang, X.; Hu, D.Z. Pharmacological targeting of MYC-regulated IRE1/XBP1 pathway suppresses MYC-driven breast cancer. J. Clin. Investig. 2018, 128, 1283–1299. [Google Scholar] [CrossRef]
- Logue, S.E.; McGrath, E.P.; Cleary, P.; Greene, S.; Mnich, K.; Almanza, A.; Chevet, E.; Dwyer, R.M.; Oommen, A.; Legembre, P. Inhibition of ire1 RNase activity modulates the tumor cell secretome and enhances response to chemotherapy. Nat. Commun. 2018, 9, 3267. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Hossain, M.M.; Miller, N.; Kerin, M.; Callagy, G.; Gupta, S. NCOA3 coactivator is a transcriptional target of XBP1 and regulates PERK–eIF2α–ATF4 signalling in breast cancer. Oncogene 2016, 35, 5860–5871. [Google Scholar] [CrossRef] [PubMed]
- Bagratuni, T.; Wu, P.; de Castro, D.G.; Davenport, E.L.; Dickens, N.J.; Walker, B.A.; Boyd, K.; Johnson, D.C.; Gregory, W.; Morgan, G.J. XBP1s levels are implicated in the biology and outcome of myeloma mediating different clinical outcomes to thalidomide-based treatments. Blood 2010, 116, 250–253. [Google Scholar] [CrossRef] [PubMed]
- Greenman, C.; Stephens, P.; Smith, R.; Dalgliesh, G.L.; Hunter, C.; Bignell, G.; Davies, H.; Teague, J.; Butler, A.; Stevens, C. Patterns of somatic mutation in human cancer genomes. Nature 2007, 446, 153–158. [Google Scholar] [CrossRef] [PubMed]
- Xue, Z.; He, Y.; Ye, K.; Gu, Z.; Mao, Y.; Qi, L. A conserved structural determinant located at the interdomain region of mammalian inositol-requiring enzyme 1α. J. Biol. Chem. 2011, 286, 30859–30866. [Google Scholar] [CrossRef] [PubMed]
- Pluquet, O.; Dejeans, N.; Bouchecareilh, M.; Lhomond, S.; Pineau, R.; Higa, A.; Delugin, M.; Combe, C.; Loriot, S.; Cubel, G. Posttranscriptional regulation of per1 underlies the oncogenic function of ireα. Cancer Res. 2013, 73, 4732–4743. [Google Scholar] [CrossRef]
- Masouleh, B.K.; Geng, H.; Hurtz, C.; Chan, L.N.; Logan, A.C.; Chang, M.S.; Huang, C.; Swaminathan, S.; Sun, H.; Paietta, E. Mechanistic rationale for targeting the unfolded protein response in pre-b acute lymphoblastic leukemia. Proc. Natl. Acad. Sci. USA 2014, 111, 2219–2228. [Google Scholar] [CrossRef]
- Mimura, N.; Fulciniti, M.; Gorgun, G.; Tai, Y.T.; Cirstea, D.; Santo, L.; Hu, Y.; Fabre, C.; Minami, J.; Ohguchi, H. Blockade of XBP1 splicing by inhibition of IRE1α is a promising therapeutic option in multiple myeloma. Blood 2012, 119, 5772–5781. [Google Scholar] [CrossRef]
- Papandreou, I.; Denko, N.C.; Olson, M.; Van Melckebeke, H.; Lust, S.; Tam, A.; Solow-Cordero, D.E.; Bouley, D.M.; Offner, F.; Niwa, M. Identification of an ire1alpha endonuclease specific inhibitor with cytotoxic activity against human multiple myeloma. Blood 2011, 117, 1311–1314. [Google Scholar] [CrossRef]
- Bi, M.; Naczki, C.; Koritzinsky, M.; Fels, D.; Blais, J.; Hu, N.; Harding, H.; Novoa, I.; Varia, M.; Raleigh, J. ER stress-regulated translation increases tolerance to extreme hypoxia and promotes tumor growth. EMBO J. 2005, 24, 3470–3481. [Google Scholar] [CrossRef]
- Blais, J.D.; Addison, C.L.; Edge, R.; Falls, T.; Zhao, H.; Wary, K.; Koumenis, C.; Harding, H.P.; Ron, D.; Holcik, M. Perk-dependent translational regulation promotes tumor cell adaptation and angiogenesis in response to hypoxic stress. Mol. Cell. Biol. 2006, 26, 9517–9532. [Google Scholar] [CrossRef] [PubMed]
- Dey, S.; Sayers, C.M.; Verginadis, I.I.; Lehman, S.L.; Cheng, Y.; Cerniglia, G.J.; Tuttle, S.W.; Feldman, M.D.; Zhang, P.J.; Fuchs, S.Y. Atf4-dependent induction of heme oxygenase 1 prevents anoikis and promotes metastasis. J. Clin. Investig. 2015, 125, 2592–2608. [Google Scholar] [CrossRef] [PubMed]
- Cullinan, S.B.; Zhang, D.; Hannink, M.; Arvisais, E.; Kaufman, R.J.; Diehl, J.A. Nrf2 is a direct perk substrate and effector of PERK-dependent cell survival. Mol. Cell. Biol. 2003, 23, 7198–7209. [Google Scholar] [CrossRef] [PubMed]
- Del Vecchio, C.A.; Feng, Y.; Sokol, E.S.; Tillman, E.J.; Sanduja, S.; Reinhardt, F.; Gupta, P.B. De-differentiation confers multidrug resistance via noncanonical PERK-Nrf2 signaling. PLoS Biol. 2014, 12, e1001945. [Google Scholar] [CrossRef] [PubMed]
- Pytel, D.; Majsterek, I.; Diehl, J.A. Tumor progression and the different faces of the PERK kinase. Oncogene 2016, 35, 1207–1215. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Alam, G.N.; Ning, Y.; Visioli, F.; Dong, Z.; Nör, J.E.; Polverini, P.J. The unfolded protein response induces the angiogenic switch in human tumor cells through the PERK/ATF4 pathway. Cancer Res. 2012, 72, 5396–5406. [Google Scholar] [CrossRef] [PubMed]
- Chang, K.C.; Chen, P.C.H.; Chen, Y.P.; Chang, Y.; Su, I.J. Dominant expression of survival signals of endoplasmic reticulum stress response in hodgkin lymphoma. Cancer Sci. 2011, 102, 275–281. [Google Scholar] [CrossRef]
- Karali, E.; Bellou, S.; Stellas, D.; Klinakis, A.; Murphy, C.; Fotsis, T. VEGF signals through ATF6 and PERK to promote endothelial cell survival and angiogenesis in the absence of ER stress. Mol. Cell 2014, 54, 559–572. [Google Scholar] [CrossRef]
- Aguirre-Ghiso, J.A. Models, mechanisms and clinical evidence for cancer dormancy. Nat. Rev. Cancer 2007, 7, 834–846. [Google Scholar] [CrossRef]
- Páez, D.; Labonte, M.J.; Bohanes, P.; Zhang, W.; Benhanim, L.; Ning, Y.; Wakatsuki, T.; Loupakis, F.; Lenz, H.J. Cancer dormancy: A model of early dissemination and late cancer recurrence. Clin. Cancer Res. 2012, 18, 645–653. [Google Scholar] [CrossRef]
- Schewe, D.M.; Aguirre-Ghiso, J.A. ATF6α-Rheb-mTOR signaling promotes survival of dormant tumor cells in vivo. Proc. Natl. Acad. Sci. USA 2008, 105, 10519–10524. [Google Scholar] [CrossRef] [PubMed]
