TYK2: An Upstream Kinase of STATs in Cancer


Abstract
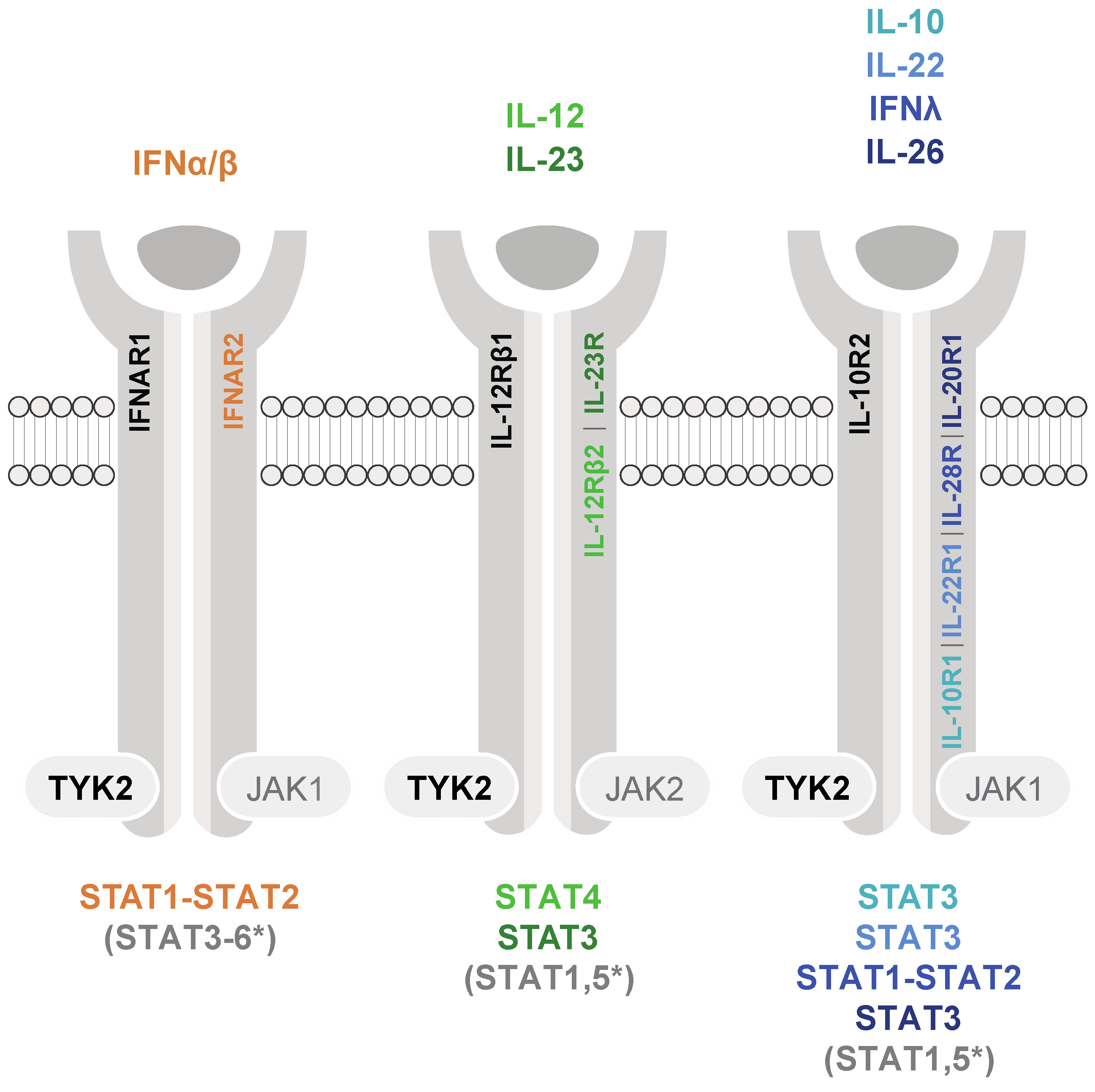
1. TYK2-Mediated Cytokine Signaling and Activation of STAT3 and STAT5
2. Aberrant Expression and/or Activity of TYK2 in Cancers
2.1. Aberrant TYK2 Levels
2.2. Aberrant Activation of TYK2
2.3. TYK2 Mutations
2.4. TYK2 Fusion Proteins
3. Tumor-Promoting Activities of (Hyper-)Active TYK2
3.1. TYK2 Activation of (Oncogenic) STAT Signaling
3.2. TYK2 Stimulation of Tumor Cell Invasion
3.3. TYK2 Prevention of Apoptosis
3.4. TYK2 Crosstalk to Oncogenes and Proto-Oncogenic Pathways
4. Deactivation and Stabilization of TYK2 under (Patho-)Physiological Conditions
5. Pharmaceutical TYK2 Inhibition
6. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Krolewski, J.J.; Lee, R.; Eddy, R.; Shows, T.B.; Dalla-Favera, R. Identification and chromosomal mapping of new human tyrosine kinase genes. Oncogene 1990, 5, 277–282. [Google Scholar] [PubMed]
- Hammaren, H.M.; Virtanen, A.T.; Raivola, J.; Silvennoinen, O. The regulation of JAKs in cytokine signaling and its breakdown in disease. Cytokine 2019, 118, 48–63. [Google Scholar] [CrossRef] [PubMed]
- Stark, G.R.; Darnell, J.E. The JAK-STAT Pathway at Twenty. Immunity 2012, 36, 503–514. [Google Scholar] [CrossRef] [PubMed]
- Ferrao, R.; Lupardus, P.J. The Janus Kinase (JAK) FERM and SH2 Domains: Bringing Specificity to JAK-Receptor Interactions. Front. Endocrinol. 2017, 8, 71. [Google Scholar] [CrossRef]
- Leitner, N.R.; Witalisz-Siepracka, A.; Strobl, B.; Muller, M. Tyrosine kinase 2-Surveillant of tumours and bona fide oncogene. Cytokine 2017, 89, 209–218. [Google Scholar] [CrossRef]
- Strobl, B.; Stoiber, D.; Sexl, V.; Mueller, M. Tyrosine kinase 2 (TYK2) in cytokine signalling and host immunity. Front. Biosci. 2011, 16, 3214–3232. [Google Scholar] [CrossRef]
- Bousoik, E.; Montazeri Aliabadi, H. “Do We Know Jack” About JAK? A Closer Look at JAK/STAT Signaling Pathway. Front. Oncol. 2018, 8, 287. [Google Scholar] [CrossRef]
- Dudakov, J.A.; Hanash, A.M.; van den Brink, M.R. Interleukin-22: Immunobiology and pathology. Annu. Rev. Immunol. 2015, 33, 747–785. [Google Scholar] [CrossRef]
- Ouyang, W.; O’Garra, A. IL-10 Family Cytokines IL-10 and IL-22: From Basic Science to Clinical Translation. Immunity 2019, 50, 871–891. [Google Scholar] [CrossRef]
- Kastelein, R.A.; Hunter, C.A.; Cua, D.J. Discovery and biology of IL-23 and IL-27: Related but functionally distinct regulators of inflammation. Annu. Rev. Immunol. 2007, 25, 221–242. [Google Scholar] [CrossRef]
- Rutz, S.; Wang, X.; Ouyang, W. The IL-20 subfamily of cytokines--from host defence to tissue homeostasis. Nat. Rev. Immunol. 2014, 14, 783–795. [Google Scholar] [CrossRef] [PubMed]
- Tait Wojno, E.D.; Hunter, C.A.; Stumhofer, J.S. The Immunobiology of the Interleukin-12 Family: Room for Discovery. Immunity 2019, 50, 851–870. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, P.; Gronke, K.; Diefenbach, A. A catch-22: Interleukin-22 and cancer. Eur. J. Immunol. 2018, 48, 15–31. [Google Scholar] [CrossRef] [PubMed]
- Lim, C.; Savan, R. The role of the IL-22/IL-22R1 axis in cancer. Cytokine Growth Factor Rev. 2014, 25, 257–271. [Google Scholar] [CrossRef] [PubMed]
- Huynh, J.; Chand, A.; Gough, D.; Ernst, M. Therapeutically exploiting STAT3 activity in cancer—Using tissue repair as a road map. Nat. Rev. Cancer 2019, 19, 82–96. [Google Scholar] [CrossRef]
- Yan, J.M.; Smyth, M.J.; Teng, M.W.L. Interleukin (IL)-12 and IL-23 and Their Conflicting Roles in Cancer. Cold Spring Harb. Perspect. Biol. 2018, 10, a028530. [Google Scholar] [CrossRef]
- Kortylewski, M.; Xin, H.; Kujawski, M.; Lee, H.; Liu, Y.; Harris, T.; Drake, C.; Pardoll, D.; Yu, H. Regulation of the IL-23 and IL-12 balance by Stat3 signaling in the tumor microenvironment. Cancer Cell 2009, 15, 114–123. [Google Scholar] [CrossRef]
- Tsai, M.H.; Pai, L.M.; Lee, C.K. Fine-Tuning of Type I Interferon Response by STAT3. Front. Immunol. 2019, 10, 1448. [Google Scholar] [CrossRef]
- Mahony, R.; Gargan, S.; Roberts, K.L.; Bourke, N.; Keating, S.E.; Bowie, A.G.; O’Farrelly, C.; Stevenson, N.J. A novel anti-viral role for STAT3 in IFN-alpha signalling responses. Cell. Mol. Life Sci. 2017, 74, 1755–1764. [Google Scholar] [CrossRef]
- Mannino, M.H.; Zhu, Z.; Xiao, H.; Bai, Q.; Wakefield, M.R.; Fang, Y. The paradoxical role of IL-10 in immunity and cancer. Cancer Lett. 2015, 367, 103–107. [Google Scholar] [CrossRef]
- Chen, Y.Y.; Li, C.F.; Yeh, C.H.; Chang, M.S.; Hsing, C.H. Interleukin-19 in breast cancer. Clin. Dev. Immunol. 2013, 2013, 294320. [Google Scholar] [CrossRef] [PubMed]
- Niess, J.H.; Hruz, P.; Kaymak, T. The Interleukin-20 Cytokines in Intestinal Diseases. Front. Immunol. 2018, 9, 1373. [Google Scholar] [CrossRef] [PubMed]
- You, W.; Tang, Q.; Zhang, C.; Wu, J.; Gu, C.; Wu, Z.; Li, X. IL-26 promotes the proliferation and survival of human gastric cancer cells by regulating the balance of STAT1 and STAT3 activation. PLoS ONE 2013, 8, e63588. [Google Scholar] [CrossRef] [PubMed]
- Wan, J.; Fu, A.K.; Ip, F.C.; Ng, H.K.; Hugon, J.; Page, G.; Wang, J.H.; Lai, K.O.; Wu, Z.; Ip, N.Y. Tyk2/STAT3 signaling mediates beta-amyloid-induced neuronal cell death: Implications in Alzheimer’s disease. J. Neurosci. 2010, 30, 6873–6881. [Google Scholar] [CrossRef] [PubMed]
- Kotenko, S.V. IFN-lambdas. Curr. Opin. Immunol. 2011, 23, 583–590. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, G. The molecular basis for differential type I interferon signaling. J. Biol. Chem. 2017, 292, 7285–7294. [Google Scholar] [CrossRef] [PubMed]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A., Jr.; Kinzler, K.W. Cancer genome landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Chen, E.; Staudt, L.M.; Green, A.R. Janus kinase deregulation in leukemia and lymphoma. Immunity 2012, 36, 529–541. [Google Scholar] [CrossRef]
