Antitumor Reactive T-Cell Responses Are Enhanced In Vivo by DAMP Prothymosin Alpha and Its C-Terminal Decapeptide

 , , , , ,
, , , , ,  ,
,  ,
,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
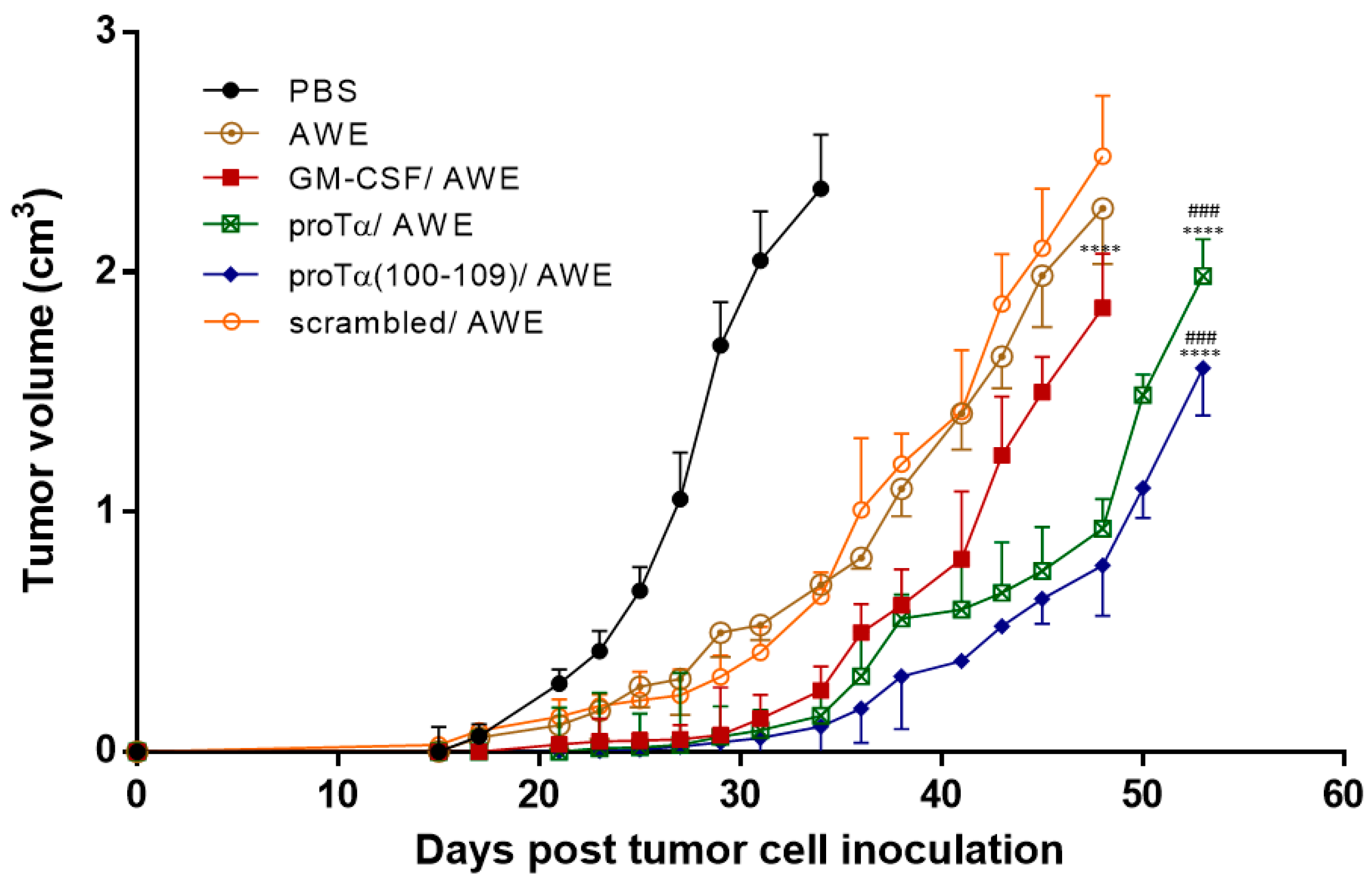
2.1. Therapeutic Administration of ProTα or ProTα(100–109) Suppressed Melanoma Tumor Growth In Vivo
2.2. Melanomas from Mice Treated with proTα/AWE or proTα(100–109)/AWE Were Infiltrated by T Cells
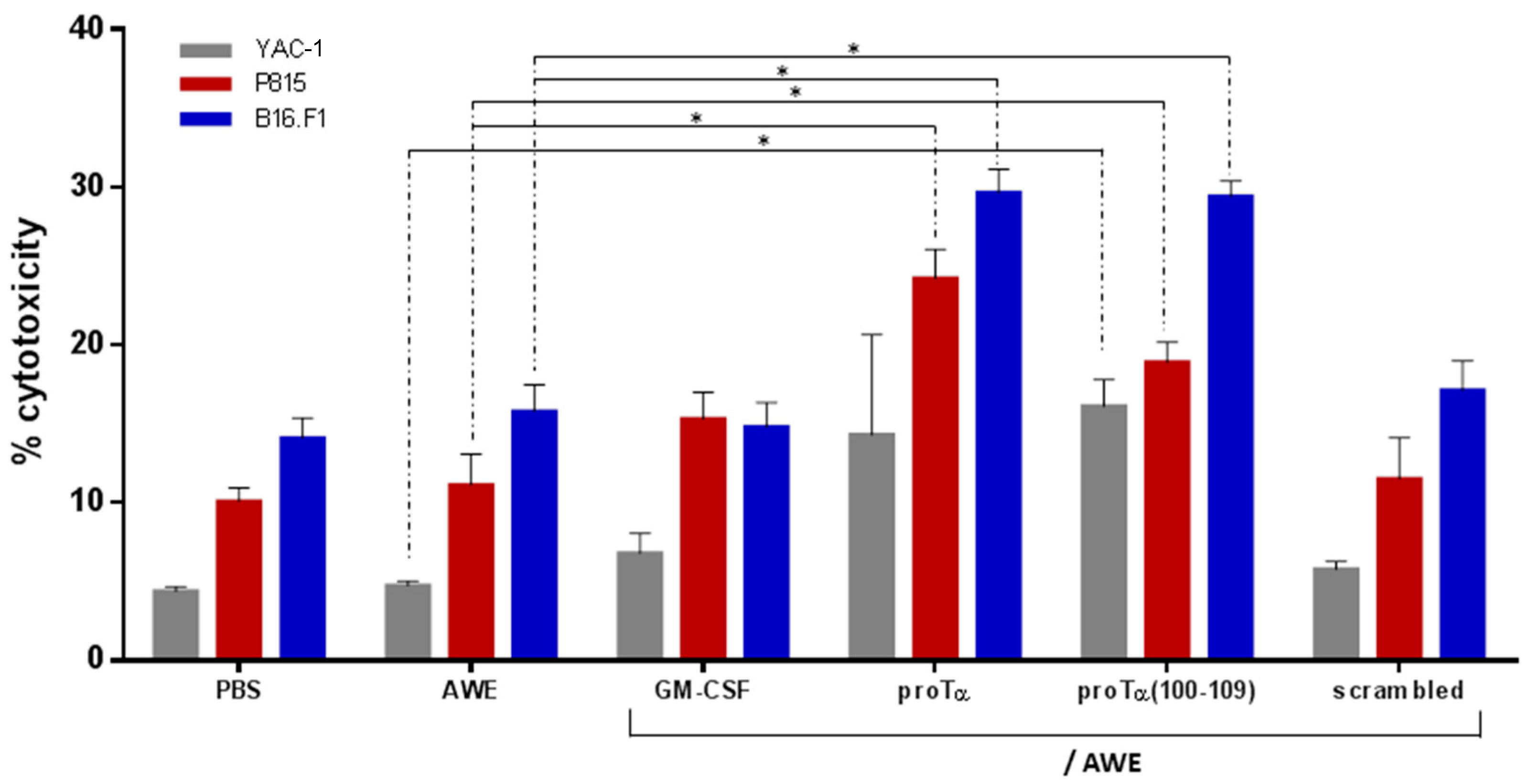
2.3. ProTα/AWE and proTα(100–109)/AWE Induced T- and NK Cell-Mediated Cytotoxic Responses In Vivo
2.4. In Vivo Administration of proTα/AWE or proTα(100–109)/AWE Enhanced Production of Th1-Polarizing Cytokines
3. Discussion
4. Materials and Methods
4.1. Proteins and Peptides
4.2. Cell Cultures
4.3. Preparation of AWE
4.4. Establishment of In Vivo Melanoma Mouse Model
4.5. Histological and Immunohistochemical Analyses of Tumor Sections
4.6. Cytotoxicity Assay
4.7. Measurement of Cytokine Levels
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Haritos, A.A.; Goodall, G.J.; Horecker, B.L. Prothymosin alpha: Isolation and properties of the major immunoreactive form of thymosin alpha 1 in rat thymus. Proc. Natl. Acad. Sci. USA 1984, 81, 1008–1011. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Márquez, J.; Rodríguez, P. Prothymosin α is a chromatin-remodelling protein in mammalian cells. Biochem. J. 1998, 333, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Eschenfeldt, W.H.; Berger, S.L. The human prothymosin alpha gene is polymorphic and induced upon growth stimulation: Evidence using a cloned cDNA. Proc. Natl. Acad. Sci. USA 1986, 83, 9403–9407. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Kim, H.E.; Shu, H.; Zhao, Y.; Zhang, H.; Kofron, J.; Donnelly, J.; Burns, D.; Ng, S.C.; Rosenberg, S.; et al. Distinctive roles of PHAP proteins and prothymosin-alpha in a death regulatory pathway. Science 2003, 299, 223–226. [Google Scholar] [CrossRef] [PubMed]
- Samara, P.; Karachaliou, C.E.; Ioannou, K.; Papaioannou, N.E.; Voutsas, I.F.; Zikos, C.; Pirmettis, I.; Papadopoulos, M.; Kalbacher, H.; Livaniou, E.; et al. Prothymosin alpha: An alarmin and more…. Curr. Med. Chem. 2017, 24, 1747–1760. [Google Scholar] [CrossRef]
