PTEN in Colorectal Cancer: Shedding Light on Its Role as Predictor and Target


 ,
,  ,
, 

Abstract
:1. Introduction
2. PTEN in CRC
3. PTEN as a Predictive Factor
4. PTEN as a Target
5. Future Perspectives
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Li, J.; Yen, C.; Liaw, D.; Podsypanina, K.; Bose, S.; Wang, S.I.; Puc, J.; Miliaresis, C.; Rodgers, L.; McCombie, R.; et al. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science 1997, 275, 1943–1947. [Google Scholar] [CrossRef] [PubMed]
- Steck, P.A.; Pershouse, M.A.; Jasser, S.A.; Yung, W.K.; Lin, H.; Ligon, A.H.; Langford, L.A.; Baumgard, M.L.; Hattier, T.; Davis, T.; et al. Identification of a candidate tumour suppressor gene, MMAC1, at chromosome 10q23.3 that is mutated in multiple advanced cancers. Nat. Genet. 1997, 15, 356–362. [Google Scholar] [CrossRef] [PubMed]
- Furnari, F.B.; Huang, H.J.; Cavenee, W.K. The phosphoinositol phosphatase activity of PTEN mediates a serum-sensitive G1 growth arrest in glioma cells. Cancer Res. 1998, 58, 5002–5008. [Google Scholar] [PubMed]
- Kotelevets, L.; van Hengel, J.; Bruyneel, E.; Mareel, M.; van Roy, F.; Chastre, E. The lipid phosphatase activity of PTEN is critical for stabilizing intercellular junctions and reverting invasiveness. J. Cell Biol. 2001, 155, 1129–1135. [Google Scholar] [CrossRef] [PubMed]
- Szado, T.; Vanderheyden, V.; Parys, J.B.; De Smedt, H.; Rietdorf, K.; Kotelevets, L.; Chastre, E.; Khan, F.; Landegren, U.; Soderberg, O.; et al. Phosphorylation of inositol 1,4,5-trisphosphate receptors by protein kinase B/Akt inhibits Ca2+ release and apoptosis. Proc. Natl. Acad. Sci. USA 2008, 105, 2427–2432. [Google Scholar] [CrossRef]
- Shen, W.H.; Balajee, A.S.; Wang, J.; Wu, H.; Eng, C.; Pandolfi, P.P.; Yin, Y. Essential role for nuclear PTEN in maintaining chromosomal integrity. Cell 2007, 128, 157–170. [Google Scholar] [CrossRef]
- Song, M.S.; Salmena, L.; Pandolfi, P.P. The functions and regulation of the PTEN tumour suppressor. Nat. Rev. Mol. Cell. Biol. 2012, 13, 283–296. [Google Scholar] [CrossRef]
- Lin, P.C.; Lin, J.K.; Lin, H.H.; Lan, Y.T.; Lin, C.C.; Yang, S.H.; Chen, W.S.; Liang, W.Y.; Jiang, J.K.; Chang, S.C. A comprehensive analysis of phosphatase and tensin homolog deleted on chromosome 10 (PTEN) loss in colorectal cancer. World J. Surg. Oncol. 2015, 13, 186. [Google Scholar] [CrossRef]
- Molinari, F.; Frattini, M. Functions and regulation of the PTEN gene in colorectal cancer. Front. Oncol. 2013, 3, 326. [Google Scholar] [CrossRef]
- Zhou, X.P.; Loukola, A.; Salovaara, R.; Nystrom-Lahti, M.; Peltomaki, P.; de la Chapelle, A.; Aaltonen, L.A.; Eng, C. PTEN mutational spectra, expression levels, and subcellular localization in microsatellite stable and unstable colorectal cancers. Am. J. Pathol. 2002, 161, 439–447. [Google Scholar] [CrossRef]
- Cancer Genome Atlas, N. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Price, T.J.; Hardingham, J.E.; Lee, C.K.; Townsend, A.R.; Wrin, J.W.; Wilson, K.; Weickhardt, A.; Simes, R.J.; Murone, C.; Tebbutt, N.C. Prognostic impact and the relevance of PTEN copy number alterations in patients with advanced colorectal cancer (CRC) receiving bevacizumab. Cancer Med. 2013, 2, 277–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dillon, L.M.; Miller, T.W. Therapeutic targeting of cancers with loss of PTEN function. Curr. Drug. Targets 2014, 15, 65–79. [Google Scholar] [CrossRef] [PubMed]
- Day, F.L.; Jorissen, R.N.; Lipton, L.; Mouradov, D.; Sakthianandeswaren, A.; Christie, M.; Li, S.; Tsui, C.; Tie, J.; Desai, J.; et al. PIK3CA and PTEN gene and exon mutation-specific clinicopathologic and molecular associations in colorectal cancer. Clin. Cancer Res. 2013, 19, 3285–3296. [Google Scholar] [CrossRef] [PubMed]