- Groner, B.; von Manstein, V. Jak Stat signaling and cancer: Opportunities, benefits and side effects of targeted inhibition. Mol. Cell. Endocrinol. 2017, 451, 1–14. [Google Scholar] [CrossRef]
- Thomas, S.J.; Snowden, J.A.; Zeidler, M.P.; Danson, S.J. The role of JAK/STAT signalling in the pathogenesis, prognosis and treatment of solid tumours. Br. J. Cancer 2015, 113, 365–371. [Google Scholar] [CrossRef] [PubMed]
- O’Shea, J.J.; Holland, S.M.; Staudt, L.M. JAKs and STATs in immunity, immunodeficiency, and cancer. N. Engl. J. Med. 2013, 368, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, D.M.; Kanno, Y.; Villarino, A.; Ward, M.; Gadina, M.; O’Shea, J.J. JAK inhibition as a therapeutic strategy for immune and inflammatory diseases. Nat. Rev. Drug Discov. 2017. [Google Scholar] [CrossRef] [PubMed]
- Knoops, L.; Hornakova, T.; Royer, Y.; Constantinescu, S.N.; Renauld, J.C. JAK kinases overexpression promotes in vitro cell transformation. Oncogene 2008, 27, 1511–1519. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ubel, C.; Mousset, S.; Trufa, D.; Sirbu, H.; Finotto, S. Establishing the role of tyrosine kinase 2 in cancer. Oncoimmunology 2013, 2, e22840. [Google Scholar] [CrossRef]
- Ide, H.; Nakagawa, T.; Terado, Y.; Kamiyama, Y.; Muto, S.; Horie, S. Tyk2 expression and its signaling enhances the invasiveness of prostate cancer cells. Biochem. Biophys. Res. Commun. 2008, 369, 292–296. [Google Scholar] [CrossRef] [PubMed]
- Santos, J.; Mesquita, D.; Barros-Silva, J.D.; Jeronimo, C.; Henrique, R.; Morais, A.; Paulo, P.; Teixeira, M.R. Uncovering potential downstream targets of oncogenic GRPR overexpression in prostate carcinomas harboring ETS rearrangements. Oncoscience 2015, 2, 497–507. [Google Scholar] [CrossRef][Green Version]
- Silver, D.L.; Naora, H.; Liu, J.; Cheng, W.; Montell, D.J. Activated signal transducer and activator of transcription (STAT) 3: Localization in focal adhesions and function in ovarian cancer cell motility. Cancer Res. 2004, 64, 3550–3558. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Lv, J.; Yu, L.; Zhu, X.; Wu, J.; Zou, S.; Jiang, S. Proteomic identification of differentially-expressed proteins in squamous cervical cancer. Gynecol. Oncol. 2009, 112, 248–256. [Google Scholar] [CrossRef] [PubMed]
- Christy, J.; Priyadharshini, L. Differential expression analysis of JAK/STAT pathway related genes in breast cancer. Meta Gene 2018, 16, 122–129. [Google Scholar] [CrossRef]
- Song, X.C.; Fu, G.; Yang, X.; Jiang, Z.; Wang, Y.; Zhou, G.W. Protein expression profiling of breast cancer cells by dissociable antibody microarray (DAMA) staining. Mol. Cell. Proteom. 2008, 7, 163–169. [Google Scholar] [CrossRef] [PubMed]
- Hirbe, A.C.; Kaushal, M.; Sharma, M.K.; Dahiya, S.; Pekmezci, M.; Perry, A.; Gutmann, D.H. Clinical genomic profiling identifies TYK2 mutation and overexpression in patients with neurofibromatosis type 1-associated malignant peripheral nerve sheath tumors. Cancer 2017, 123, 1194–1201. [Google Scholar] [CrossRef] [PubMed]
- Qin, W.J.; Godec, A.; Zhang, X.C.; Zhu, C.G.; Shao, J.Y.; Tao, Y.; Bu, X.Z.; Hirlbe, A.C. TYK2 promotes malignant peripheral nerve sheath tumor progression through inhibition of cell death. Cancer Med. 2019, 8, 5232–5241. [Google Scholar] [CrossRef] [PubMed]
- Uhlen, M.; Zhang, C.; Lee, S.; Sjostedt, E.; Fagerberg, L.; Bidkhori, G.; Benfeitas, R.; Arif, M.; Liu, Z.; Edfors, F.; et al. A pathology atlas of the human cancer transcriptome. Science 2017, 357, eaan2507. [Google Scholar] [CrossRef]
- Wang, X.; Liao, X.; Yu, T.; Gong, Y.; Zhang, L.; Huang, J.; Yang, C.; Han, C.; Yu, L.; Zhu, G.; et al. Analysis of clinical significance and prospective molecular mechanism of main elements of the JAK/STAT pathway in hepatocellular carcinoma. Int. J. Oncol. 2019, 55, 805–822. [Google Scholar] [CrossRef]
- Nemoto, M.; Hattori, H.; Maeda, N.; Akita, N.; Muramatsu, H.; Moritani, S.; Kawasaki, T.; Maejima, M.; Ode, H.; Hachiya, A.; et al. Compound heterozygous TYK2 mutations underlie primary immunodeficiency with T-cell lymphopenia. Sci. Rep. 2018, 8, 6956. [Google Scholar] [CrossRef]
- Boisson-Dupuis, S.; Ramirez-Alejo, N.; Li, Z.; Patin, E.; Rao, G.; Kerner, G.; Lim, C.K.; Krementsov, D.N.; Hernandez, N.; Ma, C.S.; et al. Tuberculosis and impaired IL-23-dependent IFN-gamma immunity in humans homozygous for a common TYK2 missense variant. Sci. Immunol. 2018, 3, eaau8714. [Google Scholar] [CrossRef]
- Dendrou, C.A.; Cortes, A.; Shipman, L.; Evans, H.G.; Attfield, K.E.; Jostins, L.; Barber, T.; Kaur, G.; Kuttikkatte, S.B.; Leach, O.A.; et al. Resolving TYK2 locus genotype-to-phenotype differences in autoimmunity. Sci. Transl. Med. 2016, 8, 363ra149. [Google Scholar] [CrossRef]
- Kerner, G.; Ramirez-Alejo, N.; Seeleuthner, Y.; Yang, R.; Ogishi, M.; Cobat, A.; Patin, E.; Quintana-Murci, L.; Boisson-Dupuis, S.; Casanova, J.L.; et al. Homozygosity for TYK2 P1104A underlies tuberculosis in about 1% of patients in a cohort of European ancestry. Proc. Natl. Acad. Sci. USA 2019, 116, 10430–10434. [Google Scholar] [CrossRef]
- Sang, Q.X.; Man, Y.G.; Sung, Y.M.; Khamis, Z.I.; Zhang, L.; Lee, M.H.; Byers, S.W.; Sahab, Z.J. Non-receptor tyrosine kinase 2 reaches its lowest expression levels in human breast cancer during regional nodal metastasis. Clin. Exp. Metastasis 2012, 29, 143–153. [Google Scholar] [CrossRef]
- Ruhe, J.E.; Streit, S.; Hart, S.; Wong, C.H.; Specht, K.; Knyazev, P.; Knyazeva, T.; Tay, L.S.; Loo, H.L.; Foo, P.; et al. Genetic alterations in the tyrosine kinase transcriptome of human cancer cell lines. Cancer Res. 2007, 67, 11368–11376. [Google Scholar] [CrossRef]
- Prutsch, N.; Gurnhofer, E.; Suske, T.; Liang, H.C.; Schlederer, M.; Roos, S.; Wu, L.C.; Simonitsch-Klupp, I.; Alvarez-Hernandez, A.; Kornauth, C.; et al. Dependency on the TYK2/STAT1/MCL1 axis in anaplastic large cell lymphoma. Leukemia 2019, 33, 696–709. [Google Scholar] [CrossRef] [PubMed]