- Ginos, M.A.; Page, G.P.; Michalowicz, B.S.; Patel, K.J.; Volker, S.E.; Pambuccian, S.E.; Ondrey, F.G.; Adams, G.L.; Gaffney, P.M. Identification of a gene expression signature associated with recurrent disease in squamous cell carcinoma of the head and neck. Cancer Res. 2004, 64, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.; Tseng, C.; Wang, M.; Chang, W.C.; Lee, C.; Chen, L.; Hung, M.C.; Su, J.L. Deacetylation of HSPA5 by HDAC6 leads to GP78-mediated HSPA5 ubiquitination at K447 and suppresses metastasis of breast cancer. Oncogene 2016, 35, 1517–1528. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.W.; Chen, H.A.; Tseng, C.F.; Hong, C.C.; Ma, J.T.; Hung, M.C.; Wu, C.H.; Huang, M.T.; Su, J.L. De-acetylation and degradation of HSPA5 is critical for E1A metastasis suppression in breast cancer cells. Oncotarget 2014, 5, 10558–10570. [Google Scholar] [CrossRef]
- Auf, G.; Jabouille, A.; Guérit, S.; Pineau, R.; Delugin, M.; Bouchecareilh, M.; Magnin, N.; Favereaux, A.; Maitre, M.; Gaiser, T. Inositol-requiring enzyme 1α is a key regulator of angiogenesis and invasion in malignant glioma. Proc. Natl. Acad. Sci. USA 2010, 107, 15553–15558. [Google Scholar] [CrossRef]
- Dejeans, N.; Pluquet, O.; Lhomond, S.; Grise, F.; Bouchecareilh, M.; Juin, A.; Meynard-Cadars, M.; Bidaud-Meynard, A.; Gentil, C.; Moreau, V. Autocrine control of glioma cells adhesion and migration through IRE1α-mediated cleavage of sparc mrna. J. Cell Sci. 2012, 125, 4278–4287. [Google Scholar] [CrossRef]
- Cubillos-Ruiz, J.R.; Bettigole, S.E.; Glimcher, L.H. Tumorigenic and immunosuppressive effects of endoplasmic reticulum stress in cancer. Cell 2017, 168, 692–706. [Google Scholar] [CrossRef]
- Mujcic, H.; Nagelkerke, A.; Rouschop, K.M.; Chung, S.; Chaudary, N.; Span, P.N.; Clarke, B.; Milosevic, M.; Sykes, J.; Hill, R.P. Hypoxic activation of the PERK/eIF2α arm of the unfolded protein response promotes metastasis through induction of LAMP3. Clin. Cancer Res. 2013, 19, 6126–6137. [Google Scholar] [CrossRef]
- Zhu, H.; Chen, X.; Chen, B.; Chen, B.; Song, W.; Sun, D.; Zhao, Y. Activating transcription factor 4 promotes esophageal squamous cell carcinoma invasion and metastasis in mice and is associated with poor prognosis in human patients. PLoS ONE 2014, 9, e103882. [Google Scholar] [CrossRef]
- Thevenot, P.T.; Sierra, R.A.; Raber, P.L.; Al-Khami, A.A.; Trillo-Tinoco, J.; Zarreii, P.; Ochoa, A.C.; Cui, Y.; Del Valle, L.; Rodriguez, P.C. The stress-response sensor chop regulates the function and accumulation of myeloid-derived suppressor cells in tumors. Immunity 2014, 41, 389–401. [Google Scholar] [CrossRef]
- Condamine, T.; Kumar, V.; Ramachandran, I.R.; Youn, J.I.; Celis, E.; Finnberg, N.; El-Deiry, W.S.; Winograd, R.; Vonderheide, R.H.; English, N.R. ER stress regulates myeloid-derived suppressor cell fate through trail-r–mediated apoptosis. J. Clin. Investig. 2014, 124, 2626–2639. [Google Scholar] [CrossRef] [PubMed]
- Cubillos-Ruiz, J.R.; Silberman, P.C.; Rutkowski, M.R.; Chopra, S.; Perales-Puchalt, A.; Song, M.; Zhang, S.; Bettigole, S.E.; Gupta, D.; Holcomb, K. ER stress sensor xbp1 controls anti-tumor immunity by disrupting dendritic cell homeostasis. Cell 2015, 161, 1527–1538. [Google Scholar] [CrossRef] [PubMed]
- Herber, D.L.; Cao, W.; Nefedova, Y.; Novitskiy, S.V.; Nagaraj, S.; Tyurin, V.A.; Corzo, A.; Cho, H.I.; Celis, E.; Lennox, B. Lipid accumulation and dendritic cell dysfunction in cancer. Nat. Med. 2010, 16, 880–886. [Google Scholar] [CrossRef] [PubMed]
- Hossain, F.; Al-Khami, A.A.; Wyczechowska, D.; Hernandez, C.; Zheng, L.; Reiss, K.; Del Valle, L.; Trillo-Tinoco, J.; Maj, T.; Zou, W. Inhibition of fatty acid oxidation modulates immunosuppressive functions of myeloid-derived suppressor cells and enhances cancer therapies. Cancer Immunol. Res. 2015, 3, 1236–1247. [Google Scholar] [CrossRef] [PubMed]
- Cao, W.; Ramakrishnan, R.; Tuyrin, V.A.; Veglia, F.; Condamine, T.; Amoscato, A.; Mohammadyani, D.; Johnson, J.J.; Zhang, L.M.; Klein-Seetharaman, J. Oxidized lipids block antigen cross-presentation by dendritic cells in cancer. J. Immunol. 2014, 192, 2920–2931. [Google Scholar] [CrossRef] [PubMed]
- Yan, D.; Wang, H.W.; Bowman, R.L.; Joyce, J.A. Stat3 and stat6 signaling pathways synergize to promote cathepsin secretion from macrophages via ire1α activation. Cell Rep. 2016, 16, 2914–2927. [Google Scholar] [CrossRef] [PubMed]
- Condamine, T.; Dominguez, G.A.; Youn, J.I.; Kossenkov, A.V.; Mony, S.; Alicea-Torres, K.; Tcyganov, E.; Hashimoto, A.; Nefedova, Y.; Lin, C. Lectin-type oxidized LDL receptor-1 distinguishes population of human polymorphonuclear myeloid-derived suppressor cells in cancer patients. Sci. Immunol. 2016, 1. [Google Scholar] [CrossRef]
- Tang, C.H.A.; Ranatunga, S.; Kriss, C.L.; Cubitt, C.L.; Tao, J.; Pinilla-Ibarz, J.A.; Del Valle, J.R.; Hu, C.C.A. Inhibition of ER stress–associated IRE-1/XBP-1 pathway reduces leukemic cell survival. J. Clin. Investig. 2014, 124, 2585–2598. [Google Scholar] [CrossRef]
- Todd, D.J.; Lee, A.H.; Glimcher, L.H. The endoplasmic reticulum stress response in immunity and autoimmunity. Nat. Rev. Immunol. 2008, 8, 663–674. [Google Scholar] [CrossRef]
- Hetz, C.; Chevet, E.; Harding, H.P. Targeting the unfolded protein response in disease. Nat. Rev. Drug Discov. 2013, 12, 703–719. [Google Scholar] [CrossRef]