- Salvin, S.B.; Horecker, B.L.; Pan, L.X.; Rabin, B.S. The effect of dietary zinc and prothymosin alpha on cellular immune responses of RF/J mice. Clin. Immunol. Immunopathol. 1987, 43, 281–288. [Google Scholar] [CrossRef]
- Mosoian, A.; Teixeira, A.; Burns, C.S.; Sander, L.E.; Gusella, G.L.; He, C.; Blander, J.M.; Klotman, P.; Klotman, M.E. Prothymosin-alpha inhibits HIV-1 via Toll-like receptor 4-mediated type I interferon induction. Proc. Natl. Acad. Sci. USA 2010, 107, 10178–10183. [Google Scholar] [CrossRef]
- Baxevanis, C.N.; Sfagos, C.; Anastasopoulos, E.; Reclos, G.J.; Papamichail, M. Prothymosin-alpha enhances HLA-DR antigen expression on monocytes from patients with multiple sclerosis. J. Neuroimmunol. 1990, 27, 141–147. [Google Scholar] [CrossRef]
- Baxevanis, C.N.; Thanos, D.; Reclos, G.J.; Anastasopoulos, E.; Tsokos, G.C.; Papamatheakis, J.; Papamichail, M. Prothymosin alpha enhances human and murine MHC class II surface antigen expression and messenger RNA accumulation. J. Immunol. 1992, 148, 1979–1984. [Google Scholar]
- Skopeliti, M.; Kratzer, U.; Altenberend, F.; Panayotou, G.; Kalbacher, H.; Stevanovic, S.; Voelter, W.; Tsitsilonis, O.E. Proteomic exploitation on prothymosin α-induced mononuclear cell activation. Proteomics 2007, 7, 1814–1824. [Google Scholar] [CrossRef]
- Baxevanis, C.N.; Frillingos, S.; Seferiadis, K.; Reclos, G.J.; Arsenis, P.; Katsiyiannis, A.; Anastasopoulos, E.; Tsolas, O.; Papamichail, M. Enhancement of human T lymphocyte function by prothymosin α: Increased production of interleukin-2 and expression of interleukin-2 receptors in normal human peripheral blood T lymphocytes. Immunopharmacol. Immunotoxicol. 1990, 12, 595–617. [Google Scholar] [CrossRef] [PubMed]
- Cordero, O.J.; Sarandeses, C.S.; López, J.L.; Cancio, E.; Regueiro, B.J.; Nogueira, M. Prothymosin α enhances interleukin 2 receptor expression in normal human T-lymphocytes. Int. J. Immunopharmacol. 1991, 13, 1059–1065. [Google Scholar] [CrossRef]
- Skopeliti, M.; Iconomidou, V.A.; Derhovanessian, E.; Pawelec, G.; Voelter, W.; Kalbacher, H.; Hamodrakas, S.J.; Tsitsilonis, O.E. Prothymosin α immunoactive carboxyl-terminal peptide TKKQKTDEDD stimulates lymphocyte reactions, induces dendritic cell maturation and adopts a β-sheet conformation in a sequence-specific manner. Mol. Immunol. 2009, 46, 784–792. [Google Scholar] [CrossRef] [PubMed]