- Sanda, T.; Tyner, J.W.; Gutierrez, A.; Ngo, V.N.; Glover, J.; Chang, B.H.; Yost, A.; Ma, W.; Fleischman, A.G.; Zhou, W.; et al. TYK2-STAT1-BCL2 pathway dependence in T-cell acute lymphoblastic leukemia. Cancer Discov. 2013, 3, 564–577. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, A.; Lahtz, C.; Nagao, T.; Song, J.Y.; Chan, W.C.; Lee, H.; Yue, C.; Look, T.; Mulfarth, R.; Li, W.; et al. CTLA4 Promotes Tyk2-STAT3-Dependent B-cell Oncogenicity. Cancer Res. 2017, 77, 5118–5128. [Google Scholar] [CrossRef] [PubMed]
- Carmo, C.R.; Lyons-Lewis, J.; Seckl, M.J.; Costa-Pereira, A.P. A Novel Requirement for Janus Kinases as Mediators of Drug Resistance Induced by Fibroblast Growth Factor-2 in Human Cancer Cells. PLoS ONE 2011, 6, e19861. [Google Scholar] [CrossRef]
- Vainchenker, W.; Constantinescu, S.N. JAK/STAT signaling in hematological malignancies. Oncogene 2013, 32, 2601–2613. [Google Scholar] [CrossRef]
- Hubbard, S.R. Mechanistic Insights into Regulation of JAK2 Tyrosine Kinase. Front. Endocrinol. 2017, 8, 361. [Google Scholar] [CrossRef]
- Morris, R.; Kershaw, N.J.; Babon, J.J. The molecular details of cytokine signaling via the JAK/STAT pathway. Protein Sci. 2018, 27, 1984–2009. [Google Scholar] [CrossRef]
- Silvennoinen, O.; Hubbard, S.R. Molecular insights into regulation of JAK2 in myeloproliferative neoplasms. Blood 2015, 125, 3388–3392. [Google Scholar] [CrossRef]
- Gakovic, M.; Ragimbeau, J.; Francois, V.; Constantinescu, S.N.; Pellegrini, S. The Stat3-activating Tyk2 V678F mutant does not up-regulate signaling through the type I interferon receptor but confers ligand hypersensitivity to a homodimeric receptor. J. Biol. Chem. 2008, 283, 18522–18529. [Google Scholar] [CrossRef]
- Staerk, J.; Kallin, A.; Demoulin, J.B.; Vainchenker, W.; Constantinescu, S.N. JAK1 and Tyk2 activation by the homologous polycythemia vera JAK2 V617F mutation: Cross-talk with IGF1 receptor. J. Biol. Chem. 2005, 280, 41893–41899. [Google Scholar] [CrossRef] [PubMed]
- Grossman, R.L.; Heath, A.P.; Ferretti, V.; Varmus, H.E.; Lowy, D.R.; Kibbe, W.A.; Staudt, L.M. Toward a Shared Vision for Cancer Genomic Data. N. Engl. Med. 2016, 375, 1109–1112. [Google Scholar] [CrossRef] [PubMed]
- Jensen, M.A.; Ferretti, V.; Grossman, R.L.; Staudt, L.M. The NCI Genomic Data Commons as an engine for precision medicine. Blood 2017, 130, 453–459. [Google Scholar] [CrossRef] [PubMed]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2019, 47, D941–D947. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal 2013, 6, pl1. [Google Scholar] [CrossRef]
- Waanders, E.; Scheijen, B.; Jongmans, M.C.; Venselaar, H.; van Reijmersdal, S.V.; van Dijk, A.H.; Pastorczak, A.; Weren, R.D.; van der Schoot, C.E.; van de Vorst, M.; et al. Germline activating TYK2 mutations in pediatric patients with two primary acute lymphoblastic leukemia occurrences. Leukemia 2017, 31, 821–828. [Google Scholar] [CrossRef]
- Kaminker, J.S.; Zhang, Y.; Waugh, A.; Haverty, P.M.; Peters, B.; Sebisanovic, D.; Stinson, J.; Forrest, W.F.; Bazan, J.F.; Seshagiri, S.; et al. Distinguishing cancer-associated missense mutations from common polymorphisms. Cancer Res. 2007, 67, 465–473. [Google Scholar] [CrossRef]
- Tomasson, M.H.; Xiang, Z.; Walgren, R.; Zhao, Y.; Kasai, Y.; Miner, T.; Ries, R.E.; Lubman, O.; Fremont, D.H.; McLellan, M.D.; et al. Somatic mutations and germline sequence variants in the expressed tyrosine kinase genes of patients with de novo acute myeloid leukemia. Blood 2008, 111, 4797–4808. [Google Scholar] [CrossRef]
- Li, Z.; Gakovic, M.; Ragimbeau, J.; Eloranta, M.L.; Ronnblom, L.; Michel, F.; Pellegrini, S. Two rare disease-associated Tyk2 variants are catalytically impaired but signaling competent. J. Immunol. 2013, 190, 2335–2344. [Google Scholar] [CrossRef]
- Gorman, J.A.; Hundhausen, C.; Kinsman, M.; Arkatkar, T.; Allenspach, E.J.; Clough, C.; West, S.E.; Thomas, K.; Eken, A.; Khim, S.; et al. The TYK2-P1104A Autoimmune Protective Variant Limits Coordinate Signals Required to Generate Specialized T Cell Subsets. Front. Immunol. 2019, 10, 44. [Google Scholar] [CrossRef] [PubMed]
- Gao, Q.S.; Liang, W.W.; Foltz, S.M.; Mutharasu, G.; Jayasinghe, R.G.; Cao, S.; Liao, W.W.; Reynolds, S.M.; Wyczalkowski, M.A.; Yao, L.J.; et al. Driver Fusions and Their Implications in the Development and Treatment of Human Cancers. Cell Rep. 2018, 23, 227–238. [Google Scholar] [CrossRef] [PubMed]
- Medves, S.; Demoulin, J.B. Tyrosine kinase gene fusions in cancer: Translating mechanisms into targeted therapies. J. Cell. Mol. Med. 2012, 16, 237–248. [Google Scholar] [CrossRef] [PubMed]
- Stransky, N.; Cerami, E.; Schalm, S.; Kim, J.L.; Lengauer, C. The landscape of kinase fusions in cancer. Nat. Commun. 2014, 5, 4846. [Google Scholar] [CrossRef] [PubMed]
- Lacronique, V.; Boureux, A.; Valle, V.D.; Poirel, H.; Quang, C.T.; Mauchauffe, M.; Berthou, C.; Lessard, M.; Berger, R.; Ghysdael, J.; et al. A TEL-JAK2 fusion protein with constitutive kinase activity in human leukemia. Science 1997, 278, 1309–1312. [Google Scholar] [CrossRef]
- Carron, C.; Cormier, F.; Janin, A.; Lacronique, V.; Giovannini, M.; Daniel, M.T.; Bernard, O.; Ghysdael, J. TEL-JAK2 transgenic mice develop T-cell leukemia. Blood 2000, 95, 3891–3899. [Google Scholar] [CrossRef]
- Lacronique, V.; Boureux, A.; Monni, R.; Dumon, S.; Mauchauffe, M.; Mayeux, P.; Gouilleux, F.; Berger, R.; Gisselbrecht, S.; Ghysdael, J.; et al. Transforming properties of chimeric TEL-JAK proteins in Ba/F3 cells. Blood 2000, 95, 2076–2083. [Google Scholar] [CrossRef]
- Ho, K.; Valdez, F.; Garcia, R.; Tirado, C.A. JAK2 Translocations in hematological malignancies: Review of the literature. J. Assoc. Genet. Technol. 2010, 36, 107–109. [Google Scholar]