- Axten, J.M.; Medina, J.S.R.; Feng, Y.; Shu, A.; Romeril, S.P.; Grant, S.W.; Li, W.H.H.; Heerding, D.A.; Minthorn, E.; Mencken, T.; et al. Discovery of 7-methyl-5-(1-{[3-(trifluoromethyl) phenyl] acetyl}-2, 3-dihydro-1 h-indol-5-yl)-7 h-pyrrolo [2, 3-d] pyrimidin-4-amine (GSK2606414), a potent and selective first-in-class inhibitor of protein kinase r (PKR)-like endoplasmic reticulum kinase (PERK). J Med. Chem. 2012, 55, 7193–7207. [Google Scholar] [PubMed]
- Atkins, C.; Liu, Q.; Minthorn, E.; Zhang, S.Y.; Figueroa, D.J.; Moss, K.; Stanley, T.B.; Sanders, B.; Goetz, A.; Gaul, N. Characterization of a novel perk kinase inhibitor with antitumor and antiangiogenic activity. Cancer Res. 2013, 73, 1993–2002. [Google Scholar] [CrossRef]
- Rouschop, K.M.; Dubois, L.J.; Keulers, T.G.; van den Beucken, T.; Lambin, P.; Bussink, J.; van der Kogel, A.J.; Koritzinsky, M.; Wouters, B.G. PERK/eIF2α signaling protects therapy resistant hypoxic cells through induction of glutathione synthesis and protection against ROS. Proc. Natl. Acad. Sci. USA 2013, 110, 4622–4627. [Google Scholar] [CrossRef] [PubMed]
- Teng, Y.; Gao, M.; Wang, J.; Kong, Q.; Hua, H.; Luo, T.; Jiang, Y. Inhibition of eIF2α dephosphorylation enhances TRAIL-induced apoptosis in hepatoma cells. Cell Death Dis. 2014, 5, e1060. [Google Scholar] [CrossRef]
- Hamamura, K.; Minami, K.; Tanjung, N.; Wan, Q.; Koizumi, M.; Matsuura, N.; Na, S.; Yokota, H. Attenuation of malignant phenotypes of breast cancer cells through eIF2α-mediated downregulation of Rac1 signaling. Int. J. Oncol. 2014, 44, 1980–1988. [Google Scholar] [CrossRef] [PubMed]
- Boyce, M.; Bryant, K.F.; Jousse, C.; Long, K.; Harding, H.P.; Scheuner, D.; Kaufman, R.J.; Ma, D.; Coen, D.M.; Ron, D. A selective inhibitor of eIF2α dephosphorylation protects cells from ER stress. Science 2005, 307, 935–939. [Google Scholar] [CrossRef] [PubMed]
- Tsaytler, P.; Harding, H.P.; Ron, D.; Bertolotti, A. Selective inhibition of a regulatory subunit of protein phosphatase 1 restores proteostasis. Science 2011, 332, 91–94. [Google Scholar] [CrossRef]
- Sidrauski, C.; McGeachy, A.M.; Ingolia, N.T.; Walter, P. The small molecule isrib reverses the effects of eIF2α phosphorylation on translation and stress granule assembly. Elife 2015, 4, e05033. [Google Scholar] [CrossRef]
- Cross, B.C.; Bond, P.J.; Sadowski, P.G.; Jha, B.K.; Zak, J.; Goodman, J.M.; Silverman, R.H.; Neubert, T.A.; Baxendale, I.R.; Ron, D. The molecular basis for selective inhibition of unconventional mrna splicing by an ire1-binding small molecule. Proc. Natl. Acad. Sci. USA 2012, 109, E869–E878. [Google Scholar] [CrossRef]
- Volkmann, K.; Lucas, J.L.; Vuga, D.; Wang, X.; Brumm, D.; Stiles, C.; Kriebel, D.; Der-Sarkissian, A.; Krishnan, K.; Schweitzer, C. Potent and selective inhibitors of the inositol-requiring enzyme 1 endoribonuclease. J. Biol. Chem. 2011, 286, 12743–12755. [Google Scholar] [CrossRef]
- Suh, D.H.; Kim, M.K.; Kim, H.S.; Chung, H.H.; Song, Y.S. Unfolded protein response to autophagy as a promising druggable target for anticancer therapy. Ann. N. Y. Acad. Sci. 2012, 1271, 20–32. [Google Scholar] [CrossRef]
- Ming, J.; Ruan, S.; Wang, M.; Ye, D.; Fan, N.; Meng, Q.; Tian, B.; Huang, T. A novel chemical, STF-083010, reverses tamoxifen-related drug resistance in breast cancer by inhibiting IRE1/XBP1. Oncotarget 2015, 6, 40692–40703. [Google Scholar] [CrossRef] [PubMed]
- Jiang, D.; Tam, A.B.; Alagappan, M.; Hay, M.P.; Gupta, A.; Kozak, M.M.; Solow-Cordero, D.E.; Lum, P.Y.; Denko, N.C.; Giaccia, A.J. Acridine derivatives as inhibitors of the ire1α–xbp1 pathway are cytotoxic to human multiple myeloma. Mol. Cancer Ther. 2016, 15, 2055–2065. [Google Scholar] [CrossRef] [PubMed]
- Sanches, M.; Duffy, N.M.; Talukdar, M.; Thevakumaran, N.; Chiovitti, D.; Canny, M.D.; Lee, K.; Kurinov, I.; Uehling, D.; Al-Awar, R. Structure and mechanism of action of the hydroxy–aryl–aldehyde class of ire1 endoribonuclease inhibitors. Nat. Commun. 2014, 5, 4202. [Google Scholar] [CrossRef] [PubMed]
- Vogelzangs, N.; Duivis, H.E.; Beekman, A.T.; Kluft, C.; Neuteboom, J.; Hoogendijk, W.; Smit, J.H.; de Jonge, P.; Penninx, B.W. Association of depressive disorders, depression characteristics and antidepressant medication with inflammation. Transl. Psychiatry 2012, 2, e79. [Google Scholar] [CrossRef]
- Wang, M.; Law, M.E.; Castellano, R.K.; Law, B.K. The unfolded protein response as a target for anticancer therapeutics. Crit. Rev. Oncol. Hematol. 2018, 127, 66–79. [Google Scholar] [CrossRef]
- Wang, L.; Perera, B.G.K.; Hari, S.B.; Bhhatarai, B.; Backes, B.J.; Seeliger, M.A.; Schürer, S.C.; Oakes, S.A.; Papa, F.R.; Maly, D.J. Divergent allosteric control of the ire1α endoribonuclease using kinase inhibitors. Nat. Chem. Biol. 2012, 8, 982–989. [Google Scholar] [CrossRef]
- Wang, F.M.; Galson, D.L.; Roodman, G.D.; Ouyang, H. Resveratrol triggers the pro-apoptotic endoplasmic reticulum stress response and represses pro-survival XBP1 signaling in human multiple myeloma cells. Exp. Hematol. 2011, 39, 999–1006. [Google Scholar] [CrossRef]
- Rojas, C.; Pan-Castillo, B.; Valls, C.; Pujadas, G.; Garcia-Vallve, S.; Arola, L.; Mulero, M. Resveratrol enhances palmitate-induced ER stress and apoptosis in cancer cells. PLoS ONE 2014, 9, e113929. [Google Scholar] [CrossRef]
- Hetz, C. The unfolded protein response: Controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 2012, 13, 89–102. [Google Scholar] [CrossRef]
- Gallagher, C.M.; Walter, P. Ceapins inhibit atf6α signaling by selectively preventing transport of ATF6α to the Golgi apparatus during ER stress. Elife 2016, 5, e11880. [Google Scholar] [CrossRef] [PubMed]