- Samara, P.; Ioannou, K.; Neagu, M.; Arnogiannaki, N.; Ardavanis, A.; Voelter, W.; Tsitsilonis, O. The C-terminal decapeptide of prothymosin α is responsible for its stimulatory effect on the functions of human neutrophils in vitro. Int. Immunopharmacol. 2013, 15, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Ioannou, K.; Derhovanessian, E.; Tsakiri, E.; Samara, P.; Kalbacher, H.; Voelter, W.; Trougakos, I.P.; Pawelec, G.; Tsitsilonis, O.E. Prothymosin α and a prothymosin α-derived peptide enhance TH1-type immune responses against defined HER-2/neu epitopes. BMC Immunol. 2013, 14, 43. [Google Scholar] [CrossRef] [PubMed]
- Karachaliou, C.E.; Triantis, C.; Liolios, C.; Palamaris, L.; Zikos, H.; Tsitsilonis, O.E.; Kalbacher, H.; Voelter, W.; Loudos, G.; Papadopoulos, M.; et al. In vivo biodistribution and imaging studies with a 99mTc-radiolabeled derivative of the C-terminus of prothymosin alpha in mice bearing experimentally-induced inflammation. Eur. J. Pharm. Biopharm. 2017, 113, 188–197. [Google Scholar] [CrossRef]
- Papanastasiou, M.; Baxevanis, C.N.; Papamichail, M. Promotion of murine antitumor activity by prothymosin alpha treatment: I. Induction of tumoricidal peritoneal cells producing high levels of tumour necrosis factor alpha. Cancer Immunol. Immunother. 1992, 35, 145–150. [Google Scholar] [CrossRef]
- Baxevanis, C.N.; Gritzapis, A.D.; Dedoussis, G.V.; Papadopoulos, N.G.; Tsolas, O.; Papamichail, M. Induction of lymphokine-activated killer activity in mice by prothymosin α. Cancer Immunol. Immunother. 1994, 38, 281–286. [Google Scholar] [CrossRef]
- Baxevanis, C.N.; Gritzapis, A.D.; Spanakos, G.; Tsitsilonis, O.E.; Papamichail, M. Induction of tumor-specific T lymphocyte responses in vivo by prothymosin α. Cancer Immunol. Immunother. 1995, 40, 410–418. [Google Scholar] [CrossRef]
- Thomas, S.; Pendergast, G.C. Cancer vaccines: A brief overview. Methods Mol. Biol. 2016, 1403, 755–761. [Google Scholar]
- Bezu, L.; Kepp, O.; Cerrato, G.; Pol, J.; Fucikova, J.; Spisek, R.; Zitvogel, L.; Kroemer, G.; Galluzzi, L. Trial watch: Peptide-based vaccines in anticancer therapy. Oncoimmunology 2018, 7, e1511506. [Google Scholar] [CrossRef] [PubMed]