- Levavi, H.; Tripodi, J.; Marcellino, B.; Mascarenhas, J.; Jones, A.V.; Cross, N.C.P.; Gruenstein, D.; Najfeld, V. A Novel t(1;9)(p36;p24.1) JAK2 Translocation and Review of the Literature. Acta Haematol. 2019, 142, 105–112. [Google Scholar] [CrossRef]
- Crescenzo, R.; Abate, F.; Lasorsa, E.; Tabbo, F.; Gaudiano, M.; Chiesa, N.; Di Giacomo, F.; Spaccarotella, E.; Barbarossa, L.; Ercole, E.; et al. Convergent mutations and kinase fusions lead to oncogenic STAT3 activation in anaplastic large cell lymphoma. Cancer Cell 2015, 27, 516–532. [Google Scholar] [CrossRef]
- Roberts, K.G.; Li, Y.; Payne-Turner, D.; Harvey, R.C.; Yang, Y.L.; Pei, D.; McCastlain, K.; Ding, L.; Lu, C.; Song, G.; et al. Targetable kinase-activating lesions in Ph-like acute lymphoblastic leukemia. N. Engl. J. Med. 2014, 371, 1005–1015. [Google Scholar] [CrossRef] [PubMed]
- Velusamy, T.; Kiel, M.J.; Sahasrabuddhe, A.A.; Rolland, D.; Dixon, C.A.; Bailey, N.G.; Betz, B.L.; Brown, N.A.; Hristov, A.C.; Wilcox, R.A.; et al. A novel recurrent NPM1-TYK2 gene fusion in cutaneous CD30-positive lymphoproliferative disorders. Blood 2014, 124, 3768–3771. [Google Scholar] [CrossRef] [PubMed]
- Roberts, K.G.; Gu, Z.; Payne-Turner, D.; McCastlain, K.; Harvey, R.C.; Chen, I.M.; Pei, D.; Iacobucci, I.; Valentine, M.; Pounds, S.B.; et al. High Frequency and Poor Outcome of Philadelphia Chromosome-Like Acute Lymphoblastic Leukemia in Adults. J. Clin. Oncol. 2017, 35, 394–401. [Google Scholar] [CrossRef] [PubMed]
- Tron, A.E.; Keeton, E.K.; Ye, M.; Casas-Selves, M.; Chen, H.; Dillman, K.S.; Gale, R.E.; Stengel, C.; Zinda, M.; Linch, D.C.; et al. Next-generation sequencing identifies a novel ELAVL1-TYK2 fusion gene in MOLM-16, an AML cell line highly sensitive to the PIM kinase inhibitor AZD1208. Leuk. Lymphoma 2016, 57, 2927–2929. [Google Scholar] [CrossRef]
- Gu, Z.; Churchman, M.; Roberts, K.; Li, Y.; Liu, Y.; Harvey, R.C.; McCastlain, K.; Reshmi, S.C.; Payne-Turner, D.; Iacobucci, I.; et al. Genomic analyses identify recurrent MEF2D fusions in acute lymphoblastic leukaemia. Nat. Commun. 2016, 7, 13331. [Google Scholar] [CrossRef]
- Roberts, K.G.; Yang, Y.L.; Payne-Turner, D.; Lin, W.; Files, J.K.; Dickerson, K.; Gu, Z.; Taunton, J.; Janke, L.J.; Chen, T.; et al. Oncogenic role and therapeutic targeting of ABL-class and JAK-STAT activating kinase alterations in Ph-like ALL. Blood Adv. 2017, 1, 1657–1671. [Google Scholar] [CrossRef]
- Prasad, A.; Rabionet, R.; Espinet, B.; Zapata, L.; Puiggros, A.; Melero, C.; Puig, A.; Sarria-Trujillo, Y.; Ossowski, S.; Garcia-Muret, M.P.; et al. Identification of gene mutations and fusion genes in patients with Sezary Syndrome. J. Investig. Dermatol. 2016, 136, 1490–1499. [Google Scholar] [CrossRef]
- Kim, P.; Zhou, X. FusionGDB: Fusion gene annotation DataBase. Nucleic Acids Res. 2019, 47, D994–D1004. [Google Scholar] [CrossRef]
- Negrini, S.; Gorgoulis, V.G.; Halazonetis, T.D. Genomic instability—An evolving hallmark of cancer. Nat. Rev. Mol. Cell Biol. 2010, 11, 220–228. [Google Scholar] [CrossRef]
- Ly, P.; Cleveland, D.W. Rebuilding Chromosomes After Catastrophe: Emerging Mechanisms of Chromothripsis. Trends Cell Biol. 2017, 27, 917–930. [Google Scholar] [CrossRef]
- Rode, A.; Maass, K.K.; Willmund, K.V.; Lichter, P.; Ernst, A. Chromothripsis in cancer cells: An update. Int. J. Cancer 2016, 138, 2322–2333. [Google Scholar] [CrossRef] [PubMed]
- Grobner, S.N.; Worst, B.C.; Weischenfeldt, J.; Buchhalter, I.; Kleinheinz, K.; Rudneva, V.A.; Johann, P.D.; Balasubramanian, G.P.; Segura-Wang, M.; Brabetz, S.; et al. The landscape of genomic alterations across childhood cancers. Nature 2018, 555, 321–327. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Kwon, M.; Mannino, M.; Yang, N.; Renda, F.; Khodjakov, A.; Pellman, D. Nuclear envelope assembly defects link mitotic errors to chromothripsis. Nature 2018, 561, 551–555. [Google Scholar] [CrossRef] [PubMed]
- Bromberg, J. Stat proteins and oncogenesis. J. Clin. Investig. 2002, 109, 1139–1142. [Google Scholar] [CrossRef]
- Wingelhofer, B.; Neubauer, H.A.; Valent, P.; Han, X.; Constantinescu, S.N.; Gunning, P.T.; Muller, M.; Moriggl, R. Implications of STAT3 and STAT5 signaling on gene regulation and chromatin remodeling in hematopoietic cancer. Leukemia 2018, 32, 1713–1726. [Google Scholar] [CrossRef]
- Avalle, L.; Pensa, S.; Regis, G.; Novelli, F.; Poli, V. STAT1 and STAT3 in tumorigenesis: A matter of balance. JAKSTAT 2012, 1, 65–72. [Google Scholar] [CrossRef]
- Meissl, K.; Macho-Maschler, S.; Muller, M.; Strobl, B. The good and the bad faces of STAT1 in solid tumours. Cytokine 2017, 89, 12–20. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, Z. STAT1 in cancer: Friend or foe? Discov. Med. 2017, 24, 19–29. [Google Scholar]
- Mori, R.; Wauman, J.; Icardi, L.; Van der Heyden, J.; De Cauwer, L.; Peelman, F.; De Bosscher, K.; Tavernier, J. TYK2-induced phosphorylation of Y640 suppresses STAT3 transcriptional activity. Sci. Rep. 2017, 7, 15919. [Google Scholar] [CrossRef]
- Koskela, H.L.; Eldfors, S.; Ellonen, P.; van Adrichem, A.J.; Kuusanmaki, H.; Andersson, E.I.; Lagstrom, S.; Clemente, M.J.; Olson, T.; Jalkanen, S.E.; et al. Somatic STAT3 mutations in large granular lymphocytic leukemia. N. Engl. J. Med. 2012, 366, 1905–1913. [Google Scholar] [CrossRef] [PubMed]
- Pilati, C.; Amessou, M.; Bihl, M.P.; Balabaud, C.; Nhieu, J.T.; Paradis, V.; Nault, J.C.; Izard, T.; Bioulac-Sage, P.; Couchy, G.; et al. Somatic mutations activating STAT3 in human inflammatory hepatocellular adenomas. J. Exp. Med. 2011, 208, 1359–1366. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.M.; Feng, L.S.; Cui, J.W. Increased expression of claudin-17 promotes a malignant phenotype in hepatocyte via Tyk2/Stat3 signaling and is associated with poor prognosis in patients with hepatocellular carcinoma. Diagn. Pathol. 2018, 13, 72. [Google Scholar] [CrossRef] [PubMed]