- Brodsky, J.L.; Wojcikiewicz, R.J. Substrate-specific mediators of ER associated degradation (ERAD). Curr. Opin. Cell Biol. 2009, 21, 516–521. [Google Scholar] [CrossRef] [PubMed]
- Hampton, R.Y. ER-associated degradation in protein quality control and cellular regulation. Curr. Opin. Cell Biol. 2002, 14, 476–482. [Google Scholar] [CrossRef]
- Hershko, A.; Ciechanover, A. The ubiquitin system. Annu. Rev. Biochem. 1998, 67, 425–479. [Google Scholar] [CrossRef]
- Suraweera, A.; Münch, C.; Hanssum, A.; Bertolotti, A. Failure of amino acid homeostasis causes cell death following proteasome inhibition. Mol. Cell 2012, 48, 242–253. [Google Scholar] [CrossRef]
- Kisselev, A.F.; van der Linden, W.A.; Overkleeft, H.S. Proteasome inhibitors: An expanding army attacking a unique target. Chem. Biol. 2012, 19, 99–115. [Google Scholar] [CrossRef]
- Begg, A.C.; Stewart, F.A.; Vens, C. Strategies to improve radiotherapy with targeted drugs. Nat. Rev. Cancer 2011, 11, 239–253. [Google Scholar] [CrossRef]
- Manasanch, E.E.; Orlowski, R.Z. Proteasome inhibitors in cancer therapy. Nat. Rev. Clin. Oncol. 2017, 14, 417–433. [Google Scholar] [CrossRef]
- Fribley, A.; Zeng, Q.; Wang, C.Y. Proteasome inhibitor PS-341 induces apoptosis through induction of endoplasmic reticulum stress-reactive oxygen species in head and neck squamous cell carcinoma cells. Mol. Cell. Biol. 2004, 24, 9695–9704. [Google Scholar] [CrossRef]
- Ciombor, K.K.; Feng, Y.; Su, Y.; Horton, L.; Short, S.P.; Kauh, J.S.W.; Staley, C.; Mulcahy, M.; Powell, M.; Amiri, K.I. Phase ii trial of bortezomib plus doxorubicin in hepatocellular carcinoma (e6202): A trial of the eastern cooperative oncology group. Investig. New Drugs 2014, 32, 1017–1027. [Google Scholar] [CrossRef]
- Nawrocki, S.T.; Carew, J.S.; Dunner, K.; Boise, L.H.; Chiao, P.J.; Huang, P.; Abbruzzese, J.L.; McConkey, D.J. Bortezomib inhibits PKR-like endoplasmic reticulum (ER) kinase and induces apoptosis via ER stress in human pancreatic cancer cells. Cancer Res. 2005, 65, 11510–11519. [Google Scholar] [CrossRef] [PubMed]
- Roccaro, A.M.; Hideshima, T.; Raje, N.; Kumar, S.; Ishitsuka, K.; Yasui, H.; Shiraishi, N.; Ribatti, D.; Nico, B.; Vacca, A. Bortezomib mediates antiangiogenesis in multiple myeloma via direct and indirect effects on endothelial cells. Cancer Res. 2006, 66, 184–191. [Google Scholar] [CrossRef] [PubMed]
- Sunwoo, J.B.; Chen, Z.; Dong, G.; Yeh, N.; Bancroft, C.C.; Sausville, E.; Adams, J.; Elliott, P.; Van Waes, C. Novel proteasome inhibitor PS-341 inhibits activation of nuclear factor-κB, cell survival, tumor growth, and angiogenesis in squamous cell carcinoma. Clin. Cancer Res. 2001, 7, 1419–1428. [Google Scholar] [PubMed]
- Politou, M.; Naresh, K.; Terpos, E.; Crawley, D.; Lampert, I.; Apperley, J.F.; Rahemtulla, A. Anti-angiogenic effect of bortezomib in patients with multiple myeloma. Acta Haematol. 2005, 114, 170–173. [Google Scholar] [CrossRef]
- Richardson, P.G.; Xie, W.; Jagannath, S.; Jakubowiak, A.; Lonial, S.; Raje, N.S.; Alsina, M.; Ghobrial, I.M.; Schlossman, R.L.; Munshi, N.C. A phase 2 trial of lenalidomide, bortezomib, and dexamethasone in patients with relapsed and relapsed/refractory myeloma. Blood 2014, 123, 1461–1469. [Google Scholar] [CrossRef]
- Roy, S.S.; Kirma, N.B.; Santhamma, B.; Tekmal, R.R.; Agyin, J.K. Effects of a novel proteasome inhibitor bu-32 on multiple myeloma cells. Cancer Chemother. Pharmacol. 2014, 73, 1263–1271. [Google Scholar] [CrossRef]
- Agyin, J.K.; Santhamma, B.; Nair, H.B.; Roy, S.S.; Tekmal, R.R. Bu-32: A novel proteasome inhibitor for breast cancer. Breast Cancer Res. 2009, 11, R74. [Google Scholar] [CrossRef]
- Ping Dou, Q.; Zonder, J.A. Overview of proteasome inhibitor-based anti-cancer therapies: Perspective on bortezomib and second generation proteasome inhibitors versus future generation inhibitors of ubiquitin-proteasome system. Curr. Cancer Drug Targets 2014, 14, 517–536. [Google Scholar]
- Garcia-Gomez, A.; Quwaider, D.; Canavese, M.; Ocio, E.M.; Tian, Z.; Blanco, J.F.; Berger, A.J.; Ortiz-de-Solorzano, C.; Hernández-Iglesias, T.; Martens, A.C. Preclinical activity of the oral proteasome inhibitor mln9708 in myeloma bone disease. Clin. Cancer Res. 2014, 20, 1542–1554. [Google Scholar] [CrossRef]
- Crawford, L.J.; Walker, B.; Irvine, A.E. Proteasome inhibitors in cancer therapy. J. Cell Commun. Signal. 2011, 5, 101–110. [Google Scholar] [CrossRef]
- Goldberg, A.L. Development of Proteasome Inhibitors as Research Tools and Cancer Drugs; Rockefeller University Press: New York, NY, USA, 2012. [Google Scholar]
- Moreau, P.; Richardson, P.G.; Cavo, M.; Orlowski, R.Z.; San Miguel, J.F.; Palumbo, A.; Harousseau, J.L. Proteasome inhibitors in multiple myeloma: 10 years later. Blood 2012, 120, 947–959. [Google Scholar] [CrossRef]
- Obrist, F.; Manic, G.; Kroemer, G.; Vitale, I.; Galluzzi, L. Trial watch: Proteasomal inhibitors for anticancer therapy. Mol. Cell. Oncol. 2015, 2, e974463. [Google Scholar] [CrossRef]
- Badin, F.B.; Chiang, A.C.; Fisher, W.B.; Orlov, S.; Harper, H.D.; Eskander, E.; Harb, W.A.; Kio, E.; Gopalan, P.K.; Haggstrom, D.E. Carfilzomib (CFZ), carboplatin and etoposide for previously untreated extensive-stage small cell lung cancer (ES-SCLC): Phase 1b results from a phase 1b/2 study. J. Clin. Oncol. 2016. [Google Scholar] [CrossRef]
- Schönthal, A.H. Endoplasmic reticulum stress: Its role in disease and novel prospects for therapy. Scientifica 2012, 2012, 857516. [Google Scholar] [CrossRef]
