STAT5A and STAT5B—Twins with Different Personalities in Hematopoiesis and Leukemia

Abstract
1. Introduction
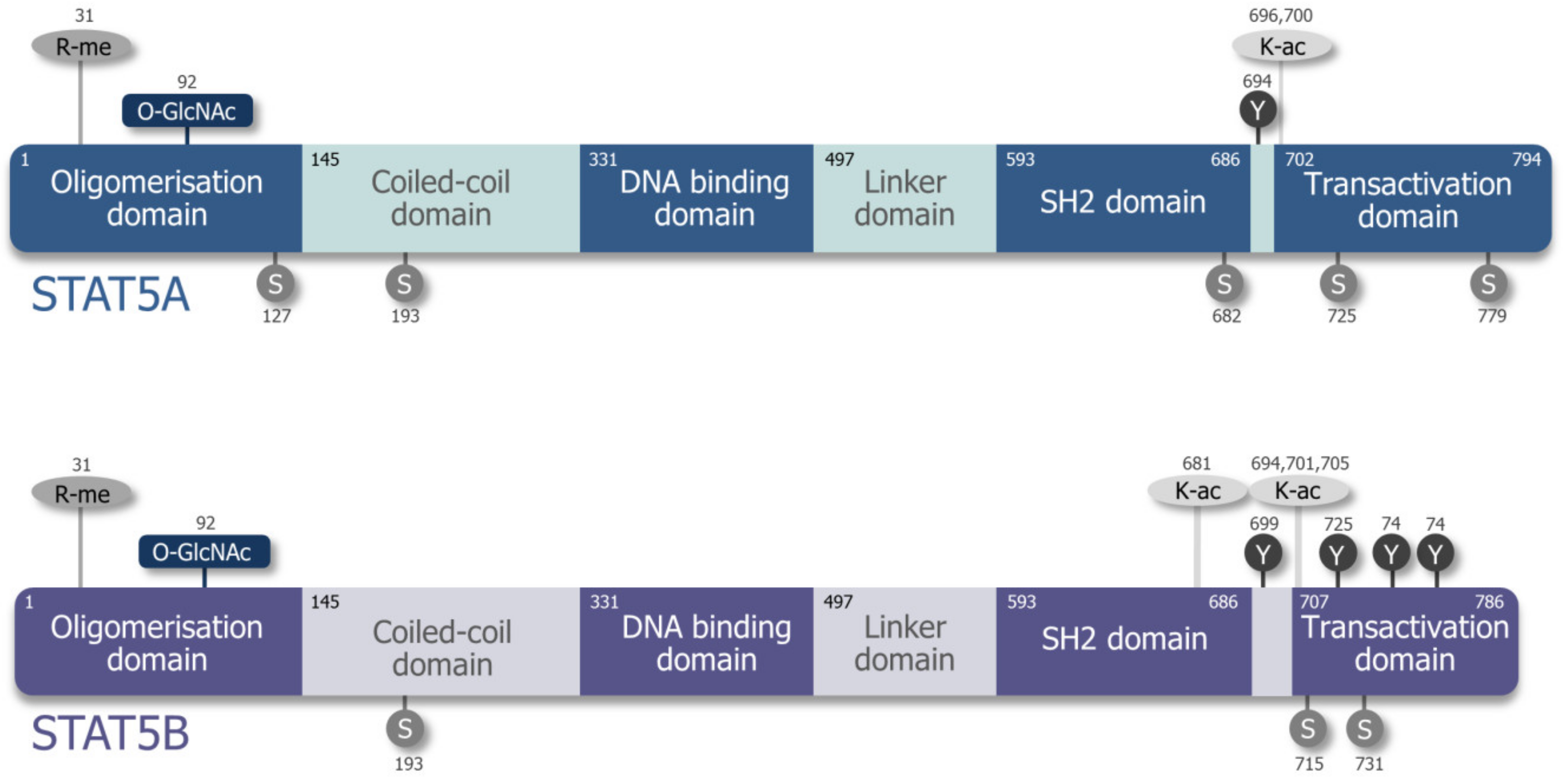
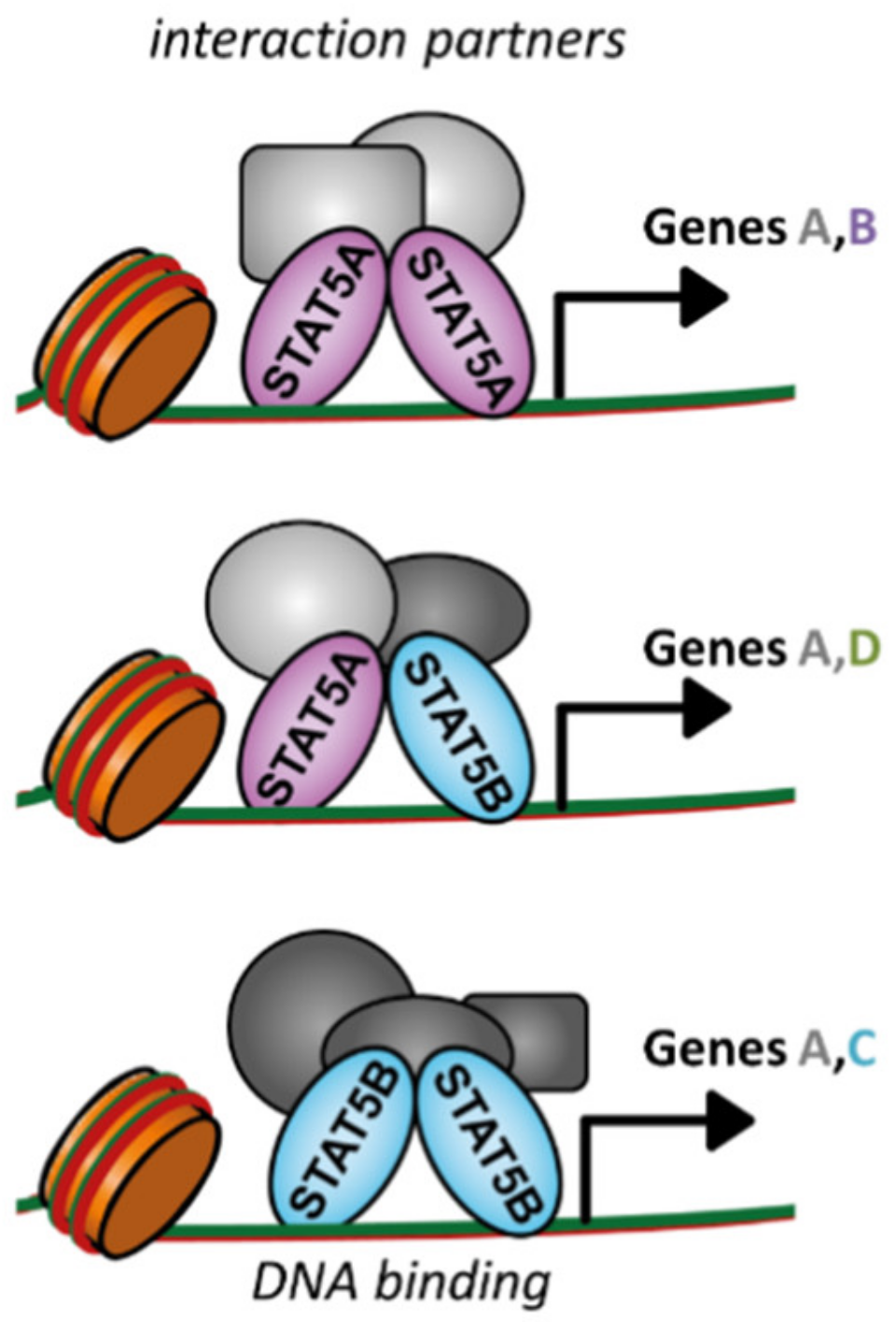
2. Differences and Similarities of STAT5A and STAT5B
3. STAT5A/B Deficiency in Mice and Men
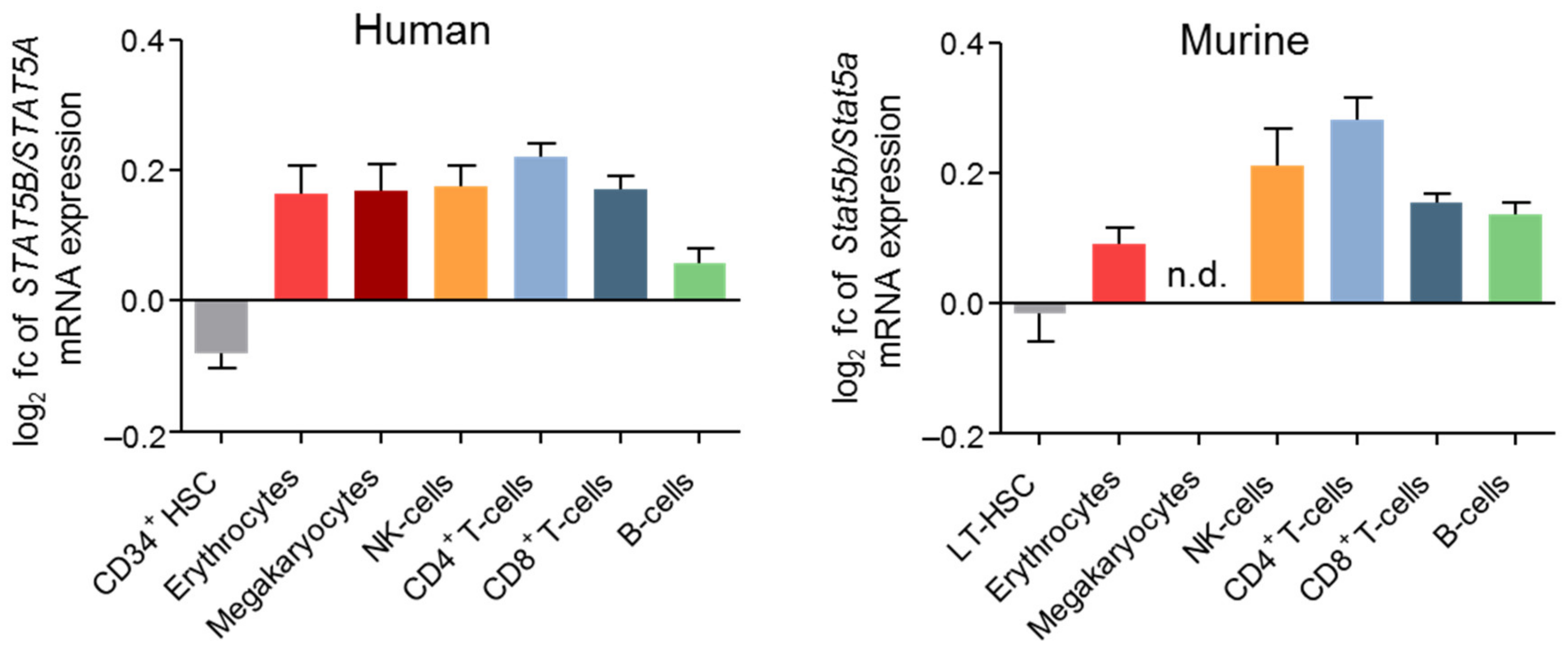
4. STAT5B as the Dominant Player in Hematopoietic Lineages
4.1. STAT5A/B as Regulators of Erythropoiesis
4.2. Megakaryopoiesis—Non-Canonical STAT5A/B Prevent Differentiation
4.3. STAT5A/B Promote Survival and Differentiation of B Cells
4.4. STAT5B is the Major Player in NK Cells
4.5. CD8+ T Cells are Sensitive to Elevated pYSTAT5A/B Levels
4.6. CD4+ T Cell Development—Quantities Matter
5. STAT5A/B are Required for Hematopoietic Stem Cell Maintenance and Self-Renewal
6. STAT5A/B as Oncogenes in Hematopoietic Cancer
6.1. STAT5A and STAT5B Mutations as Disease Drivers
6.2. STAT5B—The Major Player Downstream of BCR–ABL
6.3. STAT5A/B as Potential Opponents in NPM–ALK+ Lymphoma
7. Direct STAT5A/B Inhibition Remains Challenging
8. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Villarino, A.V.; Kanno, Y.; Ferdinand, J.R.; O’Shea, J.J. Mechanisms of Jak/STAT Signaling in Immunity and Disease. J. Immunol. 2015, 194, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Morris, R.; Kershaw, N.J.; Babon, J.J. The molecular details of cytokine signalling via the JAK/STAT pathway. Protein Sci. 2018, 27, 1984–2009. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Dutta, P.; Tsurumi, A.; Li, J.; Wang, J.; Land, H.; Li, W.X. Unphosphorylated STAT5A stabilizes heterochromatin and suppresses tumor growth. Proc. Natl. Acad. Sci. USA 2013, 110, 10213–10218. [Google Scholar] [CrossRef]
- Comoglio, F.; Park, H.J.; Schoenfelder, S.; Barozzi, I.; Bode, D.; Fraser, P.; Green, A.R. Thrombopoietin signaling to chromatin elicits rapid and pervasive epigenome remodeling within poised chromatin architectures. Genome Res. 2018, 28, 295–309. [Google Scholar] [CrossRef] [PubMed]
- Park, H.J.; Li, J.; Hannah, R.; Biddie, S.; Leal-Cervantes, A.I.; Kirschner, K.; Flores Santa Cruz, D.; Sexl, V.; Göttgens, B.; Green, A.R. Cytokine-induced megakaryocytic differentiation is regulated by genome-wide loss of a uSTAT transcriptional program. EMBO J. 2016, 35, 580–594. [Google Scholar] [CrossRef] [PubMed]
- Groner, B.; von Manstein, V. Jak Stat signaling and cancer: Opportunities, benefits and side effects of targeted inhibition. Mol. Cell. Endocrinol. 2017, 451, 1–14. [Google Scholar] [CrossRef]
- Wang, Y.; Levy, D.E. Comparative evolutionary genomics of the STAT family of transcription factors. JAK-STAT 2012, 1, 23–36. [Google Scholar] [CrossRef]
- Liongue, C.; Ward, A.C. Evolution of the JAK-STAT pathway. JAK-STAT 2013, 2, e22756. [Google Scholar] [CrossRef]
- Liu, X.; Robinson, G.W.; Wagner, K.U.; Garrett, L.; Wynshaw-Boris, A.; Hennighausen, L. Stat5a is mandatory for adult mammary gland development and lactogenesis. Genes Dev. 1997, 11, 179–186. [Google Scholar] [CrossRef]
- Liongue, C.; O’Sullivan, L.A.; Trengove, M.C.; Ward, A.C. Evolution of JAK-STAT Pathway Components: Mechanisms and Role in Immune System Development. PLoS ONE 2012, 7, e32777. [Google Scholar] [CrossRef]
- Lewis, R.S.; Ward, A.C. Conservation, duplication and divergence of the zebrafish stat5 genes. Gene 2004, 338, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Xiong, S.; Mei, J.; Huang, P.; Jing, J.; Li, Z.; Kang, J.; Gui, J.F. Essential roles of stat5.1/stat5b in controlling fish somatic growth. J. Genet. Genom. 2017, 44, 577–585. [Google Scholar] [CrossRef] [PubMed]
- Paffett-Lugassy, N.; Hsia, N.; Fraenkel, P.G.; Paw, B.; Leshinsky, I.; Barut, B.; Bahary, N.; Caro, J.; Handin, R.; Zon, L.I. Functional conservation of erythropoietin signaling in zebrafish. Blood 2007, 110, 2718–2726. [Google Scholar] [CrossRef] [PubMed]
- Wakao, H.; Gouilleux, F.; Groner, B. Mammary gland factor (MGF) is a novel member of the cytokine regulated transcription factor gene family and confers the prolactin response. EMBO J. 1994, 13, 2182–2191. [Google Scholar] [CrossRef] [PubMed]