- Muller, S.; Chen, Y.; Ginter, T.; Schafer, C.; Buchwald, M.; Schmitz, L.M.; Klitzsch, J.; Schutz, A.; Haitel, A.; Schmid, K.; et al. SIAH2 antagonizes TYK2-STAT3 signaling in lung carcinoma cells. Oncotarget 2014, 5, 3184–3196. [Google Scholar] [CrossRef]
- Liu, H.; Wang, M.; Liang, N.; Guan, L. Claudin-9 enhances the metastatic potential of hepatocytes via Tyk2/Stat3 signaling. Turk. J. Gastroenterol. 2019, 30, 722–731. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.M.; Feng, L.S.; Cui, J.W. Increased expression of claudin-12 promotes the metastatic phenotype of human bronchial epithelial cells and is associated with poor prognosis in lung squamous cell carcinoma. Exp. Ther. Med. 2019, 17, 165–174. [Google Scholar] [CrossRef] [PubMed]
- Cathcart, J.; Pulkoski-Gross, A.; Cao, J. Targeting matrix metalloproteinases in cancer: Bringing new life to old ideas. Genes Dis. 2015, 2, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Tabaries, S.; Siegel, P.M. The role of claudins in cancer metastasis. Oncogene 2017, 36, 1176–1190. [Google Scholar] [CrossRef]
- Cutler, S.J.; Doecke, J.D.; Ghazawi, I.; Yang, J.; Griffiths, L.R.; Spring, K.J.; Ralph, S.J.; Mellick, A.S. Novel STAT binding elements mediate IL-6 regulation of MMP-1 and MMP-3. Sci. Rep. 2017, 7, 8526. [Google Scholar] [CrossRef]
- Fanjul-Fernandez, M.; Folgueras, A.R.; Cabrera, S.; Lopez-Otin, C. Matrix metalloproteinases: Evolution, gene regulation and functional analysis in mouse models. Biochim. Biophys. Acta 2010, 1803, 3–19. [Google Scholar] [CrossRef]
- Costantino, G.; Egerbacher, M.; Kolbe, T.; Karaghiosoff, M.; Strobl, B.; Vogl, C.; Helmreich, M.; Muller, M. Tyk2 and signal transducer and activator of transcription 1 contribute to intestinal I/R injury. Shock 2008, 29, 238–244. [Google Scholar] [CrossRef]
- Araki, Y.; Tsuzuki Wada, T.; Aizaki, Y.; Sato, K.; Yokota, K.; Fujimoto, K.; Kim, Y.T.; Oda, H.; Kurokawa, R.; Mimura, T. Histone Methylation and STAT-3 Differentially Regulate Interleukin-6-Induced Matrix Metalloproteinase Gene Activation in Rheumatoid Arthritis Synovial Fibroblasts. Arthritis Rheumatol. 2016, 68, 1111–1123. [Google Scholar] [CrossRef] [PubMed]
- Mahmood, N.; Mihalcioiu, C.; Rabbani, S.A. Multifaceted Role of the Urokinase-Type Plasminogen Activator (uPA) and Its Receptor (uPAR): Diagnostic, Prognostic, and Therapeutic Applications. Front. Oncol. 2018, 8, 24. [Google Scholar] [CrossRef] [PubMed]
- Kusch, A.; Tkachuk, S.; Haller, H.; Dietz, R.; Gulba, D.C.; Lipp, M.; Dumler, I. Urokinase stimulates human vascular smooth muscle cell migration via a phosphatidylinositol 3-kinase-Tyk2 interaction. J. Biol. Chem. 2000, 275, 39466–39473. [Google Scholar] [CrossRef] [PubMed]
- Radwan, M.; Miller, I.; Grunert, T.; Marchetti-Deschmann, M.; Vogl, C.; O’Donoghue, N.; Dunn, M.J.; Kolbe, T.; Allmaier, G.; Gemeiner, M.; et al. The impact of tyrosine kinase 2 (Tyk2) on the proteome of murine macrophages and their response to lipopolysaccharide (LPS). Proteomics 2008, 8, 3469–3485. [Google Scholar] [CrossRef] [PubMed]
- Schuster, C.; Muller, M.; Freissmuth, M.; Sexl, V.; Stoiber, D. Commentary on H. Ide. Tyk2 expression and its signaling enhances the invasiveness of prostate cancer cells. Biochem. Biophys. Res. Commun. 2008, 366, 869–870. [Google Scholar] [CrossRef]
- Murphy, D.A.; Courtneidge, S.A. The ‘ins’ and ‘outs’ of podosomes and invadopodia: Characteristics, formation and function. Nat. Rev. Mol. Cell Biol. 2011, 12, 413–426. [Google Scholar] [CrossRef]
- Revach, O.Y.; Sandler, O.; Samuels, Y.; Geiger, B. Cross-Talk between Receptor Tyrosine Kinases AXL and ERBB3 Regulates Invadopodia Formation in Melanoma Cells. Cancer Res. 2019, 79, 2634–2648. [Google Scholar] [CrossRef]
- Aasen, T.; Leithe, E.; Graham, S.V.; Kameritsch, P.; Mayán, M.D.; Mesnil, M.; Pogoda, K.; Tabernero, A. Connexins in cancer: Bridging the gap to the clinic. Oncogene 2019, 38, 4429–4451. [Google Scholar] [CrossRef]
- Bonacquisti, E.E.; Nguyen, J. Connexin 43 (Cx43) in cancer: Implications for therapeutic approaches via gap junctions. Cancer Lett. 2019, 442, 439–444. [Google Scholar] [CrossRef]
- Li, H.; Spagnol, G.; Zheng, L.; Stauch, K.L.; Sorgen, P.L. Regulation of Connexin43 Function and Expression by Tyrosine Kinase 2. J. Biol. Chem. 2016, 291, 15867–15880. [Google Scholar] [CrossRef]
- Kotredes, K.P.; Gamero, A.M. Interferons as inducers of apoptosis in malignant cells. J. Interferon Cytokine Res. 2013, 33, 162–170. [Google Scholar] [CrossRef] [PubMed]
- Kale, J.; Osterlund, E.J.; Andrews, D.W. BCL-2 family proteins: Changing partners in the dance towards death. Cell Death Differ. 2018, 25, 65–80. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Letai, A.; Sarosiek, K. Regulation of apoptosis in health and disease: The balancing act of BCL-2 family proteins. Nat. Rev. Mol. Cell Biol. 2019, 20, 175–193. [Google Scholar] [CrossRef] [PubMed]
- Shimoda, H.K.; Shide, K.; Kameda, T.; Matsunaga, T.; Shimoda, K. Tyrosine kinase 2 interacts with the proapoptotic protein Siva-1 and augments its apoptotic functions. Biochem. Biophys. Res. Commun. 2010, 400, 252–257. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, H.; Shibata, F.; Kumagai, H.; Shimoda, K.; Kitamura, T. Tyk2 is dispensable for induction of myeloproliferative disease by mutant FLT3. Int. J. Hematol. 2006, 84, 54–59. [Google Scholar] [CrossRef]
- Yamaji, T.; Shide, K.; Kameda, T.; Sekine, M.; Kamiunten, A.; Hidaka, T.; Kubuki, Y.; Shimoda, H.; Abe, H.; Miike, T.; et al. Loss of Tyrosine Kinase 2 Does Not Affect the Severity of Jak2V617F-induced Murine Myeloproliferative Neoplasm. Anticancer Res. 2017, 37, 3841–3847. [Google Scholar] [CrossRef]