- Kazi, A.; Daniel, K.G.; Smith, D.M.; Kumar, N.B.; Dou, Q.P. Inhibition of the proteasome activity, a novel mechanism associated with the tumor cell apoptosis-inducing ability of genistein. Biochem. Pharmacol. 2003, 66, 965–976. [Google Scholar] [CrossRef]
- Chen, D.; Chen, M.S.; Cui, Q.C.; Yang, H.; Dou, Q.P. Structure-proteasome-inhibitory activity relationships of dietary flavonoids in human cancer cells. Front. Biosci. 2007, 12, 1935–1945. [Google Scholar] [CrossRef]
- Chen, D.; Landis-Piwowar, K.R.; Chen, M.S.; Dou, Q.P. Inhibition of proteasome activity by the dietary flavonoid apigenin is associated with growth inhibition in cultured breast cancer cells and xenografts. Breast Cancer Res. 2007, 9, R80. [Google Scholar] [CrossRef]
- Chen, D.; Daniel, K.G.; Chen, M.S.; Kuhn, D.J.; Landis-Piwowar, K.R.; Dou, Q.P. Dietary flavonoids as proteasome inhibitors and apoptosis inducers in human leukemia cells. Biochem. Pharmacol. 2005, 69, 1421–1432. [Google Scholar] [CrossRef]
- Jana, N.R.; Dikshit, P.; Goswami, A.; Nukina, N. Inhibition of proteasomal function by curcumin induces apoptosis through mitochondrial pathway. J. Biol. Chem. 2004, 279, 11680–11685. [Google Scholar] [CrossRef]
- Nam, S.; Smith, D.M.; Dou, Q.P. Tannic acid potently inhibits tumor cell proteasome activity, increases p27 and Bax expression, and induces g1 arrest and apoptosis. Cancer Epidemiol. Prev. Biomark. 2001, 10, 1083–1088. [Google Scholar]
- Saiko, P.; Steinmann, M.T.; Schuster, H.; Graser, G.; Bressler, S.; Giessrigl, B.; Lackner, A.; Grusch, M.; Krupitza, G.; Bago-Horvath, Z. Epigallocatechin gallate, ellagic acid, and rosmarinic acid perturb dNTP pools and inhibit de novo DNA synthesis and proliferation of human HL-60 promyelocytic leukemia cells: Synergism with arabinofuranosylcytosine. Phytomedicine 2015, 22, 213–222. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Henning, S.M.; Heber, D.; Vadgama, J.V. Sensitization to docetaxel in prostate cancer cells by green tea and quercetin. J. Nutr. Biochem. 2015, 26, 408–415. [Google Scholar] [CrossRef]
- Ho Zhi Guang, M.; Kavanagh, E.L.; Dunne, L.P.; Dowling, P.; Zhang, L.; Lindsay, S.; Bazou, D.; Goh, C.Y.; Hanley, C.; Bianchi, G. Targeting proteotoxic stress in cancer: A review of the role that protein quality control pathways play in oncogenesis. Cancers 2019, 11, 66. [Google Scholar] [CrossRef]
- Vogl, D.T.; Stadtmauer, E.A.; Tan, K.S.; Heitjan, D.F.; Davis, L.E.; Pontiggia, L.; Rangwala, R.; Piao, S.; Chang, Y.C.; Scott, E.C. Combined autophagy and proteasome inhibition: A phase 1 trial of hydroxychloroquine and bortezomib in patients with relapsed/refractory myeloma. Autophagy 2014, 10, 1380–1390. [Google Scholar] [CrossRef] [PubMed]
- Jakubowiak, A.; Offidani, M.; Pégourie, B.; De La Rubia, J.; Garderet, L.; Laribi, K.; Bosi, A.; Marasca, R.; Laubach, J.; Mohrbacher, A. Randomized phase 2 study: Elotuzumab plus bortezomib/dexamethasone vs bortezomib/dexamethasone for relapsed/refractory mm. Blood 2016, 127, 2833–2840. [Google Scholar] [CrossRef]
- Palumbo, A.; Chanan-Khan, A.; Weisel, K.; Nooka, A.K.; Masszi, T.; Beksac, M.; Spicka, I.; Hungria, V.; Munder, M.; Mateos, M.V. Daratumumab, bortezomib, and dexamethasone for multiple myeloma. N. Engl. J. Med. 2016, 375, 754–766. [Google Scholar] [CrossRef] [PubMed]
- San-Miguel, J.F.; Hungria, V.T.; Yoon, S.S.; Beksac, M.; Dimopoulos, M.A.; Elghandour, A.; Jedrzejczak, W.W.; Günther, A.; Nakorn, T.N.; Siritanaratkul, N. Panobinostat plus bortezomib and dexamethasone versus placebo plus bortezomib and dexamethasone in patients with relapsed or relapsed and refractory multiple myeloma: A multicentre, randomised, double-blind phase 3 trial. Lancet Oncol. 2014, 15, 1195–1206. [Google Scholar] [CrossRef]
- San-Miguel, J.F.; Hungria, V.T.; Yoon, S.S.; Beksac, M.; Dimopoulos, M.A.; Elghandour, A.; Jedrzejczak, W.W.; Günther, A.; Nakorn, T.N.; Siritanaratkul, N. Overall survival of patients with relapsed multiple myeloma treated with panobinostat or placebo plus bortezomib and dexamethasone (the panorama 1 trial): A randomised, placebo-controlled, phase 3 trial. Lancet Haematol. 2016, 3, e506–e515. [Google Scholar] [CrossRef]
- Moreau, P.; Chanan-Khan, A.; Roberts, A.W.; Agarwal, A.B.; Facon, T.; Kumar, S.; Touzeau, C.; Punnoose, E.A.; Cordero, J.; Munasinghe, W. Promising efficacy and acceptable safety of venetoclax plus bortezomib and dexamethasone in relapsed/refractory mm. Blood 2017, 130, 2392–2400. [Google Scholar] [CrossRef] [PubMed]
- Santo, L.; Hideshima, T.; Kung, A.L.; Tseng, J.C.; Tamang, D.; Yang, M.; Jarpe, M.; van Duzer, J.H.; Mazitschek, R.; Ogier, W.C. Preclinical activity, pharmacodynamic, and pharmacokinetic properties of a selective hdac6 inhibitor, acy-1215, in combination with bortezomib in multiple myeloma. Blood 2012, 119, 2579–2589. [Google Scholar] [CrossRef]
- Mishima, Y.; Santo, L.; Eda, H.; Cirstea, D.; Nemani, N.; Yee, A.J.; O’Donnell, E.; Selig, M.K.; Quayle, S.N.; Arastu-Kapur, S. Ricolinostat (acy-1215) induced inhibition of aggresome formation accelerates carfilzomib-induced multiple myeloma cell death. Br. J. Haematol. 2015, 169, 423–434. [Google Scholar] [CrossRef] [PubMed]
- Petrocca, F.; Altschuler, G.; Tan, S.M.; Mendillo, M.L.; Yan, H.; Jerry, D.J.; Kung, A.L.; Hide, W.; Ince, T.A.; Lieberman, J. A genome-wide sirna screen identifies proteasome addiction as a vulnerability of basal-like triple-negative breast cancer cells. Cancer Cell 2013, 24, 182–196. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.J.; Yeh, M.H.; Yu, M.C.; Wei, Y.L.; Chen, W.S.; Chen, J.Y.; Shih, C.Y.; Tu, C.Y.; Chen, C.H.; Hsia, T.C. Lapatinib–induced NF-kappaB activation sensitizes triple-negative breast cancer cells to proteasome inhibitors. Breast Cancer Res. 2013, 15, R108. [Google Scholar] [CrossRef] [PubMed]
