AXL Receptor Tyrosine Kinase as a Therapeutic Target in Hematological Malignancies: Focus on Multiple Myeloma

 , , , ,
, , , ,  ,
,
Abstract
1. Introduction
2. Normal Biology of the AXL Receptor
2.1. The Basic Function of AXL and GAS6
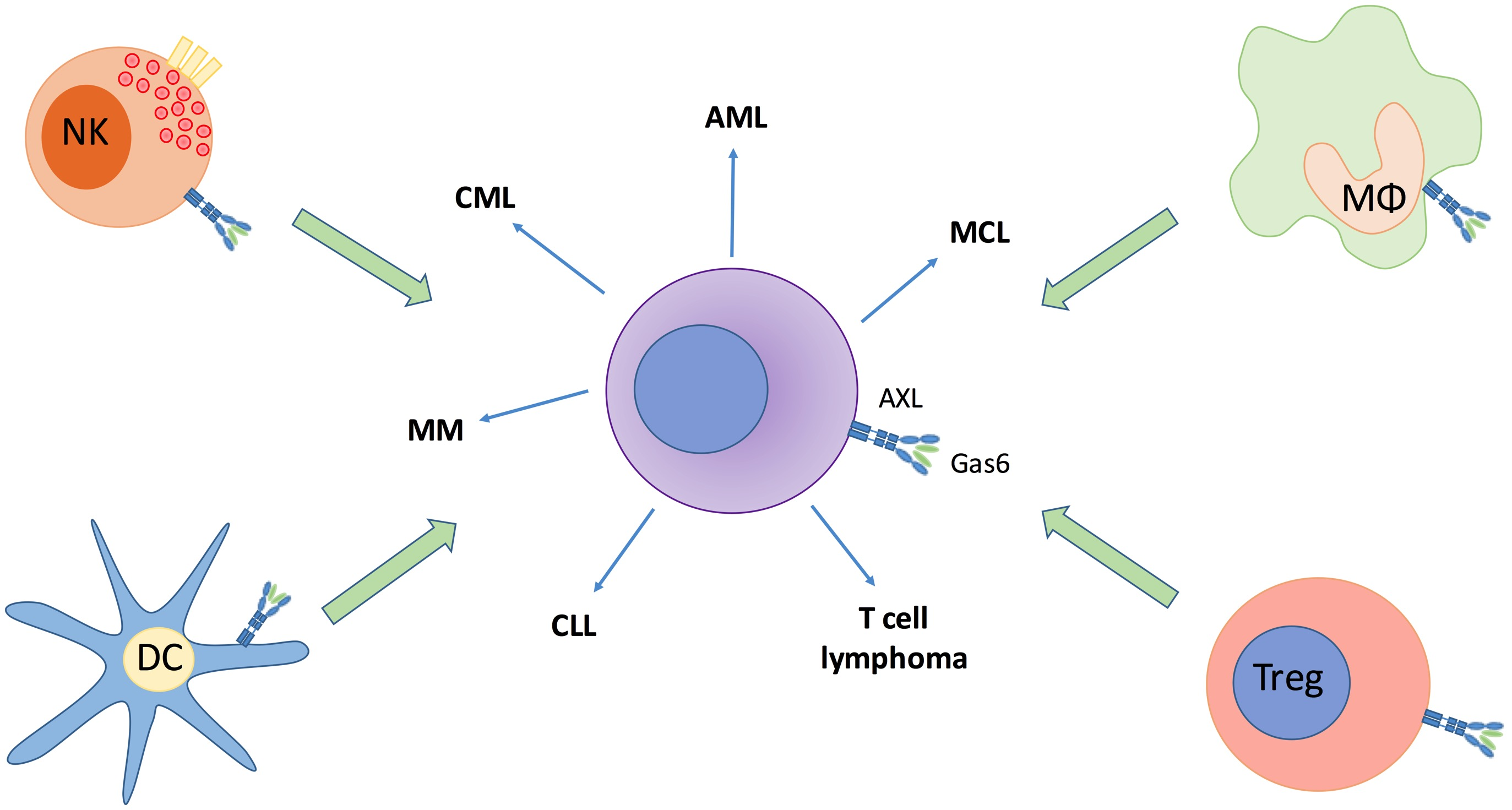
2.2. The Role of AXL in the Immune System
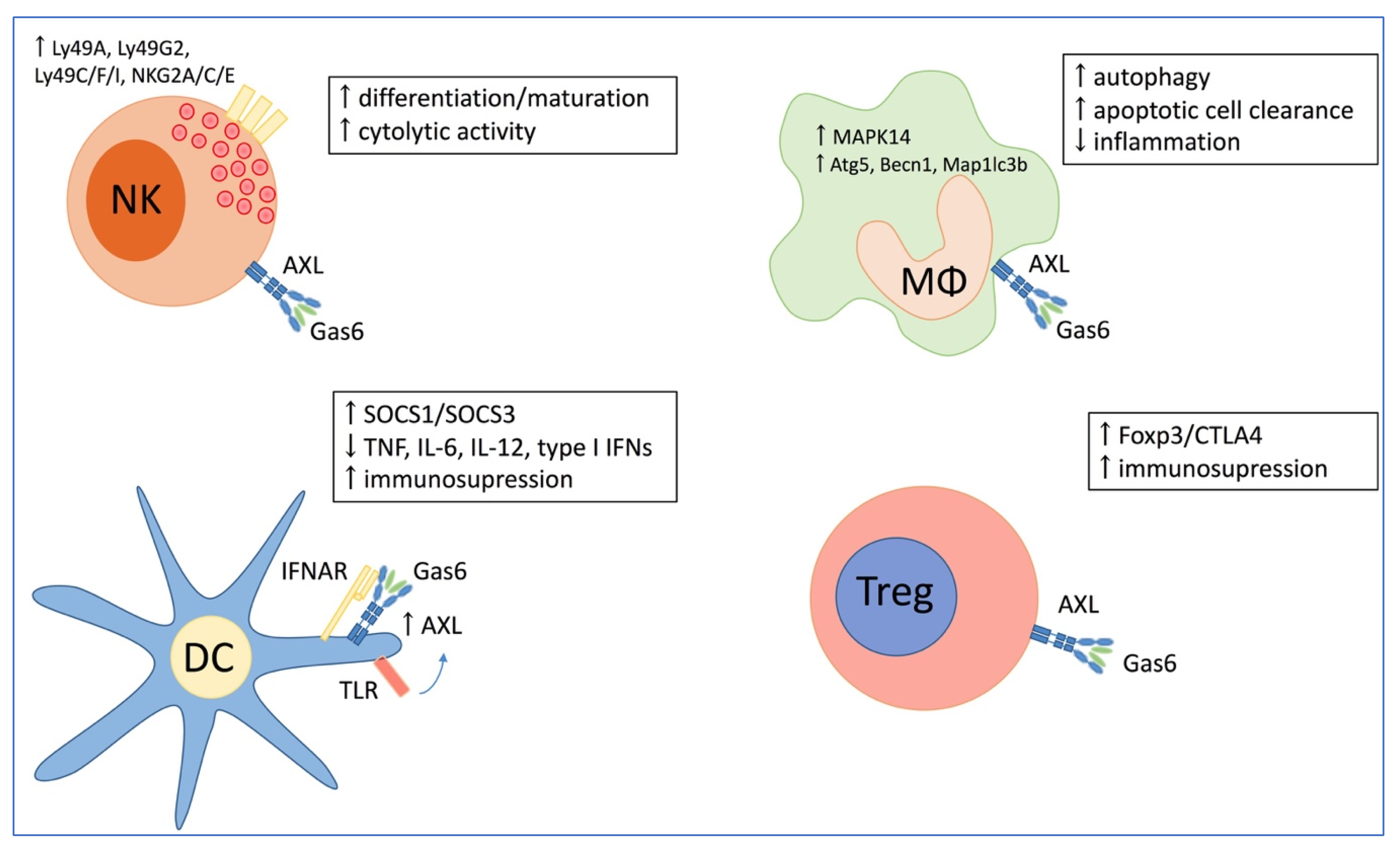
3. The Role of AXL in Solid Cancers
3.1. Direct Effect of AXL on Tumor Growth
3.2. The Role of AXL in Chemoresistance
3.3. Immune Mediated Pro-Tumoral Effect of AXL
4. The Role of AXL in Multiple Myeloma
4.1. Current Treatment Strategies in Multiple Myeloma
4.2. TAM Receptors in Multiple Myeloma
4.3. AXL Expression in Multiple Myeloma
4.4. Clinical Trials Using AXL Inhibitors in Multiple Myeloma
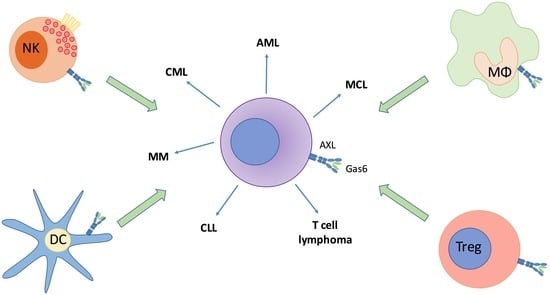
5. The Role of AXL in Other Hematological Cancers
5.1. AXL in Chronic Lymphocytic Leukemia (CLL)
5.2. AXL in Chronic Myeloid Leukemia (CML)
5.3. AXL in Acute Myeloid Leukemia (AML)
5.4. AXL in Mantle Cell Lymphoma (MCL)
5.5. AXL in T Cell Lymphoma
6. Targeting AXL and Its Therapeutic Potential in Hematological Cancers
6.1. Specific AXL Inhibitors
6.2. Multi-Target AXL Inhibitors
7. Conclusions and Future Developments
Author Contributions
Funding
Conflicts of Interest
References
- O’Bryan, J.P.; Frye, R.A.; Cogswell, P.C.; Neubauer, A.; Kitch, B.; Prokop, C.; Espinosa, R., 3rd; Le Beau, M.M.; Earp, H.S.; Liu, E.T. Axl, a transforming gene isolated from primary human myeloid leukemia cells, encodes a novel receptor tyrosine kinase. Mol. Cell. Biol. 1991, 11, 5016–5031. [Google Scholar] [CrossRef] [PubMed]
- Lai, C.; Gore, M.; Lemke, G. Structure, Expression, and Activity of Tyro-3, a Neural Adhesion-Related Receptor Tyrosine Kinase. Oncogene 1994, 9, 2567–2578. [Google Scholar] [PubMed]
- Heiring, C.; Dahlback, B.; Muller, Y.A. Ligand recognition and homophilic interactions in Tyro3: Structural insights into the Axl/Tyro3 receptor tyrosine kinase family. J. Biol. Chem. 2004, 279, 6952–6958. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, T.; Knyazev, P.G.; Clout, N.J.; Cheburkin, Y.; Gohring, W.; Ullrich, A.; Timpl, R.; Hohenester, E. Structural basis for Gas6-Axl signalling. EMBO J. 2006, 25, 80–87. [Google Scholar] [CrossRef]
- Sasaki, T.; Knyazev, P.G.; Cheburkin, Y.; Gohring, W.; Tisi, D.; Ullrich, A.; Timpl, R.; Hohenester, E. Crystal structure of a C-terminal fragment of growth arrest-specific protein Gas6. Receptor tyrosine kinase activation by laminin G-like domains. J. Biol. Chem. 2002, 277, 44164–44170. [Google Scholar] [CrossRef]
- Stitt, T.N.; Conn, G.; Gore, M.; Lai, C.; Bruno, J.; Radziejewski, C.; Mattsson, K.; Fisher, J.; Gies, D.R.; Jones, P.F.; et al. The anticoagulation factor protein S and its relative, Gas6, are ligands for the Tyro 3/Axl family of receptor tyrosine kinases. Cell 1995, 80, 661–670. [Google Scholar] [CrossRef]
- Manfioletti, G.; Brancolini, C.; Avanzi, G.; Schneider, C. The Protein Encoded by a Growth Arrest-Specific Gene (Gas6) Is a New Member of the Vitamin-K-Dependent Proteins Related to Protein-S, a Negative Coregulator in the Blood-Coagulation Cascade. Mol. Cell. Biol. 1993, 13, 4976–4985. [Google Scholar] [CrossRef]
- Avanzi, G.C.; Gallicchio, M.; Cavalloni, G.; Gammaitoni, L.; Leone, F.; Rosina, A.; Boldorini, R.; Monga, G.; Pegoraro, L.; Varnum, B.; et al. GAS6, the ligand of Axl and Rse receptors, is expressed in hematopoietic tissue but lacks mitogenic activity. Exp. Hematol. 1997, 25, 1219–1226. [Google Scholar]
- Nakano, T.; Higashino, K.; Kikuchi, N.; Kishino, J.; Nomura, K.; Fujita, H.; Ohara, O.; Arita, H. Vascular smooth muscle cell-derived, Gla-containing growth-potentiating factor for Ca(2+)-mobilizing growth factors. J. Biol. Chem. 1995, 270, 5702–5705. [Google Scholar] [CrossRef]