- Schmitt-Ney, M.; Doppler, W.; Ball, R.K.; Groner, B. Beta-casein gene promoter activity is regulated by the hormone-mediated relief of transcriptional repression and a mammary-gland-specific nuclear factor. Mol. Cell. Biol. 1991, 11, 3745–3755. [Google Scholar] [CrossRef]
- Wakao, H.; Harada, N.; Kitamura, T.; Mui, A.L.; Miyajima, A. Interleukin 2 and erythropoietin activate STAT5/MGF via distinct pathways. EMBO J. 1995, 14, 2527–2535. [Google Scholar] [CrossRef]
- Mui, A.L.; Wakao, H.; O’Farrell, A.M.; Harada, N.; Miyajima, A. Interleukin-3, granulocyte-macrophage colony stimulating factor and interleukin-5 transduce signals through two STAT5 homologs. EMBO J. 1995, 14, 1166–1175. [Google Scholar] [CrossRef]
- Gouilleux, F.; Wakao, H.; Mundt, M.; Groner, B. Prolactin induces phosphorylation of Tyr694 of Stat5 (MGF), a prerequisite for DNA binding and induction of transcription. EMBO J. 1994, 13, 4361–4369. [Google Scholar] [CrossRef]
- Liu, K.D.; Lai, S.Y.; Goldsmith, M.A.; Greene, W.C. Identification of a Variable Region within the Cytoplasmic Tail of the IL-2 Receptor β Chain That Is Required for Growth Signal Transduction. J. Biol. Chem. 1995, 270, 22176–22181. [Google Scholar] [CrossRef]
- Miyoshi, K.; Cui, Y.; Riedlinger, G.; Robinson, P.; Lehoczky, J.; Zon, L.; Oka, T.; Dewar, K.; Hennighausen, L. Structure of the Mouse Stat 3/5 Locus: Evolution from Drosophila to Zebrafish to Mouse. Genomics 2001, 71, 150–155. [Google Scholar] [CrossRef]
- Moriggl, R.; Sexl, V.; Kenner, L.; Duntsch, C.; Stangl, K.; Gingras, S.; Hoffmeyer, A.; Bauer, A.; Piekorz, R.; Wang, D.; et al. Stat5 tetramer formation is associated with leukemogenesis. Cancer Cell 2005, 7, 87–99. [Google Scholar] [CrossRef]
- Kisseleva, T.; Bhattacharya, S.; Braunstein, J.; Schindler, C.W. Signaling through the JAK/STAT pathway, recent advances and future challenges. Gene 2002, 285, 1–24. [Google Scholar] [CrossRef]
- Kornfeld, J.W.; Grebien, F.; Kerenyi, M.A.; Friedbichler, K.; Kovacic, B.; Zankl, B.; Hoelbl, A.; Nivarti, H.; Beug, H.; Sexl, V.; et al. The different functions of Stat5 and chromatin alteration through Stat5 proteins. Front. Biosci. 2008, 13, 6237–6254. [Google Scholar] [CrossRef] [PubMed]
- Grimley, P.M.; Dong, F.; Rui, H. Stat5a and Stat5b: Fraternal twins of signal transduction and transcriptional activation. Cytokine Growth Factor Rev. 1999, 10, 131–157. [Google Scholar] [CrossRef]
- Boucheron, C.; Dumon, S.; Santos, S.C.R.; Moriggl, R.; Hennighausen, L.; Gisselbrecht, S.; Gouilleux, F. A Single Amino Acid in the DNA Binding Regions of STAT5A and STAT5B Confers Distinct DNA Binding Specificities. J. Biol. Chem. 1998, 273, 33936–33941. [Google Scholar] [CrossRef]
- Lin, J.X.; Mietz, J.; Modi, W.S.; John, S.; Leonard, W.J. Cloning of Human Stat5B: RECONSTITUTION OF INTERLEUKIN-2-INDUCED Stat5A AND Stat5B DNA BINDING ACTIVITY IN COS-7 CELLS. J. Biol. Chem. 1996, 271, 10738–10744. [Google Scholar] [CrossRef]
- Kanai, T.; Seki, S.; Jenks, J.; Kohli, A.; Kawli, T.; Martin, D.; Snyder, M.; Baccetta, R.; Nadeau, K. Identification of STATA and STAT5B target genes in human T cells. PLoS ONE 2014, 9, e86790. [Google Scholar] [CrossRef]
- Basham, B.; Sathe, M.; Grein, J.; McClanahan, T.; D‘Andrea, A.; Lees, E.; Rascle, A. In vivo identification of novel STAT5 target genes. Nucleic Acids Res. 2008, 36, 3802–3818. [Google Scholar] [CrossRef]
- Meinke, A.; Barahmand-Pour, F.; Wöhrl, S.; Stoiber, D.; Decker, T. Activation of different Stat5 isoforms contributes to cell-type-restricted signaling in response to interferons. Mol. Cell. Biol. 1996, 16, 6937–6944. [Google Scholar] [CrossRef]
- Boehm, M.E.; Adlung, L.; Schilling, M.; Roth, S.; Klingmüller, U.; Lehmann, W.D. Identification of Isoform-Specific Dynamics in Phosphorylation-Dependent STAT5 Dimerization by Quantitative Mass Spectrometry and Mathematical Modeling. J. Proteome Res. 2014, 13, 5685–5694. [Google Scholar] [CrossRef]
- Imada, K.; Leonard, W.J. The Jak-STAT pathway. Mol. Immunol. 2000, 37, 1–11. [Google Scholar] [CrossRef]
- Berger, A.; Hoelbl-Kovacic, A.; Bourgeais, J.; Hoefling, L.; Warsch, W.; Grundschober, E.; Uras, I.Z.; Menzl, I.; Putz, E.M.; Hoermann, G.; et al. PAK-dependent STAT5 serine phosphorylation is required for BCR-ABL-induced leukemogenesis. Leukemia 2014, 28, 629–641. [Google Scholar] [CrossRef] [PubMed]