- Koppikar, P.; Bhagwat, N.; Kilpivaara, O.; Manshouri, T.; Adli, M.; Hricik, T.; Liu, F.; Saunders, L.M.; Mullally, A.; Abdel-Wahab, O.; et al. Heterodimeric JAK-STAT activation as a mechanism of persistence to JAK2 inhibitor therapy. Nature 2012, 489, 155–159. [Google Scholar] [CrossRef]
- Kohlhuber, F.; Rogers, N.C.; Watling, D.; Feng, J.; Guschin, D.; Briscoe, J.; Witthuhn, B.A.; Kotenko, S.V.; Pestka, S.; Stark, G.R.; et al. A JAK1/JAK2 chimera can sustain alpha and gamma interferon responses. Mol. Cell. Biol. 1997, 17, 695–706. [Google Scholar] [CrossRef]
- Briscoe, J.; Rogers, N.C.; Witthuhn, B.A.; Watling, D.; Harpur, A.G.; Wilks, A.; Stark, G.R.; Ihle, J.N.; Kerr, I.M. Kinase-negative mutants of JAK1 can sustain interferon-gamma-inducible gene expression but not an antiviral state. EMBO J. 1996, 15, 799–809. [Google Scholar] [CrossRef]
- Adam, L.; Bandyopadhyay, D.; Kumar, R. Interferon-alpha signaling promotes nucleus-to-cytoplasmic redistribution of p95Vav, and formation of a multisubunit complex involving Vav, Ku80, and Tyk2. Biochem. Biophys. Res. Commun. 2000, 267, 692–696. [Google Scholar] [CrossRef]
- Uddin, S.; Gardziola, C.; Dangat, A.; Yi, T.; Platanias, L.C. Interaction of the c-cbl proto-oncogene product with the Tyk-2 protein tyrosine kinase. Biochem. Biophys. Res. Commun. 1996, 225, 833–838. [Google Scholar] [CrossRef] [PubMed]
- Uddin, S.; Grumbach, I.M.; Yi, T.; Colamonici, O.R.; Platanias, L.C. Interferon alpha activates the tyrosine kinase Lyn in haemopoietic cells. Br. J. Haematol. 1998, 101, 446–449. [Google Scholar] [CrossRef] [PubMed]
- Uddin, S.; Sher, D.A.; Alsayed, Y.; Pons, S.; Colamonici, O.R.; Fish, E.N.; White, M.F.; Platanias, L.C. Interaction of p59(fyn) with interferon-activated Jak kinases. Biochem. Biophys. Res. Commun. 1997, 235, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Uddin, S.; Sweet, M.; Colamonici, O.R.; Krolewski, J.J.; Platanias, L.C. The vav proto-oncogene product (p95vav) interacts with the Tyk-2 protein tyrosine kinase. FEBS Lett. 1997, 403, 31–34. [Google Scholar] [CrossRef]
- Akahane, K.; Li, Z.; Etchin, J.; Berezovskaya, A.; Gjini, E.; Masse, C.E.; Miao, W.; Rocnik, J.; Kapeller, R.; Greenwood, J.R.; et al. Anti-leukaemic activity of the TYK2 selective inhibitor NDI-031301 in T-cell acute lymphoblastic leukaemia. Br. J. Haematol. 2017, 177, 271–282. [Google Scholar] [CrossRef] [PubMed]
- Muromoto, R.; Tawa, K.; Ohgakiuchi, Y.; Sato, A.; Saino, Y.; Hirashima, K.; Minoguchi, H.; Kitai, Y.; Kashiwakura, J.I.; Shimoda, K.; et al. IkappaB-zeta Expression Requires Both TYK2/STAT3 Activity and IL-17-Regulated mRNA Stabilization. Immunohorizons 2019, 3, 172–185. [Google Scholar] [CrossRef] [PubMed]
- Willems, M.; Dubois, N.; Musumeci, L.; Bours, V.; Robe, P.A. IkappaBzeta: An emerging player in cancer. Oncotarget 2016, 7, 66310–66322. [Google Scholar] [CrossRef]
- Luck, K.; Sheynkman, G.M.; Zhang, I.; Vidal, M. Proteome-Scale Human Interactomics. Trends Biochem. Sci. 2017, 42, 342–354. [Google Scholar] [CrossRef]
- Rolland, T.; Tasan, M.; Charloteaux, B.; Pevzner, S.J.; Zhong, Q.; Sahni, N.; Yi, S.; Lemmens, I.; Fontanillo, C.; Mosca, R.; et al. A proteome-scale map of the human interactome network. Cell 2014, 159, 1212–1226. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Jensen, L.J. Protein-protein interaction databases. Methods Mol. Biol. 2015, 1278, 39–56. [Google Scholar] [CrossRef]
- Lievens, S.; Gerlo, S.; Lemmens, I.; De Clercq, D.J.; Risseeuw, M.D.; Vanderroost, N.; De Smet, A.S.; Ruyssinck, E.; Chevet, E.; Van Calenbergh, S.; et al. Kinase Substrate Sensor (KISS), a mammalian in situ protein interaction sensor. Mol. Cell. Proteom. 2014, 13, 3332–3342. [Google Scholar] [CrossRef] [PubMed]
- Masschaele, D.; Gerlo, S.; Lemmens, I.; Lievens, S.; Tavernier, J. KISS: A Mammalian Two-Hybrid Method for In Situ Analysis of Protein-Protein Interactions. Methods Mol. Biol. 2018, 1794, 269–278. [Google Scholar] [CrossRef] [PubMed]
- Babon, J.J.; Lucet, I.S.; Murphy, J.M.; Nicola, N.A.; Varghese, L.N. The molecular regulation of Janus kinase (JAK) activation. Biochem. J. 2014, 462, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Murray, P.J. The JAK-STAT signaling pathway: Input and output integration. J. Immunol. 2007, 178, 2623–2629. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Huang, H.; Wu, C.H. Protein Bioinformatics Databases and Resources. Methods Mol. Biol. 2017, 1558, 3–39. [Google Scholar] [CrossRef] [PubMed]
- Hornbeck, P.V.; Kornhauser, J.M.; Latham, V.; Murray, B.; Nandhikonda, V.; Nord, A.; Skrzypek, E.; Wheeler, T.; Zhang, B.; Gnad, F. 15 years of PhosphoSitePlus(R): Integrating post-translationally modified sites, disease variants and isoforms. Nucleic Acids Res. 2019, 47, D433–D441. [Google Scholar] [CrossRef]
- Kershaw, N.J.; Murphy, J.M.; Lucet, I.S.; Nicola, N.A.; Babon, J.J. Regulation of Janus kinases by SOCS proteins. Biochem. Soc. Trans. 2013, 41, 1042–1047. [Google Scholar] [CrossRef]
- Xu, D.; Qu, C.K. Protein tyrosine phosphatases in the JAK/STAT pathway. Front. Biosci. 2008, 13, 4925–4932. [Google Scholar] [CrossRef]
- Bollu, L.R.; Mazumdar, A.; Savage, M.I.; Brown, P.H. Molecular Pathways: Targeting Protein Tyrosine Phosphatases in Cancer. Clin. Cancer Res. 2017, 23, 2136–2142. [Google Scholar] [CrossRef]
- Inagaki-Ohara, K.; Kondo, T.; Ito, M.; Yoshimura, A. SOCS, inflammation, and cancer. JAKSTAT 2013, 2, e24053. [Google Scholar] [CrossRef]
- Jiang, M.; Zhang, W.W.; Liu, P.; Yu, W.; Liu, T.; Yu, J. Dysregulation of SOCS-Mediated Negative Feedback of Cytokine Signaling in Carcinogenesis and Its Significance in Cancer Treatment. Front. Immunol. 2017, 8, 70. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Guan, Q.; Wang, Y.; Zhao, Z.J.; Zhou, G.W. SHP-1 suppresses cancer cell growth by promoting degradation of JAK kinases. J. Cell. Biochem. 2003, 90, 1026–1037. [Google Scholar] [CrossRef] [PubMed]