- Mbow, M.L.; De Gregorio, E.; Valiante, N.M.; Rappuoli, R. New adjuvants for human vaccines. Curr. Opin. Immunol. 2010, 22, 411–416. [Google Scholar] [CrossRef] [PubMed]
- Baxevanis, C.N.; Voutsas, I.F.; Tsitsilonis, O.E. Toll-like receptor agonists: Current status and future perspective on their utility as adjuvants in improving anticancer vaccination strategies. Immunotherapy 2013, 5, 497–511. [Google Scholar] [CrossRef] [PubMed]
- Maeda, S.; Sasaki, K.; Halder, S.K.; Fujita, W.; Ueda, H. Neuroprotective DAMPs member prothymosin alpha has additional beneficial actions against cerebral ischemia-induced vascular damages. J. Pharmacol. Sci. 2016, 132, 100–104. [Google Scholar] [CrossRef] [PubMed]
- Gusella, G.L.; Teixeira, A.; Aberg, J.; Uversky, V.N.; Mosoian, A. Prothymosin-α variants elicit anti-HIV-1 response via TLR4 dependent and independent pathways. PLoS ONE 2016, 11, e0156486. [Google Scholar] [CrossRef]
- Halder, S.K.; Matsunaga, H.; Ishii, K.J.; Ueda, H. Prothymosin-alpha preconditioning activates TLR4-TRIF signaling to induce protection of ischemic retina. J. Neurochem. 2015, 135, 1161–1177. [Google Scholar] [CrossRef]
- Omotuyi, O.; Matsunaga, H.; Ueda, H. Evidence for proTα-TLR4/MD-2 binding: Molecular dynamics and gravimetric assay studies. Expert Opin. Biol. Ther. 2015, 15, 223–229. [Google Scholar] [CrossRef]
- Cordero, O.J. Data on the interaction between prothymosin α and TLR4 may help to the design of new antiviral compounds. J. Acquir. Immune Defic. Syndr. 2011, 56, 110–111. [Google Scholar] [CrossRef]
- Wang, L.C.; Wu, C.L.; Cheng, Y.Y.; Tsai, K.J. Deletion of nuclear localizing signal attenuates proinflammatory activity of prothymosin-alpha and enhances its neuroprotective effect on transient ischemic stroke. Mol. Neurobiol. 2017, 54, 582–593. [Google Scholar] [CrossRef]
- Voutsas, I.F.; Pistamaltzian, N.; Tsiatas, M.L.; Skopeliti, M.; Katsila, T.; Mavrothalassiti, I.; Spyrou, S.; Dimopoulos, M.A.; Tsitsilonis, O.E.; Bamias, A. Ovarian malignant ascites-derived lymphocytes stimulated with prothymosin α or its immunoactive decapeptide lyse autologous tumour cells in vitro and retard tumour growth in SCID mice. Eur. J. Cancer 2013, 49, 1706–1714. [Google Scholar] [CrossRef]