- Friedbichler, K.; Kerenyi, M.A.; Kovacic, B.; Li, G.; Hoelbl, A.; Yahiaoui, S.; Sexl, V.; Mullner, E.W.; Fajmann, S.; Cerny-Reiterer, S.; et al. Stat5a serine 725 and 779 phosphorylation is a prerequisite for hematopoietic transformation. Blood 2010, 116, 1548–1558. [Google Scholar] [CrossRef] [PubMed]
- Clark, D.E.; Williams, C.C.; Duplessis, T.T.; Moring, K.L.; Notwick, A.R.; Long, W.; Lane, W.S.; Beuvink, I.; Hynes, N.E.; Jones, F.E. ERBB4/HER4 potentiates STAT5A transcriptional activity by regulating novel STAT5A serine phosphorylation events. J. Biol. Chem. 2005, 280, 24175–24180. [Google Scholar] [CrossRef] [PubMed]
- Schaller-Schonitz, M.; Barzan, D.; Williamson, A.J.; Griffiths, J.R.; Dallmann, I.; Battmer, K.; Ganser, A.; Whetton, A.D.; Scherr, M.; Eder, M. BCR-ABL affects STAT5A and STAT5B differentially. PLoS ONE 2014, 9, e97243. [Google Scholar] [CrossRef]
- Mitra, A.; Ross, J.A.; Rodriguez, G.; Nagy, Z.S.; Wilson, H.L.; Kirken, R.A. Signal transducer and activator of transcription 5b (Stat5b) serine 193 is a novel cytokine-induced phospho-regulatory site that is constitutively activated in primary hematopoietic malignancies. J. Biol. Chem. 2012, 287, 16596–16608. [Google Scholar] [CrossRef]
- Bartalucci, N.; Calabresi, L.; Balliu, M.; Martinelli, S.; Rossi, M.C.; Villeval, J.L.; Annunziato, F.; Guglielmelli, P.; Vannucchi, A.M. Inhibitors of the PI3K/mTOR pathway prevent STAT5 phosphorylation in JAK2V617F mutated cells through PP2A/CIP2A axis. Oncotarget 2017, 8, 96710–96724. [Google Scholar] [CrossRef]
- Kabotyanski, E.B.; Rosen, J.M. Signal transduction pathways regulated by prolactin and Src result in different conformations of activated Stat5b. J. Biol. Chem. 2003, 278, 17218–17227. [Google Scholar] [CrossRef]
- Fox, E.M.; Bernaciak, T.M.; Wen, J.; Weaver, A.M.; Shupnik, M.A.; Silva, C.M. Signal transducer and activator of transcription 5b, c-Src, and epidermal growth factor receptor signaling play integral roles in estrogen-stimulated proliferation of estrogen receptor-positive breast cancer cells. Mol. Endocrinol. 2008, 22, 1781–1796. [Google Scholar] [CrossRef][Green Version]
- Weaver, A.M.; Silva, C.M. S731 in the transactivation domain modulates STAT5b activity. Biochem. Biophys. Res. Commun. 2007, 362, 1026–1030. [Google Scholar] [CrossRef][Green Version]
- Bancerek, J.; Poss, Z.C.; Steinparzer, I.; Sedlyarov, V.; Pfaffenwimmer, T.; Mikulic, I.; Dölken, L.; Strobl, B.; Müller, M.; Taatjes, D.J.; et al. CDK8 kinase phosphorylates transcription factor STAT1 to selectively regulate the interferon response. Immunity 2013, 38, 250–262. [Google Scholar] [CrossRef] [PubMed]
- Beuvink, I.; Hess, D.; Flotow, H.; Hofsteenge, J.; Groner, B.; Hynes, N.E. Stat5a Serine Phosphorylation: SERINE 779 IS CONSTITUTIVELY PHOSPHORYLATED IN THE MAMMARY GLAND, AND SERINE 725 PHOSPHORYLATION INFLUENCES PROLACTIN-STIMULATEDIN VITRO DNA BINDING ACTIVITY. J. Biol. Chem. 2000, 275, 10247–10255. [Google Scholar] [CrossRef] [PubMed]
- Weaver, A.M.; Silva, C.M. Modulation of Signal Transducer and Activator of Transcription 5b Activity in Breast Cancer Cells by Mutation of Tyrosines within the Transactivation Domain. Mol. Endocrinol. 2006, 20, 2392–2405. [Google Scholar] [CrossRef] [PubMed]
- Kloth, M.T.; Catling, A.D.; Silva, C.M. Novel Activation of STAT5b in Response to Epidermal Growth Factor. J. Biol. Chem. 2002, 277, 8693–8701. [Google Scholar] [CrossRef] [PubMed]
- Van Nguyen, T.; Angkasekwinai, P.; Dou, H.; Lin, F.M.; Lu, L.S.; Cheng, J.; Chin, Y.E.; Dong, C.; Yeh, E.T. SUMO-specific protease 1 is critical for early lymphoid development through regulation of STAT5 activation. Mol. Cell 2012, 45, 210–221. [Google Scholar] [CrossRef]
- Yamamoto, M.; Iguchi, G.; Fukuoka, H.; Suda, K.; Bando, H.; Takahashi, M.; Nishizawa, H.; Seino, S.; Takahashi, Y. SIRT1 regulates adaptive response of the growth hormone--insulin-like growth factor-I axis under fasting conditions in liver. Proc. Natl. Acad. Sci. USA 2013, 110, 14948–14953. [Google Scholar] [CrossRef]
- Kuwabara, T.; Kasai, H.; Kondo, M. Acetylation Modulates IL-2 Receptor Signaling in T Cells. J. Immunol. 2016, 197, 4334–4343. [Google Scholar] [CrossRef]