- Braunger, J.; Schleithoff, L.; Schulz, A.S.; Kessler, H.; Lammers, R.; Ullrich, A.; Bartram, C.R.; Janssen, J.W. Intracellular signaling of the Ufo/Axl receptor tyrosine kinase is mediated mainly by a multi-substrate docking-site. Oncogene 1997, 14, 2619–2631. [Google Scholar] [CrossRef]
- Cosemans, J.M.; Van Kruchten, R.; Olieslagers, S.; Schurgers, L.J.; Verheyen, F.K.; Munnix, I.C.; Waltenberger, J.; Angelillo-Scherrer, A.; Hoylaerts, M.F.; Carmeliet, P.; et al. Potentiating role of Gas6 and Tyro3, Axl and Mer (TAM) receptors in human and murine platelet activation and thrombus stabilization. J. Thromb. Haemost. 2010, 8, 1797–1808. [Google Scholar] [CrossRef] [PubMed]
- Couchie, D.; Lafdil, F.; Martin-Garcia, N.; Laperche, Y.; Zafrani, E.S.; Mavier, P. Expression and role of Gas6 protein and of its receptor Axl in hepatic regeneration from oval cells in the rat. Gastroenterology 2005, 129, 1633–1642. [Google Scholar] [CrossRef] [PubMed]
- Park, I.K.; Giovenzana, C.; Hughes, T.L.; Yu, J.H.; Trotta, R.; Caligiuri, M.A. The Axl/Gas6 pathway is required for optimal cytokine signaling during human natural killer cell development. Blood 2009, 113, 2470–2477. [Google Scholar] [CrossRef] [PubMed]
- Pierce, A.; Bliesner, B.; Xu, M.; Nielsen-Preiss, S.; Lemke, G.; Tobet, S.; Wierman, M.E. Axl and Tyro3 modulate female reproduction by influencing gonadotropin-releasing hormone neuron survival and migration. Mol. Endocrinol. 2008, 22, 2481–2495. [Google Scholar] [CrossRef]
- Tang, H.; Chen, S.; Wang, H.; Wu, H.; Lu, Q.; Han, D. TAM receptors and the regulation of erythropoiesis in mice. Haematologica 2009, 94, 326–334. [Google Scholar] [CrossRef]
- Rothlin, C.V.; Ghosh, S.; Zuniga, E.I.; Oldstone, M.B.A.; Lemke, G. TAM receptors are pleiotropic inhibitors of the innate immune response. Cell 2007, 131, 1124–1136. [Google Scholar] [CrossRef]
- Caraux, A.; Lu, Q.X.; Fernandez, N.; Riou, S.; Di Santo, J.P.; Raulet, D.H.; Lemke, G.; Roth, C. Natural killer cell differentiation driven by Tyro3 receptor tyrosine kinases. Nat. Immunol. 2006, 7, 747–754. [Google Scholar] [CrossRef]
- Kim, E.M.; Lee, E.H.; Lee, H.Y.; Choi, H.R.; Ji, K.Y.; Kim, S.M.; Kim, K.D.; Kang, H.S. Axl signaling induces development of natural killer cells in vitro and in vivo. Protoplasma 2017, 254, 1091–1101. [Google Scholar] [CrossRef]
- Budagian, V.; Bulanova, E.; Orinska, Z.; Thon, L.; Mamat, U.; Bellosta, P.; Basilico, C.; Adam, D.; Paus, R.; Bulfone-Paus, S. A promiscuous liaison between IL-15 receptor and Axl receptor tyrosine kinase in cell death control (Retracted Article. See vol 30, pg 627, 2011). EMBO J. 2005, 24, 4260–4270. [Google Scholar] [CrossRef]
- Scutera, S.; Fraone, T.; Musso, T.; Cappello, P.; Rossi, S.; Pierobon, D.; Orinska, Z.; Paus, R.; Bulfone-Paus, S.; Giovarelli, M. Survival and Migration of Human Dendritic Cells Are Regulated by an IFN-alpha-Inducible Axl/Gas6 Pathway. J. Immunol. 2009, 183, 3004–3013. [Google Scholar] [CrossRef]
- Han, J.; Bae, J.; Choi, C.Y.; Choi, S.P.; Kang, H.S.; Jo, E.K.; Park, J.; Lee, Y.S.; Moon, H.S.; Park, C.G.; et al. Autophagy induced by AXL receptor tyrosine kinase alleviates acute liver injury via inhibition of NLRP3 inflammasome activation in mice. Autophagy 2016, 12, 2326–2343. [Google Scholar] [CrossRef] [PubMed]
- Fujimori, T.; Grabiec, A.M.; Kaur, M.; Bell, T.J.; Fujino, N.; Cook, P.C.; Svedberg, F.R.; MacDonald, A.S.; Maciewicz, R.A.; Singh, D.; et al. The Axl receptor tyrosine kinase is a discriminator of macrophage function in the inflamed lung. Mucosal Immunol. 2015, 8, 1021–1030. [Google Scholar] [CrossRef] [PubMed]
- Grabiec, A.M.; Goenka, A.; Fife, M.E.; Fujimori, T.; Hussell, T. Axl and MerTK receptor tyrosine kinases maintain human macrophage efferocytic capacity in the presence of viral triggers. Eur. J. Immunol. 2018, 48, 855–860. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.J.; Zheng, J.Y.; Bian, J.L.; Chen, L.W.; Dong, N.; Yu, Y.; Hong, G.L.; Chandoo, A.; Yao, Y.M.; Lu, Z.Q. Growth Arrest-Specific 6 Enhances the Suppressive Function of CD4(+) CD25(+) Regulatory T Cells Mainly through Axl Receptor. Mediat. Inflamm. 2017. [Google Scholar] [CrossRef] [PubMed]