- Richardson, P.G.; Sonneveld, P.; Schuster, M.W.; Stadtmauer, E.A.; Facon, T.; Harousseau, J.L.; Ben-Yehuda, D.; Lonial, S.; Goldschmidt, H.; Reece, D. Reversibility of symptomatic peripheral neuropathy with bortezomib in the phase iii apex trial in relapsed multiple myeloma: Impact of a dose-modification guideline. Br. J. Haematol. 2009, 144, 895–903. [Google Scholar] [CrossRef] [PubMed]
- Richardson, P.G.; Sonneveld, P.; Schuster, M.W.; Irwin, D.; Stadtmauer, E.A.; Facon, T.; Harousseau, J.L.; Ben-Yehuda, D.; Lonial, S.; Goldschmidt, H. Bortezomib or high-dose dexamethasone for relapsed multiple myeloma. N. Engl. J. Med. 2005, 352, 2487–2498. [Google Scholar] [CrossRef] [PubMed]
- Lonial, S.; Richardson, P.G.; San Miguel, J.; Sonneveld, P.; Schuster, M.W.; Bladé, J.; Cavenagh, J.; Rajkumar, S.V.; Jakubowiak, A.J.; Esseltine, D.L. Characterisation of haematological profiles and low risk of thromboembolic events with bortezomib in patients with relapsed multiple myeloma. Br. J. Haematol. 2008, 143, 222–229. [Google Scholar] [CrossRef]
- Chanan-Khan, A.; Sonneveld, P.; Schuster, M.W.; Stadtmauer, E.A.; Facon, T.; Harousseau, J.L.; Ben-Yehuda, D.; Lonial, S.; Goldschmidt, H.; Reece, D. Analysis of herpes zoster events among bortezomib-treated patients in the phase iii apex study. J. Clin. Oncol. 2008, 26, 4784–4790. [Google Scholar] [CrossRef]
- Grandin, E.W.; Ky, B.; Cornell, R.F.; Carver, J.; Lenihan, D.J. Patterns of cardiac toxicity associated with irreversible proteasome inhibition in the treatment of multiple myeloma. J. Card. Fail. 2015, 21, 138–144. [Google Scholar] [CrossRef]
- Danhof, S.; Schreder, M.; Rasche, L.; Strifler, S.; Einsele, H.; Knop, S. ‘Real-life’experience of preapproval carfilzomib-based therapy in myeloma–analysis of cardiac toxicity and predisposing factors. Eur. J. Haematol. 2016, 97, 25–32. [Google Scholar] [CrossRef]
- Dimopoulos, M.A.; Roussou, M.; Gavriatopoulou, M.; Psimenou, E.; Ziogas, D.; Eleutherakis-Papaiakovou, E.; Fotiou, D.; Migkou, M.; Kanellias, N.; Panagiotidis, I. Cardiac and renal complications of carfilzomib in patients with multiple myeloma. Blood Adv. 2017, 1, 449–454. [Google Scholar] [CrossRef]
- Brem, G.J.; Mylonas, I.; Brüning, A. Eeyarestatin causes cervical cancer cell sensitization to bortezomib treatment by augmenting ER stress and chop expression. Gynecol. Oncol. 2013, 128, 383–390. [Google Scholar] [CrossRef] [PubMed]
- Chou, T.F.; Brown, S.J.; Minond, D.; Nordin, B.E.; Li, K.; Jones, A.C.; Chase, P.; Porubsky, P.R.; Stoltz, B.M.; Schoenen, F.J. Reversible inhibitor of p97, dbeq, impairs both ubiquitin-dependent and autophagic protein clearance pathways. Proc. Natl. Acad. Sci. USA 2011, 108, 4834–4839. [Google Scholar] [CrossRef] [PubMed]
- Chou, T.F.; Li, K.; Frankowski, K.J.; Schoenen, F.J.; Deshaies, R.J. Structure–activity relationship study reveals ML240 and ML241 as potent and selective inhibitors of p97 ATPase. ChemMedChem 2013, 8, 297–312. [Google Scholar] [CrossRef] [PubMed]
- Polucci, P.; Magnaghi, P.; Angiolini, M.; Asa, D.; Avanzi, N.; Badari, A.; Bertrand, J.; Casale, E.; Cauteruccio, S.; Cirla, A. Alkylsulfanyl-1, 2, 4-triazoles, a new class of allosteric valosine containing protein inhibitors. Synthesis and structure–activity relationships. J. Med. Chem. 2013, 56, 437–450. [Google Scholar] [CrossRef] [PubMed]
- Valle, C.W.; Min, T.; Bodas, M.; Mazur, S.; Begum, S.; Tang, D.; Vij, N. Critical role of VCP/p97 in the pathogenesis and progression of non-small cell lung carcinoma. PLoS ONE 2011, 6, e29073. [Google Scholar] [CrossRef] [PubMed]
- Vekaria, P.H.; Home, T.; Weir, S.; Schoenen, F.J.; Rao, R. Targeting p97 to disrupt protein homeostasis in cancer. Front. Oncol. 2016, 6, 181. [Google Scholar] [CrossRef]
- Chen, O.I.; Bobak, Y.P.; Stasyk, O.V.; Kunz-Schughart, L.A. A complex scenario and underestimated challenge: The tumor microenvironment, ER stress, and cancer treatment. Curr. Med. Chem. 2018, 25, 2465–2502. [Google Scholar] [CrossRef]
- Wang, Q.; Li, L.; Ye, Y. Inhibition of p97-dependent protein degradation by eeyarestatin I. J. Biol. Chem. 2008, 283, 7445–7454. [Google Scholar] [CrossRef]
- Wang, Q.; Mora-Jensen, H.; Weniger, M.A.; Perez-Galan, P.; Wolford, C.; Hai, T.; Ron, D.; Chen, W.; Trenkle, W.; Wiestner, A. ERAD inhibitors integrate ER stress with an epigenetic mechanism to activate bh3-only protein NOXA in cancer cells. Proc. Natl. Acad. Sci. USA 2009, 106, 2200–2205. [Google Scholar] [CrossRef]
- Anderson, D.J.; Le Moigne, R.; Djakovic, S.; Kumar, B.; Rice, J.; Wong, S.; Wang, J.; Yao, B.; Valle, E.; von Soly, S.K. Targeting the AAA ATPase p97 as an approach to treat cancer through disruption of protein homeostasis. Cancer Cell 2015, 28, 653–665. [Google Scholar] [CrossRef]
- Zhou, H.J.; Wang, J.; Yao, B.; Wong, S.; Djakovic, S.; Kumar, B.; Rice, J.; Valle, E.; Soriano, F.; Menon, M.K. Discovery of a first-in-class, potent, selective, and orally bioavailable inhibitor of the p97 AAA ATPase (CB-5083). J. Med. Chem. 2015, 58, 9480–9497. [Google Scholar] [CrossRef] [PubMed]
- Le Moigne, R.; Aftab, B.T.; Djakovic, S.; Dhimolea, E.; Valle, E.; Murnane, M.; King, E.M.; Soriano, F.; Menon, M.K.; Wu, Z.Y. The p97 inhibitor CB-5083 is a unique disrupter of protein homeostasis in models of multiple myeloma. Mol. Cancer Ther. 2017, 16, 2375–2386. [Google Scholar] [CrossRef] [PubMed]