- Freund, P.; Kerenyi, M.A.; Hager, M.; Wagner, T.; Wingelhofer, B.; Pham, H.T.T.; Elabd, M.; Han, X.; Valent, P.; Gouilleux, F.; et al. O-GlcNAcylation of STAT5 controls tyrosine phosphorylation and oncogenic transcription in STAT5-dependent malignancies. Leukemia 2017, 31, 2132. [Google Scholar] [CrossRef]
- Shi, S.; Larson, K.; Guo, D.; Lim, S.J.; Dutta, P.; Yan, S.J.; Li, W.X. Drosophila STAT is required for directly maintaining HP1 localization and heterochromatin stability. Nat. Cell Biol. 2008, 10, 489–496. [Google Scholar] [CrossRef]
- Szybinski, J. Role of Unphosphorylated STAT5 in Maintenance of Acute Myeloid Leukemia Cells; Johannes-Gutenberg-Universität Mainz: Mainz, Germany, 2019. [Google Scholar]
- Wingelhofer, B.; Neubauer, H.A.; Valent, P.; Han, X.; Constantinescu, S.N.; Gunning, P.T.; Müller, M.; Moriggl, R. Implications of STAT3 and STAT5 signaling on gene regulation and chromatin remodeling in hematopoietic cancer. Leukemia 2018, 32, 1713–1726. [Google Scholar] [CrossRef]
- Hoelbl, A.; Kovacic, B.; Kerenyi, M.A.; Simma, O.; Warsch, W.; Cui, Y.; Beug, H.; Hennighausen, L.; Moriggl, R.; Sexl, V. Clarifying the role of Stat5 in lymphoid development and Abelson-induced transformation. Blood 2006, 107, 4898–4906. [Google Scholar] [CrossRef] [PubMed]
- Sexl, V.; Piekorz, R.; Moriggl, R.; Rohrer, J.; Brown, M.P.; Bunting, K.D.; Rothammer, K.; Roussel, M.F.; Ihle, J.N. Stat5a/b contribute to interleukin 7–induced B-cell precursor expansion, but abl-andbcr/abl-induced transformation are independent of Stat5. Blood 2000, 96, 2277–2283. [Google Scholar] [CrossRef] [PubMed]
- Goetz, C.A.; Harmon, I.R.; O’Neil, J.J.; Burchill, M.A.; Farrar, M.A. STAT5 Activation Underlies IL7 Receptor-Dependent B Cell Development. J. Immunol. 2004, 172, 4770–4778. [Google Scholar] [CrossRef]
- Kang, J.; DiBenedetto, B.; Narayan, K.; Zhao, H.; Der, S.D.; Chambers, C.A. STAT5 Is Required for Thymopoiesis in a Development Stage-Specific Manner. J. Immunol. 2004, 173, 2307–2314. [Google Scholar] [CrossRef]
- Teglund, S.; McKay, C.; Schuetz, E.; van Deursen, J.M.; Stravopodis, D.; Wang, D.; Brown, M.; Bodner, S.; Grosveld, G.; Ihle, J.N. Stat5a and Stat5b Proteins Have Essential and Nonessential, or Redundant, Roles in Cytokine Responses. Cell 1998, 93, 841–850. [Google Scholar] [CrossRef]
- Lin, J.X.; Li, P.; Liu, D.; Jin, H.T.; He, J.; Ata Ur Rasheed, M.; Rochman, Y.; Wang, L.; Cui, K.; Liu, C.; et al. Critical Role of STAT5 transcription factor tetramerization for cytokine responses and normal immune function. Immunity 2012, 36, 586–599. [Google Scholar] [CrossRef]
- Moriggl, R.; Topham, D.J.; Teglund, S.; Sexl, V.; McKay, C.; Wang, D.; Hoffmeyer, A.; van Deursen, J.; Sangster, M.Y.; Bunting, K.D.; et al. Stat5 Is Required for IL-2-Induced Cell Cycle Progression of Peripheral T Cells. Immunity 1999, 10, 249–259. [Google Scholar] [CrossRef]
- Cui, Y.; Riedlinger, G.; Miyoshi, K.; Tang, W.; Li, C.; Deng, C.X.; Robinson, G.W.; Hennighausen, L. Inactivation of Stat5 in Mouse Mammary Epithelium during Pregnancy Reveals Distinct Functions in Cell Proliferation, Survival, and Differentiation. Mol. Cell. Biol. 2004, 24, 8037–8047. [Google Scholar] [CrossRef]
- Wang, Z.; Medrzycki, M.; Bunting, S.T.; Bunting, K.D. Stat5-deficient hematopoiesis is permissive for Myc-induced B-cell leukemogenesis. Oncotarget 2015, 6, 28961–28972. [Google Scholar] [CrossRef]
- Zhu, B.M.; McLaughlin, S.K.; Na, R.; Liu, J.; Cui, Y.; Martin, C.; Kimura, A.; Robinson, G.W.; Andrews, N.C.; Hennighausen, L. Hematopoietic-specific Stat5-null mice display microcytic hypochromic anemia associated with reduced transferrin receptor gene expression. Blood 2008, 112, 2071–2080. [Google Scholar] [CrossRef]
- Wang, Z.; Li, G.; Tse, W.; Bunting, K.D. Conditional deletion of STAT5 in adult mouse hematopoietic stem cells causes loss of quiescence and permits efficient nonablative stem cell replacement. Blood 2009, 113, 4856–4865. [Google Scholar] [CrossRef] [PubMed]
- Udy, G.B.; Towers, R.P.; Snell, R.G.; Wilkins, R.J.; Park, S.H.; Ram, P.A.; Waxman, D.J.; Davey, H.W. Requirement of STAT5b for sexual dimorphism of body growth rates and liver gene expression. Proc. Natl. Acad. Sci. USA 1997, 94, 7239–7244. [Google Scholar] [CrossRef] [PubMed]