- Paccez, J.D.; Vasques, G.J.; Correa, R.G.; Vasconcellos, J.F.; Duncan, K.; Gu, X.; Bhasin, M.; Libermann, T.A.; Zerbini, L.F. The receptor tyrosine kinase Ax1 is an essential regulator of prostate cancer proliferation and tumor growth and represents a new therapeutic target. Oncogene 2013, 32, 689–698. [Google Scholar] [CrossRef] [PubMed]
- Shiozawa, Y.; Pedersen, E.A.; Patel, L.R.; Ziegler, A.M.; Havens, A.M.; Jung, Y.H.; Wang, J.C.; Zalucha, S.; Loberg, R.D.; Pienta, K.J.; et al. GAS6/AXL Axis Regulates Prostate Cancer Invasion, Proliferation, and Survival in the Bone Marrow Niche. Neoplasia 2010, 12, 116–127. [Google Scholar] [CrossRef]
- Martinelli, E.; Martini, G.; Cardone, C.; Troiani, T.; Liguori, G.; Vitagliano, D.; Napolitano, S.; Morgillo, F.; Rinaldi, B.; Melillo, R.M.; et al. AXL is an oncotarget in human colorectal cancer. Oncotarget 2015, 6, 23281–23296. [Google Scholar] [CrossRef]
- Sawabu, T.; Seno, H.; Kawashima, T.; Fukuda, A.; Uenoyama, Y.; Kawada, M.; Kanda, N.; Sekikawa, A.; Fukui, H.; Yanagita, M.; et al. Growth arrest-specific gene 6 and Axl signaling enhances gastric cancer cell survival via Akt pathway. Mol. Carcinogen. 2007, 46, 155–164. [Google Scholar] [CrossRef]
- Axelrod, H.; Pienta, K.J. Axl as a mediator of cellular growth and survival. Oncotarget 2014, 5, 1–35. [Google Scholar] [CrossRef]
- Woo, S.M.; Min, K.J.; Seo, S.U.; Kim, S.; Kubatka, P.; Park, J.W.; Kwon, T.K. Axl Inhibitor R428 Enhances TRAIL-Mediated Apoptosis Through Downregulation of c-FLIP and Survivin Expression in Renal Carcinoma. Int. J. Mol. Sci. 2019, 20, 3253. [Google Scholar] [CrossRef]
- Goruppi, S.; Chiaruttini, C.; Ruaro, M.E.; Varnum, B.; Schneider, C. Gas6 induces growth, beta-catenin stabilization, and T-cell factor transcriptional activation in contact-inhibited C57 mammary cells. Mol. Cell. Biol. 2001, 21, 902–915. [Google Scholar] [CrossRef] [PubMed]
- Goruppi, S.; Ruaro, E.; Schneider, C. Gas6, the ligand of Axl tyrosine kinase receptor, has mitogenic and survival activities for serum starved NIH3T3 fibroblasts. Oncogene 1996, 12, 471–480. [Google Scholar] [PubMed]
- Goruppi, S.; Ruaro, E.; Varnum, B.; Schneider, C. Requirement of phosphatidylinositol 3-kinase-dependent pathway and Src for Gas6-Axl mitogenic and survival activities in NIH 3T3 fibroblasts. Mol. Cell. Biol. 1997, 17, 4442–4453. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Tian, R.; Yong, B.C.; Luo, C.Q.; Tan, P.X.; Shen, J.N.; Peng, T.S. Gas6/Axl mediates tumor cell apoptosis, migration and invasion and predicts the clinical outcome of osteosarcoma patients. Biochem. Bioph. Res. Commun. 2013, 435, 493–500. [Google Scholar] [CrossRef] [PubMed]
- Vajkoczy, P.; Knyazev, P.; Kunkel, A.; Capelle, H.H.; Behrndt, S.; von Tengg-Kobligk, H.; Kiessling, F.; Eichelsbacher, U.; Essig, M.; Read, T.A.; et al. Dominant-negative inhibition of the Axl receptor tyrosine kinase suppresses brain tumor cell growth and invasion and prolongs survival. Proc. Natl. Acad. Sci. USA 2006, 103, 5799–5804. [Google Scholar] [CrossRef] [PubMed]
- Nakano, T.; Tani, M.; Ishibashi, Y.; Kimura, K.; Park, Y.B.; Imaizumi, N.; Tsuda, H.; Aoyagi, K.; Sasaki, H.; Ohwada, S.; et al. Biological properties and gene expression associated with metastatic potential of human osteosarcoma. Clin. Exp. Metastas. 2003, 20, 665–674. [Google Scholar] [CrossRef] [PubMed]
- Revach, O.Y.; Sandler, O.; Samuels, Y.; Geiger, B. Cross-Talk between Receptor Tyrosine Kinases AXL and ERBB3 Regulates Invadopodia Formation in Melanoma Cells. Cancer Res. 2019, 79, 2634–2648. [Google Scholar] [CrossRef]