- Nair, S.K.; Boczkowski, D.; Snyder, D.; Gilboa, E. Antigen-presenting cells pulsed with unfractionated tumor-derived peptides are potent tumor vaccines. Eur. J. Immunol. 1997, 27, 589–597. [Google Scholar] [CrossRef] [PubMed]
- Gjertsen, M.K.; Buanes, T.; Rosseland, A.R.; Bakka, A.; Gladhaug, I.; Søreide, O.; Eriksen, J.A.; Møller, M.; Baksaas, I.; Lothe, R.A.; et al. Intradermal ras peptide vaccination with granulocyte-macrophage colony-stimulating factor as adjuvant: Clinical and immunological responses in patients with pancreatic adenocarcinoma. Int. J. Cancer 2001, 92, 441–450. [Google Scholar] [CrossRef] [PubMed]
- Rahma, O.E.; Herrin, V.E.; Ibrahim, R.A.; Toubaji, A.; Bernstein, S.; Dakheel, O.; Steinberg, S.M.; Abu Eid, R.; Mkrtichyan, M.; Berzofsky, J.A.; et al. Pre-immature dendritic cells (PIDC) pulsed with HPV16 E6 or E7 peptide are capable of eliciting specific immune response in patients with advanced cervical cancer. J. Transl. Med. 2014, 12, 353. [Google Scholar] [CrossRef] [PubMed]
- Nitschke, N.J.; Bjoern, J.; Iversen, T.Z.; Andersen, M.H.; Svane, I.M. Indoleamine 2,3-dioxygenase and survivin peptide vaccine combined with temozolomide in metastatic melanoma. Stem Cell Investig. 2017, 4, 77. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Jensen, F.C.; Savary, J.R.; Diveley, J.P.; Chang, J.C. Adjuvant activity of incomplete Freund’s adjuvant. Adv. Drug Deliv. Rev. 1998, 32, 173–186. [Google Scholar]
- Yan, W.L.; Shen, K.Y.; Tien, C.Y.; Chen, Y.A.; Liu, S.J. Recent progress in GM-CSF-based cancer immunotherapy. Immunotherapy 2017, 9, 347–360. [Google Scholar] [CrossRef]
- Halder, S.K.; Sugimoto, J.; Matsunaga, H.; Ueda, H. Therapeutic benefits of 9-amino acid peptide derived from prothymosin alpha against ischemic damages. Peptides 2013, 43, 68–75. [Google Scholar] [CrossRef]
- Ali, O.A.; Verbeke, C.; Johnson, C.; Sands, R.W.; Lewin, S.A.; White, D.; Doherty, E.; Dranoff, G.; Mooney, D.J. Identification of immune factors regulating antitumor immunity using polymeric vaccines with multiple adjuvants. Cancer Res. 2014, 74, 1670–1681. [Google Scholar] [CrossRef]