- Kerenyi, M.A.; Grebien, F.; Gehart, H.; Schifrer, M.; Artaker, M.; Kovacic, B.; Beug, H.; Moriggl, R.; Müllner, E.W. Stat5 regulates cellular iron uptake of erythroid cells via IRP-2 and TfR-1. Blood 2008, 112, 3878–3888. [Google Scholar] [CrossRef] [PubMed]
- Yao, Z.; Cui, Y.; Watford, W.T.; Bream, J.H.; Yamaoka, K.; Hissong, B.D.; Li, D.; Durum, S.K.; Jiang, Q.; Bhandoola, A.; et al. Stat5a/b are essential for normal lymphoid development and differentiation. Proc. Natl. Acad. Sci. USA 2006, 103, 1000–1005. [Google Scholar] [CrossRef] [PubMed]
- Villarino, A.; Laurence, A.; Robinson, G.W.; Bonelli, M.; Dema, B.; Afzali, B.; Shih, H.Y.; Sun, H.W.; Brooks, S.R.; Hennighausen, L.; et al. Signal transducer and activator of transcription 5 (STAT5) paralog dose governs T cell effector and regulatory functions. eLife 2016, 5, e08384. [Google Scholar] [CrossRef]
- Bernasconi, A.; Marino, R.; Ribas, A.; Rossi, J.; Ciaccio, M.; Oleastro, M.; Ornani, A.; Paz, R.; Rivarola, M.A.; Zelazko, M.; et al. Characterization of Immunodeficiency in a Patient With Growth Hormone Insensitivity Secondary to a Novel STAT5b Gene Mutation. Pediatrics 2006, 118, e1584–e1592. [Google Scholar] [CrossRef]
- Cohen, A.C.; Nadeau, K.C.; Tu, W.; Hwa, V.; Dionis, K.; Bezrodnik, L.; Teper, A.; Gaillard, M.; Heinrich, J.; Krensky, A.M.; et al. Cutting Edge: Decreased Accumulation and Regulatory Function of CD4 + CD25high T Cells in Human STAT5b Deficiency. J. Immunol. 2006, 177, 2770–2774. [Google Scholar] [CrossRef]
- Kofoed, E.M.; Hwa, V.; Little, B.; Woods, K.A.; Buckway, C.K.; Tsubaki, J.; Pratt, K.L.; Bezrodnik, L.; Jasper, H.; Tepper, A.; et al. Growth Hormone Insensitivity Associated with a STAT5b Mutation. N. Engl. J. Med. 2003, 349, 1139–1147. [Google Scholar] [CrossRef]
- Bezrodnik, L.; Di Giovanni, D.; Caldirola, M.S.; Azcoiti, M.E.; Torgerson, T.; Gaillard, M.I. Long-Term Follow-up of STAT5B Deficiency in Three Argentinian Patients: Clinical and Immunological Features. J. Clin. Immunol. 2015, 35, 264–272. [Google Scholar] [CrossRef]
- Hwa, V. STAT5B deficiency: Impacts on human growth and immunity. Growth Horm. IGF Res. 2016, 28, 16–20. [Google Scholar] [CrossRef]
- Klammt, J.; Neumann, D.; Gevers, E.F.; Andrew, S.F.; Schwartz, I.D.; Rockstroh, D.; Colombo, R.; Sanchez, M.A.; Vokurkova, D.; Kowalczyk, J.; et al. Dominant-negative STAT5B mutations cause growth hormone insensitivity with short stature and mild immune dysregulation. Nat. Commun. 2018, 9, 2105. [Google Scholar] [CrossRef] [PubMed]
- Nivarthi, H.; Friedbichler, K.; Moriggl, R. Stat5 as a Hematopoietic Master Regulator for Differentiation and Neoplasia Development. In Jak-Stat Signaling: From Basics to Disease; Decker, T., Mueller, M., Eds.; Springer: Vienna, Austria, 2012; pp. 153–167. [Google Scholar]
- Villarino, A.V.; Kanno, Y.; O’Shea, J.J. Mechanisms and consequences of Jak–STAT signaling in the immune system. Nat. Immunol. 2017, 18, 374. [Google Scholar] [CrossRef] [PubMed]
- Rani, A.; Murphy, J.J. STAT5 in Cancer and Immunity. J. Interferon Cytokine Res. 2015, 36, 226–237. [Google Scholar] [CrossRef]
- Vainchenker, W.; Constantinescu, S.N. JAK/STAT signaling in hematological malignancies. Oncogene 2012, 32, 2601–2613. [Google Scholar] [CrossRef]
- Bagger, F.O.; Sasivarevic, D.; Sohi, S.H.; Laursen, L.G.; Pundhir, S.; Sønderby, C.K.; Winther, O.; Rapin, N.; Porse, B.T. BloodSpot: A database of gene expression profiles and transcriptional programs for healthy and malignant haematopoiesis. Nucleic Acids Res. 2015, 44, D917–D924. [Google Scholar] [CrossRef]
- Gillinder, K.R.; Tuckey, H.; Bell, C.C.; Magor, G.W.; Huang, S.; Ilsley, M.D.; Perkins, A.C. Direct targets of pSTAT5 signalling in erythropoiesis. PLoS ONE 2017, 12, e0180922. [Google Scholar] [CrossRef]
- Perreault, A.A.; Venters, B.J. Integrative view on how erythropoietin signaling controls transcription patterns in erythroid cells. Curr. Opin. Hematol. 2018, 25, 189–195. [Google Scholar] [CrossRef]
- Grebien, F.; Kerenyi, M.A.; Kovacic, B.; Kolbe, T.; Becker, V.; Dolznig, H.; Pfeffer, K.; Klingmüller, U.; Müller, M.; Beug, H.; et al. Stat5 activation enables erythropoiesis in the absence of EpoR and Jak2. Blood 2008, 111, 4511–4522. [Google Scholar] [CrossRef]