- Martin, S.; Lamb, H.; Brady, C.; Lefkove, B.; Bonner, M.; Thompson, P.; Lovat, P.; Arbiser, J.; Hawkins, A.; Redfern, C. Inducing apoptosis of cancer cells using small-molecule plant compounds that bind to GRP78. Br. J. Cancer 2013, 109, 433–443. [Google Scholar] [CrossRef] [PubMed]
- Paton, A.W.; Beddoe, T.; Thorpe, C.M.; Whisstock, J.C.; Wilce, M.C.; Rossjohn, J.; Talbot, U.M.; Paton, J.C. AB5 subtilase cytotoxin inactivates the endoplasmic reticulum chaperone BiP. Nature 2006, 443, 548–552. [Google Scholar] [CrossRef] [PubMed]
- Backer, J.M.; Krivoshein, A.V.; Hamby, C.V.; Pizzonia, J.; Gilbert, K.S.; Ray, Y.S.; Brand, H.; Paton, A.W.; Paton, J.C.; Backer, M.V. Chaperone-targeting cytotoxin and endoplasmic reticulum stress-inducing drug synergize to kill cancer cells. Neoplasia 2009, 11, 1165–1173. [Google Scholar] [CrossRef] [PubMed]
- Firczuk, M.; Gabrysiak, M.; Barankiewicz, J.; Domagala, A.; Nowis, D.; Kujawa, M.; Jankowska-Steifer, E.; Wachowska, M.; Glodkowska-Mrowka, E.; Korsak, B. Grp78-targeting subtilase cytotoxin sensitizes cancer cells to photodynamic therapy. Cell Death Dis. 2013, 4, e741. [Google Scholar] [CrossRef]
- Cerezo, M.; Lehraiki, A.; Millet, A.; Rouaud, F.; Plaisant, M.; Jaune, E.; Botton, T.; Ronco, C.; Abbe, P.; Amdouni, H. Compounds triggering ER stress exert anti-melanoma effects and overcome BRAF inhibitor resistance. Cancer Cell 2016, 29, 805–819. [Google Scholar] [CrossRef]
- Ermakova, S.P.; Kang, B.S.; Choi, B.Y.; Choi, H.S.; Schuster, T.F.; Ma, W.Y.; Bode, A.M.; Dong, Z. (−)−epigallocatechin gallate overcomes resistance to etoposide-induced cell death by targeting the molecular chaperone glucose-regulated protein 78. Cancer Res. 2006, 66, 9260–9269. [Google Scholar] [CrossRef]
- Matsuo, J.; Tsukumo, Y.; Sakurai, J.; Tsukahara, S.; Park, H.R.; Shin-ya, K.; Watanabe, T.; Tsuruo, T.; Tomida, A. Preventing the unfolded protein response via aberrant activation of 4E-binding protein 1 by versipelostatin. Cancer Sci. 2009, 100, 327–333. [Google Scholar] [CrossRef]
- Cook, K.L.; Clarke, R. Role of GRP78 in promoting therapeutic-resistant breast cancer. Future Med. Chem. 2015, 7, 1529–1534. [Google Scholar] [CrossRef]
- Kitao, Y.; Ozawa, K.; Miyazaki, M.; Tamatani, M.; Kobayashi, T.; Yanagi, H.; Okabe, M.; Ikawa, M.; Yamashima, T.; Stern, D.M. Expression of the endoplasmic reticulum molecular chaperone (ORP150) rescues hippocampal neurons from glutamate toxicity. J. Clin. Investig. 2001, 108, 1439–1450. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.S.; Lu, F.E.; Xu, L.J.; Dong, H. Berberine reduces endoplasmic reticulum stress and improves insulin signal transduction in Hep G2 cells. Acta Pharmacol. Sin. 2010, 31, 578–584. [Google Scholar] [CrossRef] [PubMed]
- Lawson, B.; Brewer, J.W.; Hendershot, L.M. Geldanamycin, an hsp90/GRP94-binding drug, induces increased transcription of endoplasmic reticulum (ER) chaperones via the ER stress pathway. J. Cell. Physiol. 1998, 174, 170–179. [Google Scholar] [CrossRef]
- Jones, D.T.; Addison, E.; North, J.M.; Lowdell, M.W.; Hoffbrand, A.V.; Mehta, A.B.; Ganeshaguru, K.; Folarin, N.I.; Wickremasinghe, R.G. Geldanamycin and herbimycin a induce apoptotic killing of b chronic lymphocytic leukemia cells and augment the cells’ sensitivity to cytotoxic drugs. Blood 2004, 103, 1855–1861. [Google Scholar] [CrossRef]
- Booth, L.; Roberts, J.L.; Cruickshanks, N.; Conley, A.; Durrant, D.E.; Das, A.; Fisher, P.B.; Kukreja, R.C.; Grant, S.; Poklepovic, A. Phosphodiesterase 5 inhibitors enhance chemotherapy killing in gastrointestinal/genitourinary cancer cells. Mol. Pharmacol. 2014, 85, 408–419. [Google Scholar] [CrossRef]
- Booth, L.; Roberts, J.L.; Cruickshanks, N.; Grant, S.; Poklepovic, A.; Dent, P. Regulation of osu-03012 toxicity by ER stress proteins and ER stress–inducing drugs. Mol. Cancer Ther. 2014, 13, 2384–2398. [Google Scholar] [CrossRef]
- Jhaveri, K.; Taldone, T.; Modi, S.; Chiosis, G. Advances in the clinical development of heat shock protein 90 (Hsp90) inhibitors in cancers. Biochim. Biophys. Acta BBA Mol. Cell Res. 2012, 1823, 742–755. [Google Scholar] [CrossRef]
- Neckers, L.; Workman, P. Hsp90 molecular chaperone inhibitors: Are we there yet? Clin. Cancer Res. 2012, 18, 64–76. [Google Scholar] [CrossRef]
- Davenport, E.L.; Moore, H.E.; Dunlop, A.S.; Sharp, S.Y.; Workman, P.; Morgan, G.J.; Davies, F.E. Heat shock protein inhibition is associated with activation of the unfolded protein response pathway in myeloma plasma cells. Blood 2007, 110, 2641–2649. [Google Scholar] [CrossRef]
- De Raedt, T.; Walton, Z.; Yecies, J.L.; Li, D.; Chen, Y.; Malone, C.F.; Maertens, O.; Jeong, S.M.; Bronson, R.T.; Lebleu, V. Exploiting cancer cell vulnerabilities to develop a combination therapy for ras-driven tumors. Cancer Cell 2011, 20, 400–413. [Google Scholar] [CrossRef]
- Duerfeldt, A.S.; Peterson, L.B.; Maynard, J.C.; Ng, C.L.; Eletto, D.; Ostrovsky, O.; Shinogle, H.E.; Moore, D.S.; Argon, Y.; Nicchitta, C.V. Development of a Grp94 inhibitor. J. Am. Chem. Soc. 2012, 134, 9796–9804. [Google Scholar] [CrossRef] [PubMed]
- Duerfeldt, A.S.; Brandt, G.E.; Blagg, B.S. Design, synthesis, and biological evaluation of conformationally constrained cis-amide hsp90 inhibitors. Org. Lett. 2009, 11, 2353–2356. [Google Scholar] [CrossRef] [PubMed]
- Goplen, D.; Wang, J.; Enger, P.Ø.; Tysnes, B.B.; Terzis, A.; Laerum, O.D.; Bjerkvig, R. Protein disulfide isomerase expression is related to the invasive properties of malignant glioma. Cancer Res. 2006, 66, 9895–9902. [Google Scholar] [CrossRef] [PubMed]