- Colakoglu, T.; Yildirim, S.; Kayaselcuk, F.; Nursal, T.Z.; Ezer, A.; Noyan, T.; Karakayali, H.; Haberal, M. Clinicopathological significance of PTEN loss and the phosphoinositide 3-kinase/Akt pathway in sporadic colorectal neoplasms: Is PTEN loss predictor of local recurrence? Am. J. Surg. 2008, 195, 719–725. [Google Scholar] [CrossRef] [PubMed]
- Sawai, H.; Yasuda, A.; Ochi, N.; Ma, J.; Matsuo, Y.; Wakasugi, T.; Takahashi, H.; Funahashi, H.; Sato, M.; Takeyama, H. Loss of PTEN expression is associated with colorectal cancer liver metastasis and poor patient survival. BMC Gastroenterol. 2008, 8, 56. [Google Scholar] [CrossRef]
- Lin, M.S.; Huang, J.X.; Chen, W.C.; Zhang, B.F.; Fang, J.; Zhou, Q.; Hu, Y.; Gao, H.J. Expression of PPARgamma and PTEN in human colorectal cancer: An immunohistochemical study using tissue microarray methodology. Oncol. Lett. 2011, 2, 1219–1224. [Google Scholar] [CrossRef]
- Li, X.H.; Zheng, H.C.; Takahashi, H.; Masuda, S.; Yang, X.H.; Takano, Y. PTEN expression and mutation in colorectal carcinomas. Oncol. Rep. 2009, 22, 757–764. [Google Scholar] [CrossRef] [Green Version]
- Perren, A.; Weng, L.P.; Boag, A.H.; Ziebold, U.; Thakore, K.; Dahia, P.L.; Komminoth, P.; Lees, J.A.; Mulligan, L.M.; Mutter, G.L.; et al. Immunohistochemical evidence of loss of PTEN expression in primary ductal adenocarcinomas of the breast. Am. J. Pathol. 1999, 155, 1253–1260. [Google Scholar] [CrossRef]
- Marsit, C.J.; Zheng, S.; Aldape, K.; Hinds, P.W.; Nelson, H.H.; Wiencke, J.K.; Kelsey, K.T. PTEN expression in non-small-cell lung cancer: Evaluating its relation to tumor characteristics, allelic loss, and epigenetic alteration. Hum. Pathol. 2005, 36, 768–776. [Google Scholar] [CrossRef]
- Nagata, Y.; Lan, K.H.; Zhou, X.; Tan, M.; Esteva, F.J.; Sahin, A.A.; Klos, K.S.; Li, P.; Monia, B.P.; Nguyen, N.T.; et al. PTEN activation contributes to tumor inhibition by trastuzumab, and loss of PTEN predicts trastuzumab resistance in patients. Cancer Cell 2004, 6, 117–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kokubo, Y.; Gemma, A.; Noro, R.; Seike, M.; Kataoka, K.; Matsuda, K.; Okano, T.; Minegishi, Y.; Yoshimura, A.; Shibuya, M.; et al. Reduction of PTEN protein and loss of epidermal growth factor receptor gene mutation in lung cancer with natural resistance to gefitinib (IRESSA). Br. J. Cancer 2005, 92, 1711–1719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jhawer, M.; Goel, S.; Wilson, A.J.; Montagna, C.; Ling, Y.H.; Byun, D.S.; Nasser, S.; Arango, D.; Shin, J.; Klampfer, L.; et al. PIK3CA mutation/PTEN expression status predicts response of colon cancer cells to the epidermal growth factor receptor inhibitor cetuximab. Cancer Res. 2008, 68, 1953–1961. [Google Scholar] [CrossRef] [PubMed]
- Frattini, M.; Saletti, P.; Romagnani, E.; Martin, V.; Molinari, F.; Ghisletta, M.; Camponovo, A.; Etienne, L.L.; Cavalli, F.; Mazzucchelli, L. PTEN loss of expression predicts cetuximab efficacy in metastatic colorectal cancer patients. Br. J. Cancer 2007, 97, 1139–1145. [Google Scholar] [CrossRef] [Green Version]
- Loupakis, F.; Pollina, L.; Stasi, I.; Ruzzo, A.; Scartozzi, M.; Santini, D.; Masi, G.; Graziano, F.; Cremolini, C.; Rulli, E.; et al. PTEN expression and KRAS mutations on primary tumors and metastases in the prediction of benefit from cetuximab plus irinotecan for patients with metastatic colorectal cancer. J. Clin. Oncol. 2009, 27, 2622–2629. [Google Scholar] [CrossRef]