- Olthof, S.G.; Fatrai, S.; Drayer, A.L.; Tyl, M.R.; Vellenga, E.; Schuringa, J.J. Downregulation of Signal Transducer and Activator of Transcription 5 (STAT5) in CD34+ Cells Promotes Megakaryocytic Development, Whereas Activation of STAT5 Drives Erythropoiesis. Stem Cells 2008, 26, 1732–1742. [Google Scholar] [CrossRef]
- Ogilvy, S.; Metcalf, D.; Gibson, L.; Bath, M.L.; Harris, A.W.; Adams, J.M. Promoter Elements of vav Drive Transgene Expression In Vivo Throughout the Hematopoietic Compartment. Blood 1999, 94, 1855–1863. [Google Scholar] [CrossRef] [PubMed]
- Maurer, B.; Nivarthi, H.; Wingelhofer, B.; Pham, H.T.T.; Schlederer, M.; Suske, T.; Grausenburger, R.; Schiefer, A.I.; Prchal-Murphy, M.; Chen, D.; et al. High activation of STAT5A drives peripheral T-cell lymphoma and leukemia. Haematologica 2019. [Google Scholar] [CrossRef] [PubMed]
- Pham, H.T.T.; Maurer, B.; Prchal-Murphy, M.; Grausenburger, R.; Grundschober, E.; Javaheri, T.; Nivarthi, H.; Boersma, A.; Kolbe, T.; Elabd, M.; et al. STAT5BN642H is a driver mutation for T cell neoplasia. J. Clin. Investig. 2018, 128, 387–401. [Google Scholar] [CrossRef] [PubMed]
- Kirito, K.; Watanabe, T.; Sawada, K.I.; Endo, H.; Ozawa, K.; Komatsu, N. Thrombopoietin Regulates Bcl-xL Gene Expression through Stat5 and Phosphatidylinositol 3-Kinase Activation Pathways. J. Biol. Chem. 2002, 277, 8329–8337. [Google Scholar] [CrossRef] [PubMed]
- Malin, S.; McManus, S.; Cobaleda, C.; Novatchkova, M.; Delogu, A.; Bouillet, P.; Strasser, A.; Busslinger, M. Role of STAT5 in controlling cell survival and immunoglobulin gene recombination during pro-B cell development. Nat. Immunol. 2010, 11, 171–179. [Google Scholar] [CrossRef]
- Malin, S.; McManus, S.; Busslinger, M. STAT5 in B cell development and leukemia. Curr. Opin. Immunol. 2010, 22, 168–176. [Google Scholar] [CrossRef]
- Burchill, M.A.; Goetz, C.A.; Prlic, M.; O’Neil, J.J.; Harmon, I.R.; Bensinger, S.J.; Turka, L.A.; Brennan, P.; Jameson, S.C.; Farrar, M.A. Distinct Effects of STAT5 Activation on CD4+ and CD8+ T Cell Homeostasis: Development of CD4+CD25+ Regulatory T Cells versus CD8+ Memory T Cells. J. Immunol. 2003, 171, 5853–5864. [Google Scholar] [CrossRef]
- Katerndahl, C.D.S.; Heltemes-Harris, L.M.; Willette, M.J.L.; Henzler, C.M.; Frietze, S.; Yang, R.; Schjerven, H.; Silverstein, K.A.T.; Ramsey, L.B.; Hubbard, G.; et al. Antagonism of B cell enhancer networks by STAT5 drives leukemia and poor patient survival. Nat. Immunol. 2017, 18, 694–704. [Google Scholar] [CrossRef]
- Gotthardt, D.; Sexl, V. STATs in NK-Cells: The Good, the Bad, and the Ugly. Front. Immunol. 2017, 7, 694. [Google Scholar] [CrossRef] [PubMed]
- Vargas-Hernández, A.; Forbes, L.R. JAK/STAT proteins and their biological impact on NK cell development and function. Mol. Immunol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Vargas-Hernández, A.; Witalisz-Siepracka, A.; Prchal-Murphy, M.; Klein, K.; Mahapatra, S.; Al-Herz, W.; Mace, E.M.; Carisey, A.F.; Orange, J.S.; Sexl, V.; et al. Human STAT5b mutation causes dysregulated human natural killer cell maturation and impaired lytic function. J. Allergy Clin. Immunol. 2019, S0091674919312527. [Google Scholar] [CrossRef]
- Eckelhart, E.; Warsch, W.; Zebedin, E.; Simma, O.; Stoiber, D.; Kolbe, T.; Rülicke, T.; Mueller, M.; Casanova, E.; Sexl, V. A novel Ncr1-Cre mouse reveals the essential role of STAT5 for NK-cell survival and development. Blood 2011, 117, 1565–1573. [Google Scholar] [CrossRef] [PubMed]
- Witalisz-Siepracka, A.; Klein, K.; Prinz, D.; Leidenfrost, N.; Schabbauer, G.; Dohnal, A.; Sexl, V. Loss of JAK1 Drives Innate Immune Deficiency. Front. Immunol. 2019, 9, 3108. [Google Scholar] [CrossRef] [PubMed]
- Gotthardt, D.; Putz, E.M.; Grundschober, E.; Prchal-Murphy, M.; Straka, E.; Kudweis, P.; Heller, G.; Bago-Horvath, Z.; Witalisz-Siepracka, A.; Cumaraswamy, A.A.; et al. STAT5 Is a Key Regulator in NK Cells and Acts as a Molecular Switch from Tumor Surveillance to Tumor Promotion. Cancer Discov. 2016, 6, 414–429. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.X.; Du, N.; Li, P.; Kazemian, M.; Gebregiorgis, T.; Spolski, R.; Leonard, W.J. Critical functions for STAT5 tetramers in the maturation and survival of natural killer cells. Nat. Commun. 2017, 8, 1320. [Google Scholar] [CrossRef]