- Bocchini, C.E.; Kasembeli, M.M.; Roh, S.H.; Tweardy, D.J. Contribution of chaperones to STAT pathway signaling. JAKSTAT 2014, 3, e970459. [Google Scholar] [CrossRef] [PubMed]
- Prodromou, C. Mechanisms of Hsp90 regulation. Biochem. J. 2016, 473, 2439–2452. [Google Scholar] [CrossRef] [PubMed]
- Taipale, M.; Jarosz, D.F.; Lindquist, S. HSP90 at the hub of protein homeostasis: Emerging mechanistic insights. Nat. Rev. Mol. Cell Biol. 2010, 11, 515–528. [Google Scholar] [CrossRef] [PubMed]
- Jaeger, A.M.; Whitesell, L. HSP90: Enabler of Cancer Adaptation. Annu. Rev. Cancer Biol. 2019, 3, 275–297. [Google Scholar] [CrossRef]
- Trepel, J.; Mollapour, M.; Giaccone, G.; Neckers, L. Targeting the dynamic HSP90 complex in cancer. Nat. Rev. Cancer 2010, 10, 537–549. [Google Scholar] [CrossRef]
- Caldas-Lopes, E.; Cerchietti, L.; Ahn, J.H.; Clement, C.C.; Robles, A.I.; Rodina, A.; Moulick, K.; Taldone, T.; Gozman, A.; Guo, Y.; et al. Hsp90 inhibitor PU-H71, a multimodal inhibitor of malignancy, induces complete responses in triple-negative breast cancer models. Proc. Natl. Acad. Sci. USA 2009, 106, 8368–8373. [Google Scholar] [CrossRef]
- Taipale, M.; Krykbaeva, I.; Koeva, M.; Kayatekin, C.; Westover, K.D.; Karras, G.I.; Lindquist, S. Quantitative analysis of HSP90-client interactions reveals principles of substrate recognition. Cell 2012, 150, 987–1001. [Google Scholar] [CrossRef]
- Akahane, K.; Sanda, T.; Mansour, M.R.; Radimerski, T.; DeAngelo, D.J.; Weinstock, D.M.; Look, A.T. HSP90 inhibition leads to degradation of the TYK2 kinase and apoptotic cell death in T-cell acute lymphoblastic leukemia. Leukemia 2016, 30, 219–228. [Google Scholar] [CrossRef]
- Schoof, N.; von Bonin, F.; Trumper, L.; Kube, D. HSP90 is essential for Jak-STAT signaling in classical Hodgkin Lymphoma cells. Cell Commun. Sig. 2009, 7, 17. [Google Scholar] [CrossRef] [PubMed]
- Witte, S.; Muljo, S.A. Integrating non-coding RNAs in JAK-STAT regulatory networks. JAKSTAT 2014, 3, e28055. [Google Scholar] [CrossRef] [PubMed]
- Mullany, L.E.; Herrick, J.S.; Sakoda, L.C.; Samowitz, W.; Stevens, J.R.; Wolff, R.K.; Slattery, M.L. MicroRNA-messenger RNA interactions involving JAK-STAT signaling genes in colorectal cancer. Genes Cancer 2018, 9, 232–246. [Google Scholar] [CrossRef] [PubMed]
- Pencik, J.; Pham, H.T.; Schmoellerl, J.; Javaheri, T.; Schlederer, M.; Culig, Z.; Merkel, O.; Moriggl, R.; Grebien, F.; Kenner, L. JAK-STAT signaling in cancer: From cytokines to non-coding genome. Cytokine 2016, 87, 26–36. [Google Scholar] [CrossRef]
- Li, Z.Y.; Yang, L.; Liu, X.J.; Wang, X.Z.; Pan, Y.X.; Luo, J.M. The Long Noncoding RNA MEG3 and its Target miR-147 Regulate JAK/STAT Pathway in Advanced Chronic Myeloid Leukemia. EBioMedicine 2018, 34, 61–75. [Google Scholar] [CrossRef]
- Ghoreschi, K.; Jesson, M.I.; Li, X.; Lee, J.L.; Ghosh, S.; Alsup, J.W.; Warner, J.D.; Tanaka, M.; Steward-Tharp, S.M.; Gadina, M.; et al. Modulation of innate and adaptive immune responses by tofacitinib (CP-690,550). J. Immunol. 2011, 186, 4234–4243. [Google Scholar] [CrossRef]
- Changelian, P.S.; Flanagan, M.E.; Ball, D.J.; Kent, C.R.; Magnuson, K.S.; Martin, W.H.; Rizzuti, B.J.; Sawyer, P.S.; Perry, B.D.; Brissette, W.H.; et al. Prevention of organ allograft rejection by a specific Janus kinase 3 inhibitor. Science 2003, 302, 875–878. [Google Scholar] [CrossRef]
- Harrison, C.; Kiladjian, J.J.; Al-Ali, H.K.; Gisslinger, H.; Waltzman, R.; Stalbovskaya, V.; McQuitty, M.; Hunter, D.S.; Levy, R.; Knoops, L.; et al. JAK inhibition with ruxolitinib versus best available therapy for myelofibrosis. N. Engl. J. Med. 2012, 366, 787–798. [Google Scholar] [CrossRef]
- Verstovsek, S.; Mesa, R.A.; Gotlib, J.; Levy, R.S.; Gupta, V.; DiPersio, J.F.; Catalano, J.V.; Deininger, M.; Miller, C.; Silver, R.T.; et al. A double-blind, placebo-controlled trial of ruxolitinib for myelofibrosis. N. Engl. J. Med. 2012, 366, 799–807. [Google Scholar] [CrossRef]
- Gadina, M.; Johnson, C.; Schwartz, D.; Bonelli, M.; Hasni, S.; Kanno, Y.; Changelian, P.; Laurence, A.; O’Shea, J.J. Translational and clinical advances in JAK-STAT biology: The present and future of jakinibs. J. Leukoc. Biol. 2018, 104, 499–514. [Google Scholar] [CrossRef]
- Gadina, M.; Le, M.T.; Schwartz, D.M.; Silvennoinen, O.; Nakayamada, S.; Yamaoka, K.; O’Shea, J.J. Janus kinases to jakinibs: From basic insights to clinical practice. Rheumatology 2019, 58, 4–16. [Google Scholar] [CrossRef] [PubMed]
- Vainchenker, W.; Leroy, E.; Gilles, L.; Marty, C.; Plo, I.; Constantinescu, S.N. JAK inhibitors for the treatment of myeloproliferative neoplasms and other disorders. F1000Res 2018, 7, 82. [Google Scholar] [CrossRef] [PubMed]
- Virtanen, A.T.; Haikarainen, T.; Raivola, J.; Silvennoinen, O. Selective JAKinibs: Prospects in Inflammatory and Autoimmune Diseases. Biodrugs 2019, 33, 15–32. [Google Scholar] [CrossRef] [PubMed]
- He, X.R.; Chen, X.B.; Zhang, H.C.; Xie, T.; Ye, X.Y. Selective Tyk2 inhibitors as potential therapeutic agents: A patent review (2015-2018). Expert. Opin. Ther. Pat. 2019, 29, 137–149. [Google Scholar] [CrossRef] [PubMed]
- Danese, S.; Argollo, M.; Le Berre, C.; Peyrin-Biroulet, L. JAK selectivity for inflammatory bowel disease treatment: Does it clinically matter? Gut 2019, 68, 1893–1899. [Google Scholar] [CrossRef] [PubMed]
- Papp, K.; Gordon, K.; Thaci, D.; Morita, A.; Gooderham, M.; Foley, P.; Girgis, I.G.; Kundu, S.; Banerjee, S. Phase 2 Trial of Selective Tyrosine Kinase 2 Inhibition in Psoriasis. N. Engl. J. Med. 2018, 379, 1313–1321. [Google Scholar] [CrossRef]