- He, L.Z.; Weidlick, J.; Sisson, C.; Marsh, H.C.; Keler, T. Toll-like receptor agonists shape the immune responses to a mannose receptor-targeted cancer vaccine. Cell. Mol. Immunol. 2015, 12, 719–728. [Google Scholar] [CrossRef]
- Qiu, J.T.; Alson, D.; Lee, T.H.; Tsai, C.C.; Yu, T.W.; Chen, Y.S.; Cheng, Y.F.; Lin, C.C.; Schuyler, S.C. Effect of multiple vaccinations with tumor cell-based vaccine with codon-modified GM-CSF on tumor growth in a mouse model. Cancers (Basel) 2019, 11, 368. [Google Scholar] [CrossRef]
- Dang, Y.; Wagner, W.M.; Gad, E.; Rastetter, L.; Berger, C.M.; Holt, G.E.; Disis, M.L. Dendritic cell-activating vaccine adjuvants differ in the ability to elicit antitumor immunity due to an adjuvant-specific induction of immunosuppressive cells. Clin. Cancer Res. 2012, 18, 3122–3131. [Google Scholar] [CrossRef] [PubMed]
- Lövgren, T.; Sarhan, D.; Truxová, I.; Choudhary, B.; Maas, R.; Melief, J.; Nyström, M.; Edbäck, U.; Vermeij, R.; Scurti, G.; et al. Enhanced stimulation of human tumor-specific T cells by dendritic cells matured in the presence of interferon-γ and multiple Toll-like receptor agonists. Cancer Immunol. Immunother. 2017, 66, 1333–1344. [Google Scholar] [CrossRef] [PubMed]
- Wells, J.W.; Cowled, C.J.; Farzaneh, F.; Noble, A. Combined triggering of dendritic cell receptors results in synergistic activation and potent cytotoxic immunity. J. Immunol. 2008, 181, 3422–3431. [Google Scholar] [CrossRef] [PubMed]
- Davila, E.; Kennedy, R.; Celis, E. Generation of antitumor immunity by cytotoxic T lymphocyte epitope peptide vaccination, CpG-oligodeoxynucleotide adjuvant, and CTLA-4 blockade. Cancer Res. 2003, 63, 3281–3288. [Google Scholar]
- Tian, H.; Shi, G.; Wang, Q.; Li, Y.; Yang, Q.; Li, C.; Yang, G.; Wu, M.; Xie, Q.; Zhang, S.; et al. A novel cancer vaccine with the ability to simultaneously produce anti-PD-1 antibody and GM-CSF in cancer cells and enhance Th1-biased antitumor immunity. Signal Transduct. Target. Ther. 2016, 1, 16025. [Google Scholar] [CrossRef]
- Nagato, T.; Lee, Y.-R.; Harabuchi, Y.; Celis, E. Combinatorial immunotherapy of polyinosinic-polycytidylic acid and blockade of programmed death-ligand 1 induce effective CD8 T-cell responses against established tumors. Clin. Cancer Res. 2014, 20, 1223–1234. [Google Scholar] [CrossRef]