- Li, W.J.; Xiong, X.H.; Abdalla, A.; Alejo, S.; Zhu, L.Y.; Lu, F.; Sun, H. HGF-induced formation of the MET-AXL-ELMO2-DOCK180 complex promotes RAC1 activation, receptor clustering, and cancer cell migration and invasion. J. Biol. Chem. 2018, 293, 15397–15418. [Google Scholar] [CrossRef]
- Duan, Y.T.; Luo, L.L.; Qiao, C.X.; Li, X.Y.; Wang, J.; Liu, H.; Zhou, T.T.; Shen, B.F.; Lv, M.; Feng, J.N. A novel human anti-AXL monoclonal antibody attenuates tumour cell migration. Scand. J. Immunol. 2019, 90. [Google Scholar] [CrossRef]
- Hong, C.C.; Lay, J.D.; Huang, J.S.; Cheng, A.L.; Tang, J.L.; Lin, M.T.; Lai, G.M.; Chuang, S.E. Receptor tyrosine kinase AXL is induced by chemotherapy drugs and overexpression of AXL confers drug resistance in acute myeloid leukemia. Cancer Lett. 2008, 268, 314–324. [Google Scholar] [CrossRef]
- Wang, C.; Jin, H.; Wang, N.; Fan, S.; Wang, Y.; Zhang, Y.; Wei, L.; Tao, X.; Gu, D.; Zhao, F.; et al. Gas6/Axl Axis Contributes to Chemoresistance and Metastasis in Breast Cancer through Akt/GSK-3beta/beta-catenin Signaling. Theranostics 2016, 6, 1205–1219. [Google Scholar] [CrossRef] [PubMed]
- Quinn, J.M.; Greenwade, M.M.; Palisoul, M.L.; Opara, G.; Massad, K.; Zhao, P.N.; Beck-Noia, H.; Hagemann, I.S.; Hagemann, A.R.; McCourt, C.K.; et al. Therapeutic Inhibition of the Receptor Tyrosine Kinase AXL Improves Sensitivity to Platinum and Taxane in Ovarian Cancer. Mol. Cancer Ther. 2019, 18, 389–398. [Google Scholar] [CrossRef] [PubMed]
- D’Errico, G.; Alonso-Nocelo, M.; Vallespinos, M.; Hermann, P.C.; Alcala, S.; Garcia, C.P.; Martin-Hijano, L.; Valle, S.; Earl, J.; Cassiano, C.; et al. Tumor-associated macrophage-secreted 14-3-3 zeta signals via AXL to promote pancreatic cancer chemoresistance. Oncogene 2019, 38, 5469–5485. [Google Scholar] [CrossRef] [PubMed]
- Park, I.K.; Mishra, A.; Chandler, J.; Whitman, S.P.; Marcucci, G.; Caligiuri, M.A. Inhibition of the receptor tyrosine kinase Axl impedes activation of the FLT3 internal tandem duplication in human acute myeloid leukemia: Implications for Axl as a potential therapeutic target. Blood 2013, 121, 2064–2073. [Google Scholar] [CrossRef]
- Badarni, M.; Prasad, M.; Balaban, N.; Zorea, J.; Yegodayev, K.M.; Joshua, B.Z.; Dinur, A.B.; Grenman, R.; Rotblat, B.; Cohen, L.; et al. Repression of AXL expression by AP-1/JNK blockage overcomes resistance to PI3Ka therapy. JCI Insight 2019, 5. [Google Scholar] [CrossRef]
- Elkabets, M.; Pazarentzos, E.; Juric, D.; Sheng, Q.; Pelossof, R.A.; Brook, S.; Benzaken, A.O.; Rodon, J.; Morse, N.; Yan, J.J.; et al. AXL mediates resistance to PI3Kalpha inhibition by activating the EGFR/PKC/mTOR axis in head and neck and esophageal squamous cell carcinomas. Cancer Cell 2015, 27, 533–546. [Google Scholar] [CrossRef]
- Brand, T.M.; Iida, M.; Stein, A.P.; Corrigan, K.L.; Braverman, C.M.; Luthar, N.; Toulany, M.; Gill, P.S.; Salgia, R.; Kimple, R.J.; et al. AXL mediates resistance to cetuximab therapy. Cancer Res. 2014, 74, 5152–5164. [Google Scholar] [CrossRef]
- Taniguchi, H.; Yamada, T.; Wang, R.; Tanimura, K.; Adachi, Y.; Nishiyama, A.; Tanimoto, A.; Takeuchi, S.; Araujo, L.H.; Boroni, M.; et al. AXL confers intrinsic resistance to osimertinib and advances the emergence of tolerant cells. Nat. Commun. 2019, 10. [Google Scholar] [CrossRef]
- Zhang, Z.F.; Lee, J.C.; Lin, L.P.; Olivas, V.; Au, V.; LaFramboise, T.; Abdel-Rahman, M.; Wang, X.Q.; Levine, A.D.; Rho, J.K.; et al. Activation of the AXL kinase causes resistance to EGFR-targeted therapy in lung cancer. Nat. Genet. 2012, 44, 852. [Google Scholar] [CrossRef]
- Yumoto, K.; Eber, M.R.; Wang, J.C.; Cackowski, F.C.; Decker, A.M.; Lee, E.; Nobre, A.R.; Aguirre-Ghiso, J.A.; Jung, Y.H.; Taichman, R.S. Axl is required for TGF-beta 2-induced dormancy of prostate cancer cells in the bone marrow. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef]