- Villarino, A.V.; Sciumè, G.; Davis, F.P.; Iwata, S.; Zitti, B.; Robinson, G.W.; Hennighausen, L.; Kanno, Y.; O’Shea, J.J. Subset- and tissue-defined STAT5 thresholds control homeostasis and function of innate lymphoid cells. J. Exp. Med. 2017, 214, 2999–3014. [Google Scholar] [CrossRef]
- Imada, K.; Bloom, E.T.; Nakajima, H.; Horvath-Arcidiacono, J.A.; Udy, G.B.; Davey, H.W.; Leonard, W.J. Stat5b Is Essential for Natural Killer Cell–mediated Proliferation and Cytolytic Activity. J. Exp. Med. 1998, 188, 2067–2074. [Google Scholar] [CrossRef]
- Lorenzini, T.; Dotta, L.; Giacomelli, M.; Vairo, D.; Badolato, R. STAT mutations as program switchers: Turning primary immunodeficiencies into autoimmune diseases. J. Leukoc. Biol. 2017, 101, 29–38. [Google Scholar] [CrossRef]
- Chehboun, S.; Leiva-Torres, G.A.; Charbonneau, B.; Eveleigh, R.; Bourque, G.; Vidal, S.M. A point mutation in the linker domain of mouse STAT5A is associated with impaired NK-cell regulation. Genes Immun. 2019. [CrossRef]
- Heltemes-Harris, L.M.; Farrar, M.A. The role of STAT5 in lymphocyte development and transformation. Curr. Opin. Immunol. 2012, 24, 146–152. [Google Scholar] [CrossRef]
- Ermakova, O.; Piszczek, L.; Luciani, L.; Cavalli, F.M.; Ferreira, T.; Farley, D.; Rizzo, S.; Paolicelli, R.C.; Al-Banchaabouchi, M.; Nerlov, C.; et al. Sensitized phenotypic screening identifies gene dosage sensitive region on chromosome 11 that predisposes to disease in mice. EMBO Mol. Med. 2011, 3, 50–66. [Google Scholar] [CrossRef]
- Nivarthi, H.; Prchal-Murphy, M.; Swoboda, A.; Hager, M.; Schlederer, M.; Kenner, L.; Tuckermann, J.; Sexl, V.; Moriggl, R.; Ermakova, O. Stat5 gene dosage in T cells modulates CD8+ T-cell homeostasis and attenuates contact hypersensitivity response in mice. Allergy 2015, 70, 67–79. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Adoro, S.; Guinter, T.; Erman, B.; Alag, A.S.; Catalfamo, M.; Kimura, M.Y.; Cui, Y.; Lucas, P.J.; Gress, R.E.; et al. Signaling by intrathymic cytokines, not T cell antigen receptors, specifies CD8 lineage choice and promotes the differentiation of cytotoxic-lineage T cells. Nat. Immunol. 2010, 11, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Kelly, J.; Spolski, R.; Imada, K.; Bollenbacher, J.; Lee, S.; Leonard, W.J. A Role for Stat5 in CD8+ T Cell Homeostasis. J. Immunol. 2003, 170, 210–217. [Google Scholar] [CrossRef] [PubMed]
- Hand, T.W.; Cui, W.; Jung, Y.W.; Sefik, E.; Joshi, N.S.; Chandele, A.; Liu, Y.; Kaech, S.M. Differential effects of STAT5 and PI3K/AKT signaling on effector and memory CD8 T-cell survival. Proc. Natl. Acad. Sci. USA 2010, 107, 16601–16606. [Google Scholar] [CrossRef]
- Grange, M.; Buferne, M.; Verdeil, G.; Leserman, L.; Schmitt-Verhulst, A.; Auphan-Anezin, N. Activated STAT5 Promotes Long-Lived Cytotoxic CD8+ T Cells That Induce Regression of Autochthonous Melanoma. Cancer Res. 2012, 72, 76–87. [Google Scholar] [CrossRef]
- Owen, D.L.; Farrar, M.A. STAT5 and CD4 (+) T Cell Immunity. F1000Research 2017, 6, 32. [Google Scholar] [CrossRef]
- Jenks, J.A.; Seki, S.; Kanai, T.; Huang, J.; Morgan, A.A.; Scalco, R.C.; Nath, R.; Bucayu, R.; Wit, J.M.; Al-Herz, W.; et al. Differentiating the roles of STAT5B and STAT5A in human CD4+ T cells. Clin. Immunol. 2013, 148, 227–236. [Google Scholar] [CrossRef]
- Li, G.; Miskimen, K.L.; Wang, Z.; Xie, X.Y.; Brenzovich, J.; Ryan, J.J.; Tse, W.; Moriggl, R.; Bunting, K.D. STAT5 requires the N-domain for suppression of miR15/16, induction of bcl-2, and survival signaling in myeloproliferative disease. Blood 2010, 115, 1416–1424. [Google Scholar] [CrossRef]
- Li, G.; Wang, Z.; Zhang, Y.; Kang, Z.; Haviernikova, E.; Cui, Y.; Hennighausen, L.; Moriggl, R.; Wang, D.; Tse, W.; et al. STAT5 requires the N-domain to maintain hematopoietic stem cell repopulating function and appropriate lymphoid-myeloid lineage output. Exp. Hematol. 2007, 35, 1684–1694. [Google Scholar] [CrossRef]