- Laurent-Puig, P.; Cayre, A.; Manceau, G.; Buc, E.; Bachet, J.B.; Lecomte, T.; Rougier, P.; Lievre, A.; Landi, B.; Boige, V.; et al. Analysis of PTEN, BRAF, and EGFR status in determining benefit from cetuximab therapy in wild-type KRAS metastatic colon cancer. J. Clin. Oncol. 2009, 27, 5924–5930. [Google Scholar] [CrossRef]
- Agoston, E.I.; Micsik, T.; Acs, B.; Fekete, K.; Hahn, O.; Baranyai, Z.; Dede, K.; Bodoky, G.; Bursics, A.; Kulka, J.; et al. In depth evaluation of the prognostic and predictive utility of PTEN immunohistochemistry in colorectal carcinomas: Performance of three antibodies with emphasis on intracellular and intratumoral heterogeneity. Diagn. Pathol. 2016, 11, 61. [Google Scholar] [CrossRef]
- Karapetis, C.S.; Jonker, D.; Daneshmand, M.; Hanson, J.E.; O’Callaghan, C.J.; Marginean, C.; Zalcberg, J.R.; Simes, J.; Moore, M.J.; Tebbutt, N.C.; et al. PIK3CA, BRAF, and PTEN status and benefit from cetuximab in the treatment of advanced colorectal cancer—results from NCIC CTG/AGITG CO.17. Clin. Cancer Res. 2014, 20, 744–753. [Google Scholar] [CrossRef]
- Sveen, A.; Kopetz, S.; Lothe, R.A. Biomarker-guided therapy for colorectal cancer: Strength in complexity. Nat. Rev. Clin. Oncol. 2019. [Google Scholar] [CrossRef]
- Jiang, Y.A.; Fan, L.F.; Jiang, C.Q.; Zhang, Y.Y.; Luo, H.S.; Tang, Z.J.; Xia, D.; Wang, M. Expression and significance of PTEN, hypoxia-inducible factor-1 alpha in colorectal adenoma and adenocarcinoma. World J. Gastroenterol. 2003, 9, 491–494. [Google Scholar] [CrossRef]
- Karar, J.; Maity, A. PI3K/AKT/mTOR Pathway in Angiogenesis. Front. Mol. Neurosci. 2011, 4, 51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, B.H.; Liu, L.Z. AKT signaling in regulating angiogenesis. Curr Cancer Drug Targets 2008, 8, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Guertin, D.A.; Sabatini, D.M. Defining the role of mTOR in cancer. Cancer Cell 2007, 12, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Hudson, C.C.; Liu, M.; Chiang, G.G.; Otterness, D.M.; Loomis, D.C.; Kaper, F.; Giaccia, A.J.; Abraham, R.T. Regulation of hypoxia-inducible factor 1alpha expression and function by the mammalian target of rapamycin. Mol. Cell. Biol. 2002, 22, 7004–7014. [Google Scholar] [CrossRef]
- Kara, O.; Duman, B.B.; Kara, B.; Erdogan, S.; Parsak, C.K.; Sakman, G. Analysis of PTEN, VEGF, HER2 and P53 status in determining colorectal cancer benefit from bevacizumab therapy. Asian Pac. J. Cancer Prev. 2012, 13, 6397–6401. [Google Scholar] [CrossRef]
- Sclafani, F.; Rimassa, L.; Colombo, P.; Destro, A.; Stinco, S.; Lutman, F.R.; Carnaghi, C.; Beretta, G.; Zanello, A.; Roncalli, M.; et al. An exploratory biomarker study in metastatic tumors from colorectal cancer patients treated with bevacizumab. Int. J. Biol. Markers 2015, 30, 73–80. [Google Scholar] [CrossRef]
- Di Nicolantonio, F.; Arena, S.; Tabernero, J.; Grosso, S.; Molinari, F.; Macarulla, T.; Russo, M.; Cancelliere, C.; Zecchin, D.; Mazzucchelli, L.; et al. Deregulation of the PI3K and KRAS signaling pathways in human cancer cells determines their response to everolimus. J. Clin. Investig. 2010, 120, 2858–2866. [Google Scholar] [CrossRef] [Green Version]
- Janku, F.; Hong, D.S.; Fu, S.Q.; Piha-Paul, S.A.; Naing, A.; Falchook, G.S.; Tsimberidou, A.M.; Stepanek, V.M.; Moulder, S.L.; Lee, J.J.; et al. Assessing PIK3CA and PTEN in early-phase trials with PI3K/AKT/mTOR inhibitors. Cell Rep. 2014, 6, 377–387. [Google Scholar] [CrossRef]
- Tabernero, J.; Rojo, F.; Calvo, E.; Burris, H.; Judson, I.; Hazell, K.; Martinelli, E.; Cajal, S.R.Y.; Jones, S.; Vidal, L.; et al. Dose- and schedule-dependent inhibition of the mammalian target of rapamycin pathway with everolimus: A phase I tumor pharmacodynamic study in patients with advanced solid tumors. J. Clin. Oncol. 2008, 26, 1603–1610. [Google Scholar] [CrossRef]