- Cho, H.C.; Kim, B.H.; Kim, K.; Park, J.Y.; Chang, J.H.; Kim, S.K. Cancer immunotherapeutic effects of novel CpG ODN in murine tumor model. Int. Immunopharmacol. 2008, 8, 1401–1407. [Google Scholar] [CrossRef]
- Jang, J.K.; Khawli, L.A.; Canter, D.C.; Hu, P.; Zhu, T.H.; Wu, B.W.; Angell, T.E.; Li, Z.; Epstein, A.L. Systemic delivery of chTNT-3/CpG immunoconjugates for immunotherapy in murine solid tumor models. Cancer Immunol. Immunother. 2016, 65, 511–523. [Google Scholar] [CrossRef]
- Wang, X.Y.; Sun, X.; Chen, X.; Facciponte, J.; Repasky, E.A.; Kane, J.; Subjeck, J.R. Superior antitumor response induced by large stress protein chaperoned protein antigen compared with peptide antigen. J. Immunol. 2010, 184, 6309–6319. [Google Scholar] [CrossRef]
- Saenz, R.; Messmer, B.; Futalan, D.; Tor, Y.; Larsson, M.; Daniels, G.; Esener, S.; Messmer, D. Activity of the HMGB1-derived immunostimulatory peptide Hp91 resides in the helical C-terminal portion and is enhanced by dimerization. Mol. Immunol. 2014, 57, 191–199. [Google Scholar] [CrossRef]
- Fu, Q.; Chen, N.; Ge, C.; Li, R.; Li, Z.; Zeng, B.; Li, C.; Wang, Y.; Xue, Y.; Song, X.; et al. Prognostic value of tumor-infiltrating lymphocytes in melanoma: A systematic review and meta-analysis. Oncoimmunology 2019, 8, e1593806. [Google Scholar] [CrossRef] [PubMed]
- Samara, P.; Ioannou, K.; Tsitsilonis, O.E. Prothymosin alpha and immune responses: Are we close to potential clinical applications? Vitam. Horm. 2016, 102, 179–207. [Google Scholar] [PubMed]
- Eckert, K.; Grünberg, E.; Garbin, F.; Maurer, H.R. Preclinical studies with prothumosin α1 on mononuclear cells from tumor patients. Int. J. Immunopharmacol. 1997, 19, 493–500. [Google Scholar] [CrossRef]
- Cordero, O.J.; Sarandeses, C.; López-Rodriguez, J.L.; Nogueira, M. The presence and cytotoxicity of CD16+ CD2- subset from PBL and NK cells in long-term IL-2 cultures enhanced by prothymosin-α. Immunopharmacology 1995, 29, 215–223. [Google Scholar] [CrossRef]
- Schaller, T.H.; Batich, K.A.; Suryadevara, C.M.; Desai, R.; Sampson, J.H. Chemokines as adjuvants for immunotherapy: Implications for immune activation with CCL3. Expert Rev. Clin. Immunol. 2017, 13, 1049–1060. [Google Scholar] [CrossRef]