- Axelrod, H.D.; Valkenburg, K.C.; Amend, S.R.; Hicks, J.L.; Parsana, P.; Torga, G.; DeMarzo, A.M.; Pienta, K.J. AXL Is a Putative Tumor Suppressor and Dormancy Regulator in Prostate Cancer. Mol. Cancer Res. 2019, 17, 356–369. [Google Scholar] [CrossRef] [PubMed]
- Chiu, K.C.; Lee, C.H.; Liu, S.Y.; Chou, Y.T.; Huang, R.Y.; Huang, S.M.; Shieh, Y.S. Polarization of tumor-associated macrophages and Gas6/Axl signaling in oral squamous cell carcinoma. Oral Oncol. 2015, 51, 683–689. [Google Scholar] [CrossRef] [PubMed]
- Lai, S.; Li, R.; Raha, P.; Hu, Y.; Yan, J.; Zhang, H.; Marotta, A.; Zhang, Z. Abstract B148: Activity of the TAM kinase-targeting compound, SLC-391, is mediated by the engagement of the immune system in CT-26 syngeneic mouse model. Mol. Cancer Ther. 2018, 17, B148. [Google Scholar]
- Aguilera, T.A.; Rafat, M.; Castellini, L.; Shehade, H.; Kariolis, M.S.; Hui, A.B.Y.; Stehr, H.; von Eyben, R.; Jiang, D.; Ellies, L.G.; et al. Reprogramming the immunological microenvironment through radiation and targeting Axl. Nat. Commun. 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Tsukita, Y.; Fujino, N.; Miyauchi, E.; Saito, R.; Fujishima, F.; Itakura, K.; Kyogoku, Y.; Okutomo, K.; Yamada, M.; Okazaki, T.; et al. Axl kinase drives immune checkpoint and chemokine signalling pathways in lung adenocarcinomas. Mol. Cancer 2019, 18. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.Q.; Li, Y.; Zhang, D.D.; Ma, J.Y. Axl inhibition induces the antitumor immune response which can be further potentiated by PD-1 blockade in the mouse cancer models. Oncotarget 2017, 8, 89761–89774. [Google Scholar] [CrossRef]
- Yokoyama, Y.; Lew, E.D.; Seelige, R.; Tindall, E.A.; Walsh, C.; Fagan, P.C.; Lee, J.Y.; Nevarez, R.; Oh, J.; Tucker, K.D.; et al. Immuno-oncological Efficacy of RXDX-106, a Novel TAM (TYRO3, AXL, MER) Family Small-Molecule Kinase Inhibitor. Cancer Res. 2019, 79, 1996–2008. [Google Scholar] [CrossRef]
- Kumar, S.K.; Rajkumar, V.; Kyle, R.A.; van Duin, M.; Sonneveld, P.; Mateos, M.V.; Gay, F.; Anderson, K.C. Multiple myeloma. Nat. Rev. Dis. Primers 2017, 3. [Google Scholar] [CrossRef]
- Dhodapkar, M.V. MGUS to myeloma: A mysterious gammopathy of underexplored significance. Blood 2016, 128, 2599–2606. [Google Scholar] [CrossRef]
- Chim, C.S.; Kumar, S.K.; Orlowski, R.Z.; Cook, G.; Richardson, P.G.; Gertz, M.A.; Giralt, S.; Mateos, M.V.; Leleu, X.; Anderson, K.C. Management of relapsed and refractory multiple myeloma: Novel agents, antibodies, immunotherapies and beyond. Leukemia 2018, 32, 252–262. [Google Scholar] [CrossRef]
- Waizenegger, J.S.; Ben-Batalla, I.; Weinhold, N.; Meissner, T.; Wroblewski, M.; Janning, M.; Riecken, K.; Binder, M.; Atanackovic, D.; Taipaleenmaeki, H.; et al. Role of Growth arrest-specific gene 6-Mer axis in multiple myeloma. Leukemia 2015, 29, 696–704. [Google Scholar] [CrossRef] [PubMed]
- Engelmann, J.; Ben Batalla, I.; Taipaleenmaki, H.; Riecken, K.; Gensch, V.; Paesler, S.; Berenbrok, N.; Vargas, M.E.; Waizenegger, J.; Fehse, B.; et al. Blockade of Mer By the Small Molecule Inhibitor R992 Inhibits Multiple Myeloma and Its Associated Bone Disease By Restoring the Perturbed Bone Homeostasis. Blood 2018, 132. [Google Scholar] [CrossRef]
- Khoo, W.H.; Ledergor, G.; Weiner, A.; Roden, D.L.; Terry, R.L.; McDonald, M.M.; Chai, R.C.; De Veirman, K.; Owen, K.L.; Opperman, K.S.; et al. A niche-dependent myeloid transcriptome signature defines dormant myeloma cells. Blood 2019, 134, 30–43. [Google Scholar] [CrossRef] [PubMed]
- Lendvai, N.; Yee, A.J.; Tsakos, I.; Alexander, A.; Devlin, S.M.; Hassoun, H.; Korde, N.; Lesokhin, A.M.; Landau, H.; Mailankody, S.; et al. Phase IB study of cabozantinib in patients with relapsed and/or refractory multiple myeloma. Blood 2016, 127, 2355–2356. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.K.; Secreto, C.; Boysen, J.; Sassoon, T.; Shanafelt, T.D.; Mukhopadhyay, D.; Kay, N.E. The novel receptor tyrosine kinase Axl is constitutively active in B-cell chronic lymphocytic leukemia and acts as a docking site of nonreceptor kinases: Implications for therapy. Blood 2011, 117, 1928–1937. [Google Scholar] [CrossRef] [PubMed]