- O’Donnell, A.; Faivre, S.; Burris, H.A.; Rea, D.; Papadimitrakopoulou, V.; Shand, N.; Lane, H.A.; Hazell, K.; Zoellner, U.; Kovarik, J.M.; et al. Phase I pharmacokinetic and pharmacodynamic study of the oral mammalian target of rapamycin inhibitor everolimus in patients with advanced solid tumors. J. Clin. Oncol. 2008, 26, 1588–1595. [Google Scholar] [CrossRef]
- Ng, K.; Tabernero, J.; Hwang, J.; Bajetta, E.; Sharma, S.; Del Prete, S.A.; Arrowsmith, E.R.; Ryan, D.P.; Sedova, M.; Jin, J.; et al. Phase II Study of Everolimus in Patients with Metastatic Colorectal Adenocarcinoma Previously Treated with Bevacizumab-, Fluoropyrimidine-, Oxaliplatin-, and Irinotecan-Based Regimens. Clin. Cancer Res. 2013, 19, 3987–3995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Majumder, P.K.; Febbo, P.G.; Bikoff, R.; Berger, R.; Xue, Q.; McMahon, L.M.; Manola, J.; Brugarolas, J.; McDonnell, T.J.; Golub, T.R.; et al. mTOR inhibition reverses Akt-dependent prostate intraepithelial neoplasia through regulation of apoptotic and HIF-1-dependent pathways. Nat. Med. 2004, 10, 594–601. [Google Scholar] [CrossRef] [PubMed]
- Loges, S.; Mazzone, M.; Hohensinner, P.; Carmeliet, P. Silencing or fueling metastasis with VEGF inhibitors: Antiangiogenesis revisited. Cancer Cell 2009, 15, 167–170. [Google Scholar] [CrossRef]
- Grepin, R.; Pages, G. Molecular mechanisms of resistance to tumour anti-angiogenic strategies. J. Oncol 2010, 2010, 835680. [Google Scholar] [CrossRef] [PubMed]
- Negrier, S.; Gravis, G.; Perol, D.; Chevreau, C.; Delva, R.; Bay, J.O.; Blanc, E.; Ferlay, C.; Geoffrois, L.; Rolland, F.; et al. Temsirolimus and bevacizumab, or sunitinib, or interferon alfa and bevacizumab for patients with advanced renal cell carcinoma (TORAVA): A randomised phase 2 trial. Lancet Oncol. 2011, 12, 673–680. [Google Scholar] [CrossRef]
- Weldon Gilcrease, G.; Stenehjem, D.D.; Wade, M.L.; Weis, J.; McGregor, K.; Whisenant, J.; Boucher, K.M.; Thorne, K.; Orgain, N.; Garrido-Laguna, I.; et al. Phase I/II study of everolimus combined with mFOLFOX-6 and bevacizumab for first-line treatment of metastatic colorectal cancer. Investig. New Drugs 2019, 37, 482–489. [Google Scholar] [CrossRef]
- He, C.; Sun, X.P.; Qiao, H.; Jiang, X.; Wang, D.; Jin, X.; Dong, X.; Wang, J.; Jiang, H.; Sun, X. Downregulating hypoxia-inducible factor-2alpha improves the efficacy of doxorubicin in the treatment of hepatocellular carcinoma. Cancer Sci. 2012, 103, 528–534. [Google Scholar] [CrossRef]
- Moroney, J.; Fu, S.; Moulder, S.; Falchook, G.; Helgason, T.; Levenback, C.; Hong, D.; Naing, A.; Wheler, J.; Kurzrock, R. Phase I study of the antiangiogenic antibody bevacizumab and the mTOR/hypoxia-inducible factor inhibitor temsirolimus combined with liposomal doxorubicin: Tolerance and biological activity. Clin. Cancer Res. 2012, 18, 5796–5805. [Google Scholar] [CrossRef]
- Corcoran, R.B.; Atreya, C.E.; Falchook, G.S.; Kwak, E.L.; Ryan, D.P.; Bendell, J.C.; Hamid, O.; Messersmith, W.A.; Daud, A.; Kurzrock, R.; et al. Combined BRAF and MEK inhibition with Dabrafenib and trametinib in BRAF V600-mutant colorectal cancer. J. Clin. Oncol. 2015, 33, 4023–4031. [Google Scholar] [CrossRef]
- Pishvaian, M.J.; Slack, R.S.; Jiang, W.; He, A.R.; Hwang, J.J.; Hankin, A.; Dorsch-Vogel, K.; Kukadiya, D.; Weiner, L.M.; Marshall, J.L.; et al. A phase 2 study of the PARP inhibitor veliparib plus temozolomide in patients with heavily pretreated metastatic colorectal cancer. Cancer 2018, 124, 2337–2346. [Google Scholar] [CrossRef]
- Minami, D.; Takigawa, N.; Takeda, H.; Takata, M.; Ochi, N.; Ichihara, E.; Hisamoto, A.; Hotta, K.; Tanimoto, M.; Kiura, K. Synergistic effect of olaparib with combination of cisplatin on PTEN-deficient lung cancer cells. Mol. Cancer Res. 2013, 11, 140–148. [Google Scholar] [CrossRef] [PubMed]