- Baxevanis, C.N.; Voutsas, I.F.; Tsitsilonis, O.E.; Gritzapis, A.D.; Sotiriadou, R.; Papamichail, M. Tumor-specific CD4+ T lymphocytes from cancer patients are required for optimal induction of cytotoxic T cells against the autologous tumor. J. Immunol. 2000, 164, 3902–3912. [Google Scholar] [CrossRef]
- Ioannou, K.; Cheng, K.F.; Crichlow, G.V.; Birmpilis, A.I.; Lolis, E.J.; Tsitsilonis, O.E.; Al-Abed, Y. ISO-66, a novel inhibitor of macrophage migration inhibitory factor, shows efficacy in melanoma and colon cancer models. Int. J. Oncol. 2014, 45, 1457–1468. [Google Scholar] [CrossRef]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Birmpilis, A.I.; Karachaliou, C.-E.; Samara, P.; Ioannou, K.; Selemenakis, P.; Kostopoulos, I.V.; Kavrochorianou, N.; Kalbacher, H.; Livaniou, E.; Haralambous, S.; et al. Antitumor Reactive T-Cell Responses Are Enhanced In Vivo by DAMP Prothymosin Alpha and Its C-Terminal Decapeptide. Cancers 2019, 11, 1764. https://doi.org/10.3390/cancers11111764
Birmpilis AI, Karachaliou C-E, Samara P, Ioannou K, Selemenakis P, Kostopoulos IV, Kavrochorianou N, Kalbacher H, Livaniou E, Haralambous S, et al. Antitumor Reactive T-Cell Responses Are Enhanced In Vivo by DAMP Prothymosin Alpha and Its C-Terminal Decapeptide. Cancers. 2019; 11(11):1764. https://doi.org/10.3390/cancers11111764
Chicago/Turabian StyleBirmpilis, Anastasios I., Chrysoula-Evangelia Karachaliou, Pinelopi Samara, Kyriaki Ioannou, Platon Selemenakis, Ioannis V. Kostopoulos, Nadia Kavrochorianou, Hubert Kalbacher, Evangelia Livaniou, Sylva Haralambous, and et al. 2019. "Antitumor Reactive T-Cell Responses Are Enhanced In Vivo by DAMP Prothymosin Alpha and Its C-Terminal Decapeptide" Cancers 11, no. 11: 1764. https://doi.org/10.3390/cancers11111764
APA StyleBirmpilis, A. I., Karachaliou, C.-E., Samara, P., Ioannou, K., Selemenakis, P., Kostopoulos, I. V., Kavrochorianou, N., Kalbacher, H., Livaniou, E., Haralambous, S., Kotsinas, A., Farzaneh, F., Trougakos, I. P., Voelter, W., Dimopoulos, M.-A., Bamias, A., & Tsitsilonis, O. (2019). Antitumor Reactive T-Cell Responses Are Enhanced In Vivo by DAMP Prothymosin Alpha and Its C-Terminal Decapeptide. Cancers, 11(11), 1764. https://doi.org/10.3390/cancers11111764