- Lovat, P.E.; Corazzari, M.; Armstrong, J.L.; Martin, S.; Pagliarini, V.; Hill, D.; Brown, A.M.; Piacentini, M.; Birch-Machin, M.A.; Redfern, C.P. Increasing melanoma cell death using inhibitors of protein disulfide isomerases to abrogate survival responses to endoplasmic reticulum stress. Cancer Res. 2008, 68, 5363–5369. [Google Scholar] [CrossRef] [PubMed]
- Garg, A.D.; Nowis, D.; Golab, J.; Vandenabeele, P.; Krysko, D.V.; Agostinis, P. Immunogenic cell death, damps and anticancer therapeutics: An emerging amalgamation. Biochim. Biophys. Acta BBA Rev. Cancer 2010, 1805, 53–71. [Google Scholar] [CrossRef] [PubMed]
- Garg, A.D.; Martin, S.; Golab, J.; Agostinis, P. Danger signalling during cancer cell death: Origins, plasticity and regulation. Cell Death Differ. 2014, 21, 26–38. [Google Scholar] [CrossRef]
- Krysko, D.V.; Garg, A.D.; Kaczmarek, A.; Krysko, O.; Agostinis, P.; Vandenabeele, P. Immunogenic cell death and damps in cancer therapy. Nat. Rev. Cancer 2012, 12, 860–875. [Google Scholar] [CrossRef]
- Zitvogel, L.; Kepp, O.; Senovilla, L.; Menger, L.; Chaput, N.; Kroemer, G. Immunogenic tumor cell death for optimal anticancer therapy: The calreticulin exposure pathway. Clin. Cancer Res. 2010, 16, 3100–3104. [Google Scholar] [CrossRef]
- Van Vliet, A.; Martin, S.; Garg, A.; Agostinis, P. The perks of damage-associated molecular patterns mediating cancer immunogenicity: From sensor to the plasma membrane and beyond. Semin. Cancer Biol. 2015, 33, 74–85. [Google Scholar] [CrossRef]
- Garg, A.D.; Krysko, D.V.; Verfaillie, T.; Kaczmarek, A.; Ferreira, G.B.; Marysael, T.; Rubio, N.; Firczuk, M.; Mathieu, C.; Roebroek, A.J. A novel pathway combining calreticulin exposure and ATP secretion in immunogenic cancer cell death. EMBO J. 2012, 31, 1062–1079. [Google Scholar] [CrossRef]
- Panaretakis, T.; Kepp, O.; Brockmeier, U.; Tesniere, A.; Bjorklund, A.C.; Chapman, D.C.; Durchschlag, M.; Joza, N.; Pierron, G.; Van Endert, P. Mechanisms of pre-apoptotic calreticulin exposure in immunogenic cell death. EMBO J. 2009, 28, 578–590. [Google Scholar] [CrossRef] [PubMed]
- Garg, A.D.; Dudek, A.M.; Ferreira, G.B.; Verfaillie, T.; Vandenabeele, P.; Krysko, D.V.; Mathieu, C.; Agostinis, P. Ros-induced autophagy in cancer cells assists in evasion from determinants of immunogenic cell death. Autophagy 2013, 9, 1292–1307. [Google Scholar] [CrossRef] [PubMed]
- Garg, A.D.; Dudek, A.M.; Agostinis, P. Calreticulin surface exposure is abrogated in cells lacking, chaperone-mediated autophagy-essential gene, LAMP2A. Cell Death Dis. 2013, 4, e826. [Google Scholar] [CrossRef] [PubMed]
- Michaud, M.; Martins, I.; Sukkurwala, A.Q.; Adjemian, S.; Ma, Y.; Pellegatti, P.; Shen, S.; Kepp, O.; Scoazec, M.; Mignot, G. Autophagy-dependent anticancer immune responses induced by chemotherapeutic agents in mice. Science 2011, 334, 1573–1577. [Google Scholar] [CrossRef] [PubMed]
- Garg, A.D.; Maes, H.; van Vliet, A.R.; Agostinis, P. Targeting the hallmarks of cancer with therapy-induced endoplasmic reticulum (ER) stress. Mol. Cell. Oncol. 2015, 2, e975089. [Google Scholar] [CrossRef] [PubMed]
- Fucikova, J.; Becht, E.; Iribarren, K.; Goc, J.; Remark, R.; Damotte, D.; Alifano, M.; Devi, P.; Biton, J.; Germain, C. Calreticulin expression in human non–small cell lung cancers correlates with increased accumulation of antitumor immune cells and favorable prognosis. Cancer Res. 2016, 76, 1746–1756. [Google Scholar] [CrossRef]
- Shoulders, M.D.; Ryno, L.M.; Genereux, J.C.; Moresco, J.J.; Tu, P.G.; Wu, C.; Yates, J.R., III; Su, A.I.; Kelly, J.W.; Wiseman, R.L. Stress-independent activation of XBP1s and/or ATF6 reveals three functionally diverse ER proteostasis environments. Cell Rep. 2013, 3, 1279–1292. [Google Scholar] [CrossRef]
- Kepp, O.; Menger, L.; Vacchelli, E.; Locher, C.; Adjemian, S.; Yamazaki, T.; Martins, I.; Sukkurwala, A.Q.; Michaud, M.; Senovilla, L. Crosstalk between ER stress and immunogenic cell death. Cytokine Growth Factor Rev. 2013, 24, 311–318. [Google Scholar] [CrossRef]
- Wang, S.; Kaufman, R.J. The impact of the unfolded protein response on human disease. J. Cell Biol. 2012, 197, 857–867. [Google Scholar] [CrossRef]
- Hetz, C.; Chevet, E.; Oakes, S.A. Proteostasis control by the unfolded protein response. Nat. Cell Biol. 2015, 17, 829–838. [Google Scholar] [CrossRef]
- Field-Smith, A.; Morgan, G.J.; Davies, F.E. Bortezomib (velcade™) in the treatment of multiple myeloma. Ther. Clin. Risk Manag. 2006, 2, 271–279. [Google Scholar] [CrossRef] [PubMed]
- Obeng, E.A.; Carlson, L.M.; Gutman, D.M.; Harrington, W.J.; Lee, K.P.; Boise, L.H. Proteasome inhibitors induce a terminal unfolded protein response in multiple myeloma cells. Blood 2006, 107, 4907–4916. [Google Scholar] [CrossRef] [PubMed]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nam, S.M.; Jeon, Y.J. Proteostasis in the Endoplasmic Reticulum: Road to Cure. Cancers 2019, 11, 1793. https://doi.org/10.3390/cancers11111793
Nam SM, Jeon YJ. Proteostasis in the Endoplasmic Reticulum: Road to Cure. Cancers. 2019; 11(11):1793. https://doi.org/10.3390/cancers11111793
Chicago/Turabian StyleNam, Su Min, and Young Joo Jeon. 2019. "Proteostasis in the Endoplasmic Reticulum: Road to Cure" Cancers 11, no. 11: 1793. https://doi.org/10.3390/cancers11111793
APA StyleNam, S. M., & Jeon, Y. J. (2019). Proteostasis in the Endoplasmic Reticulum: Road to Cure. Cancers, 11(11), 1793. https://doi.org/10.3390/cancers11111793