- Mendes-Pereira, A.M.; Martin, S.A.; Brough, R.; McCarthy, A.; Taylor, J.R.; Kim, J.S.; Waldman, T.; Lord, C.J.; Ashworth, A. Synthetic lethal targeting of PTEN mutant cells with PARP inhibitors. EMBO Mol. Med. 2009, 1, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Dedes, K.J.; Wetterskog, D.; Mendes-Pereira, A.M.; Natrajan, R.; Lambros, M.B.; Geyer, F.C.; Vatcheva, R.; Savage, K.; Mackay, A.; Lord, C.J.; et al. PTEN deficiency in endometrioid endometrial adenocarcinomas predicts sensitivity to PARP inhibitors. Sci. Transl. Med. 2010, 2, 53ra75. [Google Scholar] [CrossRef] [PubMed]
- Forster, M.D.; Dedes, K.J.; Sandhu, S.; Frentzas, S.; Kristeleit, R.; Ashworth, A.; Poole, C.J.; Weigelt, B.; Kaye, S.B.; Molife, L.R. Treatment with olaparib in a patient with PTEN-deficient endometrioid endometrial cancer. Nat. Rev. Clin. Oncol. 2011, 8, 302–306. [Google Scholar] [CrossRef] [PubMed]
- Therkildsen, C.; Bergmann, T.K.; Henrichsen-Schnack, T.; Ladelund, S.; Nilbert, M. The predictive value of KRAS, NRAS, BRAF, PIK3CA and PTEN for anti-EGFR treatment in metastatic colorectal cancer: A systematic review and meta-analysis. Acta Oncol. 2014, 53, 852–864. [Google Scholar] [CrossRef] [PubMed]
- Kotelevets, L.; Scott, M.G.H.; Chastre, E. Targeting PTEN in colorectal cancers. Adv. Exp. Med. Biol 2018, 1110, 55–73. [Google Scholar] [CrossRef]
- Leslie, N.R.; Foti, M. Non-genomic loss of PTEN function in cancer: Not in my genes. Trends Pharm. Sci. 2011, 32, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Garrido-Laguna, I.; McGregor, K.A.; Wade, M.; Weis, J.; Gilcrease, W.; Burr, L.; Soldi, R.; Jakubowski, L.; Davidson, C.; Morrell, G.; et al. A phase I/II study of decitabine in combination with panitumumab in patients with wild-type (wt) KRAS metastatic colorectal cancer. Investig. New Drugs 2013, 31, 1257–1264. [Google Scholar] [CrossRef]
- Jansen, Y.J.L.; Verset, G.; Schats, K.; Van Dam, P.J.; Seremet, T.; Kockx, M.; Van Laethem, J.B.; Neyns, B. Phase I clinical trial of decitabine (5-aza-2’-deoxycytidine) administered by hepatic arterial infusion in patients with unresectable liver-predominant metastases. ESMO Open 2019, 4, e000464. [Google Scholar] [CrossRef]
- Li, H.; Chiappinelli, K.B.; Guzzetta, A.A.; Easwaran, H.; Yen, R.W.; Vatapalli, R.; Topper, M.J.; Luo, J.; Connolly, R.M.; Azad, N.S.; et al. Immune regulation by low doses of the DNA methyltransferase inhibitor 5-azacitidine in common human epithelial cancers. Oncotarget 2014, 5, 587–598. [Google Scholar] [CrossRef]
- Coronel-Hernandez, J.; Lopez-Urrutia, E.; Contreras-Romero, C.; Delgado-Waldo, I.; Figueroa-Gonzalez, G.; Campos-Parra, A.D.; Salgado-Garcia, R.; Martinez-Gutierrez, A.; Rodriguez-Morales, M.; Jacobo-Herrera, N.; et al. Cell migration and proliferation are regulated by miR-26a in colorectal cancer via the PTEN-AKT axis. Cancer Cell Int. 2019, 19, 80. [Google Scholar] [CrossRef] [PubMed]
- Noorolyai, S.; Mokhtarzadeh, A.; Baghbani, E.; Asadi, M.; Baghbanzadeh Kojabad, A.; Mogaddam, M.M.; Baradaran, B. The role of microRNAs involved in PI3-kinase signaling pathway in colorectal cancer. J. Cell. Physiol. 2019, 234, 5664–5673. [Google Scholar] [CrossRef] [PubMed]
- Kudinov, A.E.; Karanicolas, J.; Golemis, E.A.; Boumber, Y. Musashi RNA-binding proteins as cancer drivers and novel therapeutic targets. Clin. Cancer Res. 2017, 23, 2143–2153. [Google Scholar] [CrossRef] [PubMed]
- Xue, Q.; Sun, K.; Deng, H.J.; Lei, S.T.; Dong, J.Q.; Li, G.X. Anti-miRNA-221 sensitizes human colorectal carcinoma cells to radiation by upregulating PTEN. World J. Gastroenterol. 2013, 19, 9307–9317. [Google Scholar] [CrossRef] [PubMed]