- Sinha, S.; Boysen, J.; Nelson, M.; Warner, S.L.; Bearss, D.; Kay, N.E.; Ghosh, A.K. Axl activates fibroblast growth factor receptor pathway to potentiate survival signals in B-cell chronic lymphocytic leukemia cells. Leukemia 2016, 30, 1431–1436. [Google Scholar] [CrossRef] [PubMed]
- Sinha, S.; Boysen, J.; Nelson, M.; Secreto, C.; Warner, S.L.; Bearss, D.J.; Lesnick, C.; Shanafelt, T.D.; Kay, N.E.; Ghosh, A.K. Targeted Axl Inhibition Primes Chronic Lymphocytic Leukemia B Cells to Apoptosis and Shows Synergistic/Additive Effects in Combination with BTK Inhibitors. Clin. Cancer Res. 2015, 21, 2115–2126. [Google Scholar] [CrossRef]
- Boysen, J.; Sinha, S.; Price-Troska, T.; Warner, S.L.; Bearss, D.J.; Viswanatha, D.; Shanafelt, T.D.; Kay, N.E.; Ghosh, A.K. The tumor suppressor axis p53/miR-34a regulates Axl expression in B-cell chronic lymphocytic leukemia: Implications for therapy in p53-defective CLL patients. Leukemia 2014, 28, 451–455. [Google Scholar] [CrossRef]
- Sinha, S.; Boysen, J.C.; Chaffee, K.G.; Kabat, B.F.; Slager, S.L.; Parikh, S.A.; Secreto, C.R.; Call, T.; Shanafelt, T.D.; Leis, J.F.; et al. Chronic lymphocytic leukemia cells from ibrutinib treated patients are sensitive to Axl receptor tyrosine kinase inhibitor therapy. Oncotarget 2018, 9, 37173–37184. [Google Scholar] [CrossRef]
- Patel, V.; Keating, M.J.; Wierda, W.G.; Gandhi, V. Preclinical combination of TP-0903, an AXL inhibitor and B-PAC-1, a procaspase-activating compound with ibrutinib in chronic lymphocytic leukemia. Leuk. Lymphoma 2016, 57, 1494–1497. [Google Scholar] [CrossRef]
- Ben-Batalla, I.; Erdmann, R.; Jorgensen, H.; Mitchell, R.; Ernst, T.; Von Amsberg, G.; Schafhausen, P.; Velthaus, J.L.; Rankin, S.; Clark, R.E.; et al. Axl Blockade by BGB324 Inhibits BCR-ABL Tyrosine Kinase Inhibitor-Sensitive and -Resistant Chronic Myeloid Leukemia. Clin. Cancer Res. 2017, 23, 2289–2300. [Google Scholar] [CrossRef]
- Gioia, R.; Tregoat, C.; Dumas, P.Y.; Lagarde, V.; Prouzet-Mauleon, V.; Desplat, V.; Sirvent, A.; Praloran, V.; Lippert, E.; Villacreces, A.; et al. CBL controls a tyrosine kinase network involving AXL, SYK and LYN in nilotinib-resistant chronic myeloid leukaemia. J. Pathol. 2015, 237, 14–24. [Google Scholar] [CrossRef]
- Sodaro, G.; Blasio, G.; Fiorentino, F.; Auberger, P.; Costanzo, P.; Cesaro, E. ZNF224 is a transcriptional repressor of AXL in chronic myeloid leukemia cells. Biochimie 2018, 154, 127–131. [Google Scholar] [CrossRef]
- Jin, Y.L.; Nie, D.N.; Li, J.; Du, X.; Lu, Y.H.; Li, Y.Q.; Liu, C.; Zhou, J.F.; Pan, J.X. Gas6/AXL Signaling Regulates Self-Renewal of Chronic Myelogenous Leukemia Stem Cells by Stabilizing beta-Catenin. Clin. Cancer Res. 2017, 23, 2842–2855. [Google Scholar] [CrossRef]
- Neubauer, A.; Fiebeler, A.; Graham, D.K.; Obryan, J.P.; Schmidt, C.A.; Barckow, P.; Serke, S.; Siegert, W.; Snodgrass, H.R.; Huhn, D.; et al. Expression of Axl, a Transforming Receptor Tyrosine Kinase, in Normal and Malignant Hematopoiesis. Blood 1994, 84, 1931–1941. [Google Scholar] [CrossRef]
- Rochlitz, C.; Lohri, A.; Bacchi, M.; Schmidt, M.; Nagel, S.; Fopp, M.; Fey, M.F.; Herrmann, R.; Neubauer, A. Axl expression is associated with adverse prognosis and with expression of Bcl-2 and CD34 in de novo acute myeloid leukemia (AML): Results from a multicenter trial of the Swiss Group for Clinical Cancer Research (SAKK). Leukemia 1999, 13, 1352–1358. [Google Scholar] [CrossRef]
- Ben-Batalla, I.; Schultze, A.; Wroblewski, M.; Erdmann, R.; Heuser, M.; Waizenegger, J.S.; Riecken, K.; Binder, M.; Schewe, D.; Sawall, S.; et al. Axl, a prognostic and therapeutic target in acute myeloid leukemia mediates paracrine crosstalk of leukemia cells with bone marrow stroma. Blood 2013, 122, 2443–2452. [Google Scholar] [CrossRef]