- Burke, J.R.; Cheng, L.; Gillooly, K.M.; Strnad, J.; Zupa-Fernandez, A.; Catlett, I.M.; Zhang, Y.; Heimrich, E.M.; McIntyre, K.W.; Cunningham, M.D.; et al. Autoimmune pathways in mice and humans are blocked by pharmacological stabilization of the TYK2 pseudokinase domain. Sci. Transl. Med. 2019, 11, eaaw1736. [Google Scholar] [CrossRef]
- Liu, C.; Lin, J.; Moslin, R.; Tokarski, J.S.; Muckelbauer, J.; Chang, C.; Tredup, J.; Xie, D.; Park, H.; Li, P.; et al. Identification of Imidazo[1,2-b]pyridazine Derivatives as Potent, Selective, and Orally Active Tyk2 JH2 Inhibitors. ACS Med. Chem. Lett. 2019, 10, 383–388. [Google Scholar] [CrossRef]
- Moslin, R.; Gardner, D.; Santella, J.; Zhang, Y.; Duncia, J.V.; Liu, C.; Lin, J.; Tokarski, J.S.; Strnad, J.; Pedicord, D.; et al. Identification of imidazo[1,2-b] pyridazine TYK2 pseudokinase ligands as potent and selective allosteric inhibitors of TYK2 signalling. Medchemcomm 2017, 8, 700–712. [Google Scholar] [CrossRef]
- Moslin, R.; Zhang, Y.; Wrobleski, S.T.; Lin, S.; Mertzman, M.; Spergel, S.; Tokarski, J.S.; Strnad, J.; Gillooly, K.; McIntyre, K.W.; et al. Identification of N-Methyl Nicotinamide and N-Methyl Pyridazine-3-Carboxamide Pseudokinase Domain Ligands as Highly Selective Allosteric Inhibitors of Tyrosine Kinase 2 (TYK2). J. Med. Chem. 2019, 62, 8953–8972. [Google Scholar] [CrossRef]
- Wrobleski, S.T.; Moslin, R.; Lin, S.; Zhang, Y.; Spergel, S.; Kempson, J.; Tokarski, J.S.; Strnad, J.; Zupa-Fernandez, A.; Cheng, L.; et al. Highly Selective Inhibition of Tyrosine Kinase 2 (TYK2) for the Treatment of Autoimmune Diseases: Discovery of the Allosteric Inhibitor BMS-986165. J. Med. Chem. 2019, 62, 8973–8995. [Google Scholar] [CrossRef] [PubMed]
- Villanueva, M.T. TYK2 inhibition shows promise. Nat. Rev. Drug Discov. 2019, 18, 668. [Google Scholar] [CrossRef] [PubMed]
- Prchal-Murphy, M.; Semper, C.; Lassnig, C.; Wallner, B.; Gausterer, C.; Teppner-Klymiuk, I.; Kobolak, J.; Muller, S.; Kolbe, T.; Karaghiosoff, M.; et al. TYK2 kinase activity is required for functional type I interferon responses in vivo. PLoS ONE 2012, 7, e39141. [Google Scholar] [CrossRef] [PubMed]
- Raje, V.; Derecka, M.; Cantwell, M.; Meier, J.; Szczepanek, K.; Sisler, J.D.; Strobl, B.; Gamero, A.; Harris, T.E.; Larner, A.C. Kinase Inactive Tyrosine Kinase (Tyk2) Supports Differentiation of Brown Fat Cells. Endocrinology 2017, 158, 148–157. [Google Scholar] [CrossRef] [PubMed]
- Lesgidou, N.; Eliopoulos, E.; Goulielmos, G.N.; Vlassi, M. Insights on the alteration of functionality of a tyrosine kinase 2 variant: A molecular dynamics study. Bioinformatics 2018, 34, i781–i786. [Google Scholar] [CrossRef] [PubMed]
- Lupardus, P.J.; Skiniotis, G.; Rice, A.J.; Thomas, C.; Fischer, S.; Walz, T.; Garcia, K.C. Structural snapshots of full-length Jak1, a transmembrane gp130/IL-6/IL-6Ralpha cytokine receptor complex, and the receptor-Jak1 holocomplex. Structure 2011, 19, 45–55. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| TYK2 Status | Disease | Activated STAT | Ref. |
|---|---|---|---|
| Activating somatic mutations (GOF) | |||
| TYK2-G36D; -S47N | T-ALL | STAT1, STAT3 | [53] (3) (2) (2 *) |
| TYK2-731I | T-ALL | STAT1, STAT3, STAT4 | [53] (3) (2) (2 *) |
| TYK2-E957D | T-ALL | STAT1, STAT3, STAT5 | [53] (3) (2) (2 *) |
| TYK2-R1027H | T-ALL | STAT1, STAT3 | [53] (3) (2) (2 *) |
| TYK2-V678F | — | STAT3, STAT5 | [61] (2 *) |
| Inactivating germline mutations (LOF) | |||
| TYK2-P1104A | MPNST | n.d. | [42] (4 *) (1) |
| TYK2-P1104A | Breast-, colon-, stomach-cancer | n.d. | [68] (1) |
| TYK2-P1104V | AML | n.d. | [69] (5) (1) (2 *) |
| Activating germline mutations (GOF) | |||
| TYK2-P760L | B-ALL | STAT1, STAT3, STAT5 | [67] (3) (1) (2 *) |
| TYK2-G761V | T-ALL | STAT1, STAT3, STAT5 | [67] (3) (1) (2 *) |
| Oncogenic fusion proteins (GOF) | |||
| NPM1-TYK2 | CD30-positive LPDs | STAT1, STAT3, STAT5 | [82] (3) (1) (2) (2 *) |
| NFkB2-TYK2 | ALCL | STAT1, STAT3, STAT5 | [80] (3) (1) (2 *) |
| ELAVL1-TYK2 | AML | STAT3, STAT5 | [84] (2) |
| PABPC4-TYK2 | ALCL | n.d. | [80] (1) |
| TEL-TYK2 | — | STAT1, STAT3, STAT5 | [77] (2 *) |
| MYB-TYK2 | Ph-like ALL | n.d. | [81] (1) |
| High wildtype TYK2 levels | |||
| TYK2 WT | T-ALL | STAT1, STAT3, STAT4, STAT5 | [53] (1) (2) (2 *) |
| TYK2 WT | ALCL | STAT1, STAT3 | [52] (4) (1) (2) |
| TYK2 WT | Hepatocarcinoma | STAT1, STAT3 | [102] (3 *) (2 **) |
| TYK2 WT | MPNST | STAT1, STAT3 | [43] (4) (?) (1) (2) |
| TYK2 WT | B-cell lymphoma | STAT3 | [54] (3 *) (2) |
| TYK2 WT | Lung cancer | STAT3 | [103] (3 *) (1) (2) (2 *) |
| TYK2 WT | Hepatocarcinoma | STAT3 | [104] (3 *) (2 **) |
| TYK2 WT | Ovarian cancer | STAT3 | [38] (3 *) (2) |
| TYK2 WT | Prostate cancer | n.d. | [36] (4) (1) (2) |
| TYK2 WT | Prostate cancer | n.d. | [37] (4) (1) (2) |
| TYK2 WT | Osteosarcoma | no | [55] (3 *) (2) |
| TYK2 WT | Breast cancer | n.d. | [40,41] (4) (1) (2) |
| TYK2 WT | Squamous cervical carcinoma | n.d. | [39] (4) (1) |
| TYK2 WT | MPNST | n.d. | [42] (4) (?) (1) |
| TYK2 WT | Lung cancer | STAT1 | [105] (2 **) |
| Low wildtype TYK2 levels | |||
| TYK2 WT | Breast cancer (metastatic) | n.d. | [50] (6) (1) (2) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wöss, K.; Simonović, N.; Strobl, B.; Macho-Maschler, S.; Müller, M. TYK2: An Upstream Kinase of STATs in Cancer. Cancers 2019, 11, 1728. https://doi.org/10.3390/cancers11111728
Wöss K, Simonović N, Strobl B, Macho-Maschler S, Müller M. TYK2: An Upstream Kinase of STATs in Cancer. Cancers. 2019; 11(11):1728. https://doi.org/10.3390/cancers11111728
Chicago/Turabian StyleWöss, Katharina, Natalija Simonović, Birgit Strobl, Sabine Macho-Maschler, and Mathias Müller. 2019. "TYK2: An Upstream Kinase of STATs in Cancer" Cancers 11, no. 11: 1728. https://doi.org/10.3390/cancers11111728
APA StyleWöss, K., Simonović, N., Strobl, B., Macho-Maschler, S., & Müller, M. (2019). TYK2: An Upstream Kinase of STATs in Cancer. Cancers, 11(11), 1728. https://doi.org/10.3390/cancers11111728