- Matarlo, J.S.; Krumpe, L.R.H.; Heinz, W.F.; Oh, D.; Shenoy, S.R.; Thomas, C.L.; Goncharova, E.I.; Lockett, S.J.; O’Keefe, B.R. The natural product butylcycloheptyl prodiginine binds Pre-miR-21, inhibits dicer-mediated processing of Pre-miR-21, and blocks cellular proliferation. Cell Chem. Biol. 2019, 26, 1133–1142 e1134. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Zhou, J.; Dong, M. Down-regulation of miR-543 expression increases the sensitivity of colorectal cancer cells to 5-Fluorouracil through the PTEN/PI3K/AKT pathway. Biosci. Rep. 2019, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, R.; Tomosugi, M.; Sakai, T.; Sowa, Y. MEK inhibitor suppresses expression of the miR-17-92 cluster with G1-phase arrest in HT-29 human colon cancer cells and MIA PaCa-2 pancreatic cancer cells. Anticancer. Res. 2016, 36, 4537–4543. [Google Scholar] [CrossRef]
- Poliseno, L.; Salmena, L.; Zhang, J.; Carver, B.; Haveman, W.J.; Pandolfi, P.P. A coding-independent function of gene and pseudogene mRNAs regulates tumour biology. Nature 2010, 465, 1033–1038. [Google Scholar] [CrossRef] [Green Version]
- Sonpavde, G.; Matveev, V.; Burke, J.M.; Caton, J.R.; Fleming, M.T.; Hutson, T.E.; Galsky, M.D.; Berry, W.R.; Karlov, P.; Holmlund, J.T.; et al. Randomized phase II trial of docetaxel plus prednisone in combination with placebo or AT-101, an oral small molecule Bcl-2 family antagonist, as first-line therapy for metastatic castration-resistant prostate cancer. Ann. Oncol. 2012, 23, 1803–1808. [Google Scholar] [CrossRef]
- Ready, N.; Karaseva, N.A.; Orlov, S.V.; Luft, A.V.; Popovych, O.; Holmlund, J.T.; Wood, B.A.; Leopold, L. Double-blind, placebo-controlled, randomized phase 2 study of the proapoptotic agent AT-101 plus docetaxel, in second-line non-small cell lung cancer. J. Thorac. Oncol. 2011, 6, 781–785. [Google Scholar] [CrossRef]
- Hopkins, B.D.; Fine, B.; Steinbach, N.; Dendy, M.; Rapp, Z.; Shaw, J.; Pappas, K.; Yu, J.S.; Hodakoski, C.; Mense, S.; et al. A secreted PTEN phosphatase that enters cells to alter signaling and survival. Science 2013, 341, 399–402. [Google Scholar] [CrossRef] [PubMed]
- Lavictoire, S.J.; Gont, A.; Julian, L.M.; Stanford, W.L.; Vlasschaert, C.; Gray, D.A.; Jomaa, D.; Lorimer, I.A.J. Engineering PTEN-L for cell-mediated delivery. Mol. Ther. Methods Clin. Dev. 2018, 9, 12–22. [Google Scholar] [CrossRef] [PubMed]
- Zou, J.; Luo, H.; Zeng, Q.; Dong, Z.; Wu, D.; Liu, L. Protein kinase CK2alpha is overexpressed in colorectal cancer and modulates cell proliferation and invasion via regulating EMT-related genes. J. Transl. Med. 2011, 9, 97. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Feng, Y.; Peng, W.; Ji, D.; Zhang, Z.; Qian, W.; Li, J.; Gu, Q.; Zhang, D.; Tang, J.; et al. Long noncoding RNA Linc02023 regulates PTEN stability and suppresses tumorigenesis of colorectal cancer in a PTEN-dependent pathway. Cancer Lett. 2019, 451, 68–78. [Google Scholar] [CrossRef] [PubMed]
- Fine, B.; Hodakoski, C.; Koujak, S.; Su, T.; Saal, L.H.; Maurer, M.; Hopkins, B.; Keniry, M.; Sulis, M.L.; Mense, S.; et al. Activation of the PI3K pathway in cancer through inhibition of PTEN by exchange factor P-REX2a. Science 2009, 325, 1261–1265. [Google Scholar] [CrossRef] [PubMed]
- Rabinovsky, R.; Pochanard, P.; McNear, C.; Brachmann, S.M.; Duke-Cohan, J.S.; Garraway, L.A.; Sellers, W.R. p85 Associates with unphosphorylated PTEN and the PTEN-associated complex. Mol. Cell. Biol. 2009, 29, 5377–5388. [Google Scholar] [CrossRef]