- Whitman, S.P.; Kohlschmidt, J.; Maharry, K.; Volinia, S.; Mrozek, K.; Nicolet, D.; Schwind, S.; Becker, H.; Metzeler, K.H.; Mendler, J.H.; et al. GAS6 expression identifies high-risk adult AML patients: Potential implications for therapy. Leukemia 2014, 28, 1252–1258. [Google Scholar] [CrossRef]
- Gilliland, D.G.; Griffin, J.D. The roles of FLT3 in hematopoiesis and leukemia. Blood 2002, 100, 1532–1542. [Google Scholar] [CrossRef]
- Dumas, P.Y.; Naudin, C.; Martin-Lanneree, S.; Izac, B.; Casetti, L.; Mansier, O.; Rousseau, B.; Artus, A.; Dufossee, M.; Giese, A.; et al. Hematopoietic niche drives FLT3-ITD acute myeloid leukemia resistance to quizartinib via STAT5-and hypoxia-dependent upregulation of AXL. Haematologica 2019, 104, 2017–2027. [Google Scholar] [CrossRef]
- Gelebart, P.; Han, J.; Karlsen, I.; Gjerstad, M.E.; Helgeland, L.; Baran-Marszak, F.; Papp, B.; Cormack, E.M. The AXL tyrosine kinase inhibitor, BGB324, induces cell death in mantle cell lymphoma. In Proceedings of the EHA23 Library, Stockholm, Sweden, 14–17 June 2018; p. PF635. [Google Scholar]
- Sakemura, R.; Yang, N.; Cox, M.J.; Sinha, S.; Hefazi, M.; Hansen, M.J.; Schick, K.J.; Forsman, C.L.; Boysen, J.C.; Tschumper, R.C.; et al. Axl-RTK Inhibition Modulates T Cell Functions and Synergizes with Chimeric Antigen Receptor T Cell Therapy in B Cell Malignancies. Blood 2018, 132. [Google Scholar] [CrossRef]
- Lee, E.H.; Kim, E.M.; Ji, K.Y.; Park, A.R.; Choi, H.R.; Lee, H.Y.; Kim, S.M.; Chung, B.Y.; Park, C.H.; Choi, H.J.; et al. Axl acts as a tumor suppressor by regulating LIGHT expression in T lymphoma. Oncotarget 2017, 8, 20645–20655. [Google Scholar] [CrossRef] [PubMed]
- Sheridan, C. First Axl inhibitor enters clinical trials. Nat. Biotechnol. 2013, 31, 775–776. [Google Scholar] [CrossRef]
- Holland, S.J.; Pan, A.; Franci, C.; Hu, Y.M.; Chang, B.; Li, W.Q.; Duan, M.; Torneros, A.; Yu, J.X.; Heckrodt, T.J.; et al. R428, a Selective Small Molecule Inhibitor of Axl Kinase, Blocks Tumor Spread and Prolongs Survival in Models of Metastatic Breast Cancer. Cancer Res. 2010, 70, 1544–1554. [Google Scholar] [CrossRef]
- Chen, F.F.; Song, Q.L.; Yu, Q. Axl inhibitor R428 induces apoptosis of cancer cells by blocking lysosomal acidification and recycling independent of Axl inhibition. Am. J. Cancer Res. 2018, 8, 1466. [Google Scholar]
- Loges, S.; Gjertsen, B.T.; Heuser, M.; Ben-Batalla, I.; Micklem, D.; Jorg, C.; Kebenko, M.; Fiedler, W.M.; Cortes, J.E. A first-in-patient phase I study of BGB324, a selective Axl kinase inhibitor in patients with refractory/relapsed AML and high-risk MDS. J. Clin. Oncol. 2016, 34. [Google Scholar] [CrossRef]
- Jeon, J.Y.; Park, I.K.; Buelow, D.R.; Whatcott, C.; Warner, S.L.; Blum, W.; Baker, S. TP-0903, a Novel Axl Inhibitor with Activity in Drug Resistant FLT3-ITD+ AML through a Mechanism That Includes FLT3 Inhibition. Blood 2017, 130, 2522. [Google Scholar]
- Kariolis, M.S.; Miao, Y.R.; Ii, D.S.J.; Kapur, S.; Mathews, I.I.; Giaccia, A.J.; Cochran, J.R. An engineered Axl ‘decoy receptor’ effectively silences the Gas6-Axl signaling axis. Nat. Chem. Biol. 2014, 10, 977–983. [Google Scholar] [CrossRef]
- Kariolis, M.S.; Miao, Y.R.; Diep, A.; Nash, S.E.; Olcina, M.M.; Jiang, D.; Jones, D.S.; Kapur, S.; Mathews, I.I.; Koong, A.C.; et al. Inhibition of the GAS6/AXL pathway augments the efficacy of chemotherapies. J. Clin. Investig. 2017, 127, 183–198. [Google Scholar] [CrossRef]
- Dhillon, S. Gilteritinib: First Global Approval. Drugs 2019, 79, 331–339. [Google Scholar] [CrossRef]
- Yan, S.B.; Peek, V.L.; Ajamie, R.; Buchanan, S.G.; Graff, J.R.; Heidler, S.A.; Hui, Y.H.; Huss, K.L.; Konicek, B.W.; Manro, J.R.; et al. LY2801653 is an orally bioavailable multi-kinase inhibitor with potent activity against MET, MST1R, and other oncoproteins, and displays anti-tumor activities in mouse xenograft models. Invest. New Drugs 2013, 31, 833–844. [Google Scholar] [CrossRef]