- He, L.; Ingram, A.; Rybak, A.P.; Tang, D. Shank-interacting protein-like 1 promotes tumorigenesis via PTEN inhibition in human tumor cells. J. Clin. Investig. 2010, 120, 2094–2108. [Google Scholar] [CrossRef]
- Shang, H.; Wang, T.; Shang, F.; Li, M.; Luo, Y.; Huang, K.M. Over-expression of DJ-1 attenuates effects of curcumin on colorectal cancer cell proliferation and apoptosis. Eur. Rev. Med. Pharm. Sci. 2019, 23, 3080–3087. [Google Scholar] [CrossRef]
- Tang, Y.; Liu, P.; Tian, Y.; Xu, Y.; Ren, F.; Cui, X.; Fan, J. Overexpression of ribonuclease inhibitor defines good prognosis and suppresses proliferation and metastasis in human colorectal cancer cells via PI3K/AKT pathway. Clin. Transl. Oncol. 2015, 17, 306–313. [Google Scholar] [CrossRef]
- McEllin, B.; Camacho, C.V.; Mukherjee, B.; Hahm, B.; Tomimatsu, N.; Bachoo, R.M.; Burma, S. PTEN loss compromises homologous recombination repair in astrocytes: Implications for glioblastoma therapy with temozolomide or poly(ADP-ribose) polymerase inhibitors. Cancer Res. 2010, 70, 5457–5464. [Google Scholar] [CrossRef]
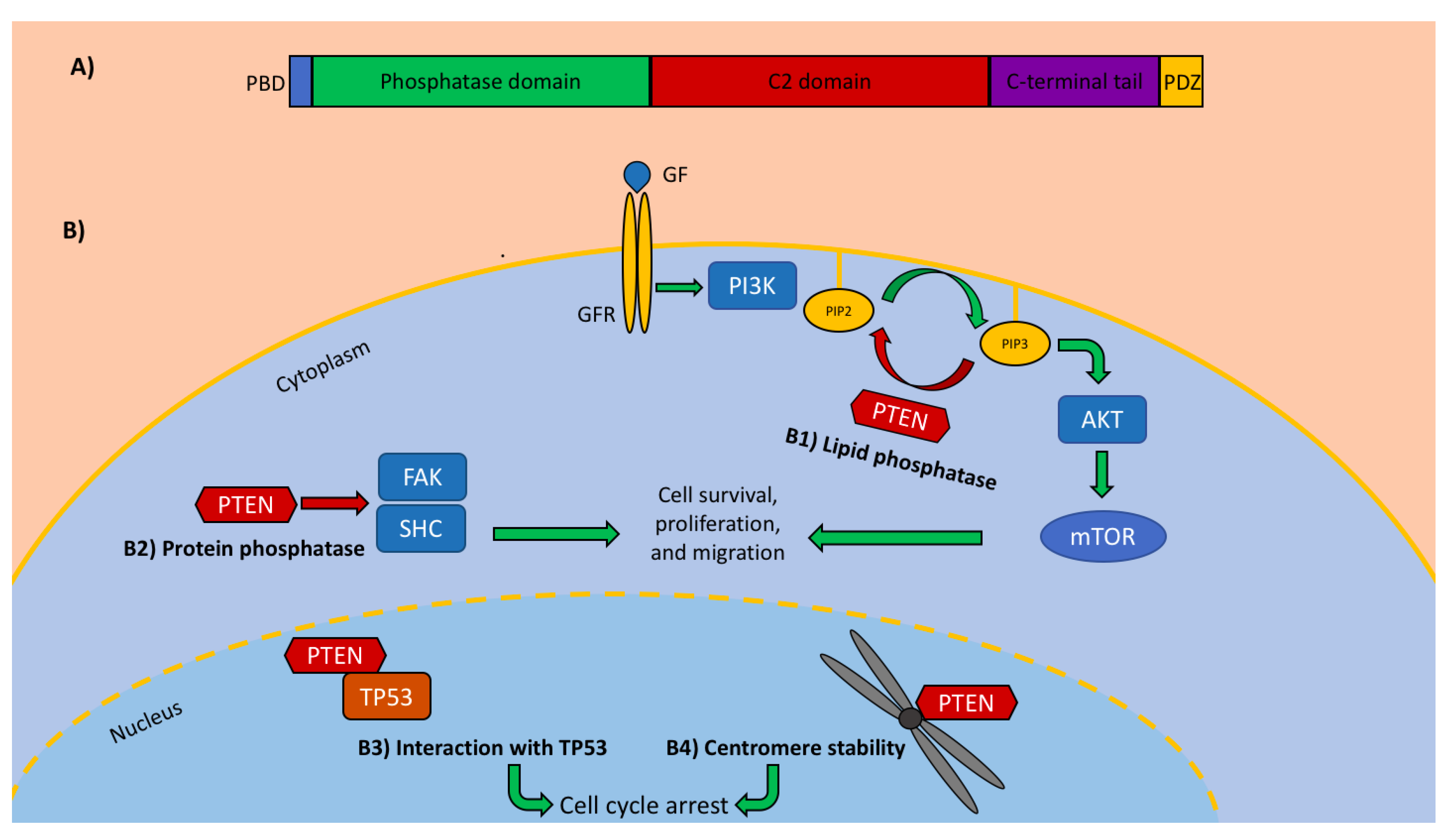

{kind=link}
{kind=link}
| Study | No. of Patients | Treatment | PTEN Assessment | RR | PFS | OS | |
|---|---|---|---|---|---|---|---|
| Frattini et al. 2007 [24] | Prospective | 27 | Cet-based | IHC | PTEN+ vs. PTEN− 62.5% vs. 0% (p > 0.001) | - | - |
| Loupakis et al. 2009 [25] | Retrospective | 59 | Iri + Cet | IHC | PTEN+ vs. PTEN− Higher RR (p = 0.007) | PTEN+ vs. PTEN- 4.7 vs. 3.3 m (HR = 0.49; p = 0.005) | - |
| Laurent-Puig et al. 2009 [26] | Retrospective | 162 | Cet-based | IHC | - | - | PTEN- associated with shorter OS (p = 0.013) |
| Therkildsen et al. 2014 [55] | Meta-analysis | 100 (9 studies) | Anti-EGFR based | Protein expression (7 studies) Mutational status (2 studies) | PTEN- Odds Ratio = 0.41 (95%CI = 0.20–0.85) | PTEN- associated with shorter PFS (HR 1.88, 95%CI = 1.35–2.61) | PTEN- associated with shorter OS (HR = 2.09, 95%CI = 1.36–3.19) |
| Karapetis et al. 2014 [28] | CO.17 trial Prespecified subgroup analysis | 205 | Cet | IHC | PTEN+ vs. PTEN− 21% vs. 15% | - | No association between PTEN status and OS Among PTEN+ OS 9.9 vs. 5.4 months for Cet vs. BSC (HR = 0.66; p = 0.32) |
| Agoston et al. 2016 [27] | Retrospective | 55 | Anti-EGFR based | IHC | - | - | No association between PTEN status and OS |
| Kara et al. 2012 [35] | Retrospective | 34 | Bev based | IHC | PTEN+ vs. PTEN− p = 0.832 | - | PTEN+ vs. PTEN− p = 0.6 |
| Price et al. 2013 [12] | AGITG MAX trial, post hoc analysis | 302 | Bev based | CNV | PTEN+ vs. PTEN− p = 0.36 | PTEN+ vs. PTEN− p = 0.26 | PTEN+ vs. PTEN− p = 0.35 |
| Sclafani et al. 2015 [36] | Retrospective | 42 | Bev based | IHC | PTEN− vs. PTEN+ 71.4% vs. 32.1% p = 0.02 | PTEN− vs. PTEN+ 9.2 vs. 8.7 months p = 0.968 | PTEN− vs. PTEN+ 21.1 vs. 17.3 months p = 0.628 |
| Weldone Gilcrease et al. 2019 [46] | Post hoc analysis (phase I/II) | 24 | Eve+mFOLFOX6-Bev | IHC | PTEN+ vs. PTEN− 40% vs. 86% p = 0.03 | - | - |
| Moroney J et al. 2012 [48] | Prospective (phase I) | 136 (including 17 with mCRC) | Tem+Bev+liposomial doxo | PCR and IHC | PIK3CA MT and/or PTEN loss/MT vs. WT 39% vs. 16%, p = 0.018 | ||
| Corcoran RB et al. 2015 [49] | Prospective (phase I/II) | 19 | Dabrafenib+ Trametinib | IHC | PTEN− vs. PTEN+ 21% vs. 0% | PTEN− vs. PTEN+ 3.48 vs. 3.61 months p = 0.35 | - |
| Pishvaian et al. 2018 [50] | Prospective (phase II) | 49 | Veli+Temo | IHC | PTEN− vs. PTEN+ 13.3% vs. 21.1% | PTEN− vs. PTEN+ 1.7 vs. 1.8 months | PTEN- vs. PTEN+ 6.2 vs. 6.3 months |
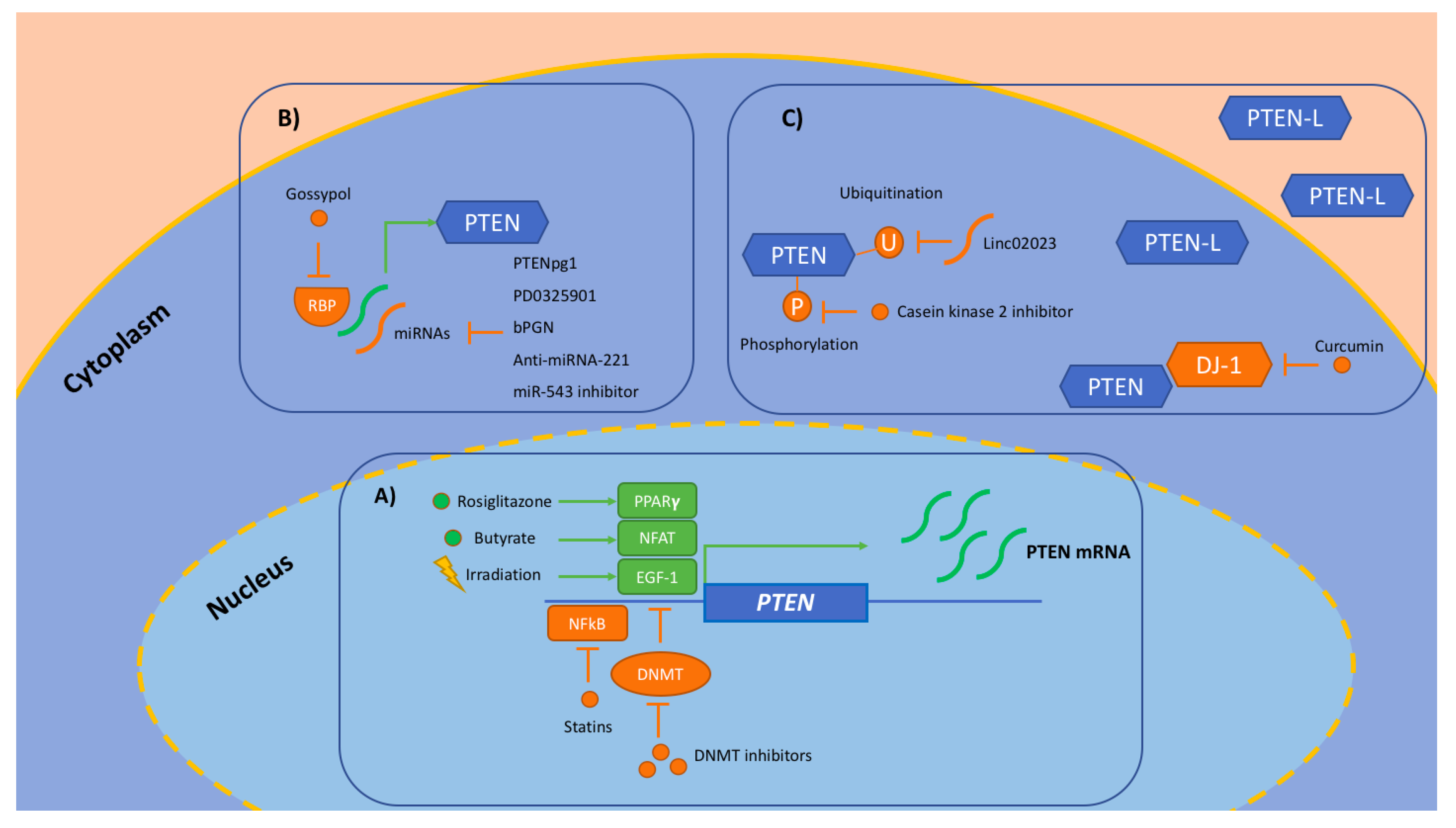
| Level | Strategy | Agents | Evidences | Reference |
|---|---|---|---|---|
| Transcriptional level | Removing epigenetic inhibition | DNA methyltransferase inhibitors | Decitabine proved to be safe and active in combination with panitumumab in KRAS wt mCRC patients previously treated with cetuximab. Decitabine proved to be safe when administered by hepatic arterial infusion in liver limited mCRC patients. | [58] [59] |
| Increasing exposure to activating transcription factors | Rosiglitazone Irradiation Butyrate | Some transcription factors can be pharmacologically stimulated: PPARγ (via rosiglitazone), EGR-1 (via irradiation), NFAT (via butyrate). | [56] | |
| Reducing exposure to inhibiting transcription factors | Statins NF-κB selective inhibitors | The inhibiting transcription factor NF-κB can be repressed through statins or selective inhibitors. | [56] | |
| Post-transcriptional level | Inhibiting miRNAs and RNA binding proteins | Anti-miRNA-221 | Anti-miRNA-221 showed to increase PTEN expression, sensitizing CRC cells to radiation. | [64] |
| Butylcycloheptyl prodiginine | Butylcycloheptyl prodiginine showed to suppress miR-21 and consequently cellular growth in CRC lines. | [65] | ||
| miR-543 inhibitor | A miR-543 inhibitor proved to reverse chemoresistance to 5-fluorouracil (5-FU), obtained by this oncomir through reduction of PTEN expression, enhancing cellular sensitivity to 5-FU. | [66] | ||
| PD0325901 | PD0325901 (a MEK inhibitor) proved to upregulate PTEN by suppressing miR-17-92 cluster. | [67] | ||
| PTENpg1 | PTENpg1, a long, non-coding RNA transcripted by the PTEN pseudogene (PTENpg1) was shown to saturate miRNAs. | [68] | ||
| Gossypol | Gossypol showed to inhibit Musashi-1/2 proteins and demonstrated antitumoral activity in a xenograft model. | [63] | ||
| Post-translational level | Targeting enzymes involved in post-translational modification or reverting post-translational modification | Casein kinase 2 inhibitor | Inhibitor of casein kinase 2 (a serine/threonine kinase, which phosphorylates PTEN, causing repression of its catalytic activity) showed to reduce cell growth and invasiveness in CRC lines. | [73] |
| Linc02023 | Linc02023 (a long non coding RNA) was shown to impair PTEN ubiquitination and subsequent degradation, inhibiting CRC cell proliferation and in vitro and in vivo survival. | [74] | ||
| Paracrine function | PTEN-L | PTEN-L, an PTEN isoform with a paracrine function, showed to counteract the PI3K/Akt pathway both in vitro and in vivo (through intraperitoneal infusion in xenograft models). | [71] | |
| Target protein–protein interaction | Curcumin | Curcumin was shown to inhibit proliferation and promote apoptosis via the downregulation of DJ-1 (a PTEN negative modulator) in CRC cell lines. | [78] | |
| Ribonuclease inhibitor | Upregulation of a ribonuclease inhibitor was shown to stimulate PTEN expression, leading to PI3K/Akt pathway suppression in CRC cell lines. | [79] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salvatore, L.; Calegari, M.A.; Loupakis, F.; Fassan, M.; Di Stefano, B.; Bensi, M.; Bria, E.; Tortora, G. PTEN in Colorectal Cancer: Shedding Light on Its Role as Predictor and Target. Cancers 2019, 11, 1765. https://doi.org/10.3390/cancers11111765
Salvatore L, Calegari MA, Loupakis F, Fassan M, Di Stefano B, Bensi M, Bria E, Tortora G. PTEN in Colorectal Cancer: Shedding Light on Its Role as Predictor and Target. Cancers. 2019; 11(11):1765. https://doi.org/10.3390/cancers11111765
Chicago/Turabian StyleSalvatore, Lisa, Maria Alessandra Calegari, Fotios Loupakis, Matteo Fassan, Brunella Di Stefano, Maria Bensi, Emilio Bria, and Giampaolo Tortora. 2019. "PTEN in Colorectal Cancer: Shedding Light on Its Role as Predictor and Target" Cancers 11, no. 11: 1765. https://doi.org/10.3390/cancers11111765