- Kosciuczuk, E.M.; Saleiro, D.; Kroczynska, B.; Beauchamp, E.M.; Eckerdt, F.; Blyth, G.T.; Abedin, S.M.; Giles, F.J.; Altman, J.K.; Platanias, L.C. Merestinib blocks Mnk kinase activity in acute myeloid leukemia progenitors and exhibits antileukemic effects in vitro and in vivo. Blood 2016, 128, 410–414. [Google Scholar] [CrossRef]
- Elisei, R.; Schlumberger, M.J.; Muller, S.P.; Schoffski, P.; Brose, M.S.; Shah, M.H.; Licitra, L.; Jarzab, B.; Medvedev, V.; Kreissl, M.C.; et al. Cabozantinib in Progressive Medullary Thyroid Cancer. J. Clin. Oncol. 2013, 31, 3639. [Google Scholar] [CrossRef]
- Lu, J.W.; Wang, A.N.; Liao, H.A.; Chen, C.Y.; Hou, H.A.; Hu, C.Y.; Tien, H.F.; Ou, D.L.; Lin, L.I. Cabozantinib is selectively cytotoxic in acute myeloid leukemia cells with FLT3-internal tandem duplication (FLT3-ITD). Cancer Lett. 2016, 376, 218–225. [Google Scholar] [CrossRef]
- Fathi, A.T.; Blonquist, T.M.; Hernandez, D. Cabozantinib is well tolerated in acute myeloid leukemia and effectively inhibits the resistance-conferring FLT3/tyrosine kinase domain/F691 mutation (vol 124, pg 306, 2018). Cancer Am. Cancer Soc. 2018, 124, 2258. [Google Scholar] [CrossRef]
- Shen, Y.Y.; Chen, X.G.; He, J.; Liao, D.F.; Zu, X.Y. Axl inhibitors as novel cancer therapeutic agents. Life Sci. 2018, 198, 99–111. [Google Scholar] [CrossRef]
- Vouri, M.; An, Q.; Birt, M.; Pilkington, G.J.; Hafizi, S. Small molecule inhibition of Axl receptor tyrosine kinase potently suppresses multiple malignant properties of glioma cells. Oncotarget 2015, 6, 16183–16197. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Drug | Targets | IC50 AXL/MERTK | Disease | Phase |
|---|---|---|---|---|
| TP-0903 | Axl [70,97] (Aurr A and B, Jak2, Alk, Abl) | 27 nM/− | Previously Treated CLL | I/II |
| BGB324 | Axl [85,98] (Abl, Mertk, Tyro3, Her-2, EGFR, InsR, PDGFR-β) | 14 nM/− | AML or MDS | Ib/II |
| MYD1-72 | Axl [90] | 0.7 nM/− | AML | Preclinical |
| Merestinib | Met/Tek/ROS/Axl/DDR1/2/Flt3 [92,93] | 11 nM/2 nM | Relapsed/Refractory AML | I |
| Cabozantinib | VEGFR2, Flt3, Met, KIT, Ret, and Axl [64] | 7 nM/1.3 nM | Relapsed/Refractory AML Refractory MM | I/II |
| Gilteritinib | Flt3/Axl [93] | 0.73 nM/− | AML (FLT3 mutated/relapsed/refractory/newly diagnosed) | I/II/III |
| BMS777607 | Met, Ron, Axl, Tyro3 [93] | 1.1 nM/3.9 nM | MM | Preclinical |
| XL880 | Met, Axl, Vegfr2, Pdgfrβ and Tie2 [44] | 11 nM/0.4 nM | AML | Preclinical |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yan, S.; Vandewalle, N.; De Beule, N.; Faict, S.; Maes, K.; De Bruyne, E.; Menu, E.; Vanderkerken, K.; De Veirman, K. AXL Receptor Tyrosine Kinase as a Therapeutic Target in Hematological Malignancies: Focus on Multiple Myeloma. Cancers 2019, 11, 1727. https://doi.org/10.3390/cancers11111727
Yan S, Vandewalle N, De Beule N, Faict S, Maes K, De Bruyne E, Menu E, Vanderkerken K, De Veirman K. AXL Receptor Tyrosine Kinase as a Therapeutic Target in Hematological Malignancies: Focus on Multiple Myeloma. Cancers. 2019; 11(11):1727. https://doi.org/10.3390/cancers11111727
Chicago/Turabian StyleYan, Siyang, Niels Vandewalle, Nathan De Beule, Sylvia Faict, Ken Maes, Elke De Bruyne, Eline Menu, Karin Vanderkerken, and Kim De Veirman. 2019. "AXL Receptor Tyrosine Kinase as a Therapeutic Target in Hematological Malignancies: Focus on Multiple Myeloma" Cancers 11, no. 11: 1727. https://doi.org/10.3390/cancers11111727
APA StyleYan, S., Vandewalle, N., De Beule, N., Faict, S., Maes, K., De Bruyne, E., Menu, E., Vanderkerken, K., & De Veirman, K. (2019). AXL Receptor Tyrosine Kinase as a Therapeutic Target in Hematological Malignancies: Focus on Multiple Myeloma. Cancers, 11(11), 1727. https://doi.org/10.3390/cancers11111727