- Snow, J.W.; Abraham, N.; Ma, M.C.; Abbey, N.W.; Herndier, B.; Goldsmith, M.A. STAT5 promotes multilineage hematolymphoid development in vivo through effects on early hematopoietic progenitor cells. Blood 2002, 99, 95–101. [Google Scholar] [CrossRef]
- Schepers, H.; van Gosliga, D.; Wierenga, A.T.J.; Eggen, B.J.L.; Schuringa, J.J.; Vellenga, E. STAT5 is required for long-term maintenance of normal and leukemic human stem/progenitor cells. Blood 2007, 110, 2880–2888. [Google Scholar] [CrossRef]
- Scherr, M.; Chaturvedi, A.; Battmer, K.; Dallmann, I.; Schultheis, B.; Ganser, A.; Eder, M. Enhanced sensitivity to inhibition of SHP2, STAT5, and Gab2 expression in chronic myeloid leukemia (CML). Blood 2006, 107, 3279–3287. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wang, Z.; Bunting, K.D. Stat5 deficiency decreases transcriptional heterogeneity and supports emergence of hematopoietic sub-populations. Oncotarget 2017, 8, 22477–22482. [Google Scholar] [CrossRef] [PubMed]
- Fatrai, S.; Wierenga, A.T.J.; Daenen, S.M.G.J.; Vellenga, E.; Schuringa, J.J. Identification of HIF2α as an important STAT5 target gene in human hematopoietic stem cells. Blood 2011, 117, 3320–3330. [Google Scholar] [CrossRef]
- Wierenga, A.T.J.; Vellenga, E.; Schuringa, J.J. Down-regulation of GATA1 uncouples STAT5-induced erythroid differentiation from stem/progenitor cell proliferation. Blood 2010, 115, 4367–4376. [Google Scholar] [CrossRef][Green Version]
- Haetscher, N.; Feuermann, Y.; Wingert, S.; Rehage, M.; Thalheimer, F.B.; Weiser, C.; Bohnenberger, H.; Jung, K.; Schroeder, T.; Serve, H.; et al. STAT5-regulated microRNA-193b controls haematopoietic stem and progenitor cell expansion by modulating cytokine receptor signalling. Nat. Commun. 2015, 6, 8928. [Google Scholar] [CrossRef]
- Grundler, R.; Brault, L.; Gasser, C.; Bullock, A.N.; Dechow, T.; Woetzel, S.; Pogacic, V.; Villa, A.; Ehret, S.; Berridge, G.; et al. Dissection of PIM serine/threonine kinases in FLT3-ITD-induced leukemogenesis reveals PIM1 as regulator of CXCL12-CXCR4-mediated homing and migration. J. Exp. Med. 2009, 206, 1957–1970. [Google Scholar] [CrossRef]
- Ghanem, S.; Friedbichler, K.; Boudot, C.; Bourgeais, J.; Gouilleux-Gruart, V.; Régnier, A.; Herault, O.; Moriggl, R.; Gouilleux, F. STAT5A/5B-specific expansion and transformation of hematopoietic stem cells. Blood Cancer J. 2017, 7, e514. [Google Scholar] [CrossRef]
- Essers, M.A.G.; Offner, S.; Blanco-Bose, W.E.; Waibler, Z.; Kalinke, U.; Duchosal, M.A.; Trumpp, A. IFNα activates dormant haematopoietic stem cells in vivo. Nature 2009, 458, 904. [Google Scholar] [CrossRef]
- Baldridge, M.T.; King, K.Y.; Boles, N.C.; Weksberg, D.C.; Goodell, M.A. Quiescent haematopoietic stem cells are activated by IFN-γ in response to chronic infection. Nature 2010, 465, 793. [Google Scholar] [CrossRef]
- Morales-Mantilla, D.E.; King, K.Y. The Role of Interferon-Gamma in Hematopoietic Stem Cell Development, Homeostasis, and Disease. Curr. Stem Cell Rep. 2018, 4, 264–271. [Google Scholar] [CrossRef] [PubMed]
- Hirche, C.; Frenz, T.; Haas, S.F.; Döring, M.; Borst, K.; Tegtmeyer, P.K.; Brizic, I.; Jordan, S.; Keyser, K.; Chhatbar, C.; et al. Systemic Virus Infections Differentially Modulate Cell Cycle State and Functionality of Long-Term Hematopoietic Stem Cells In Vivo. Cell Rep. 2017, 19, 2345–2356. [Google Scholar] [CrossRef] [PubMed]
- Kato, Y.; Iwama, A.; Tadokoro, Y.; Shimoda, K.; Minoguchi, M.; Akira, S.; Tanaka, M.; Miyajima, A.; Kitamura, T.; Nakauchi, H. Selective activation of STAT5 unveils its role in stem cell self-renewal in normal and leukemic hematopoiesis. J. Exp. Med. 2005, 202, 169–179. [Google Scholar] [CrossRef] [PubMed]
- De Bruin, A.M.; Demirel, Ö.; Hooibrink, B.; Brandts, C.H.; Nolte, M.A. Interferon-γ impairs proliferation of hematopoietic stem cells in mice. Blood 2013, 121, 3578–3585. [Google Scholar] [CrossRef]
- Yoshihara, H.; Arai, F.; Hosokawa, K.; Hagiwara, T.; Takubo, K.; Nakamura, Y.; Gomei, Y.; Iwasaki, H.; Matsuoka, S.; Miyamoto, K.; et al. Thrombopoietin/MPL Signaling Regulates Hematopoietic Stem Cell Quiescence and Interaction with the Osteoblastic Niche. Cell Stem Cell 2007, 1, 685–697. [Google Scholar] [CrossRef]
- Kollmann, S.; Grundschober, E.; Maurer, B.; Warsch, W.; Grausenburger, R.; Edlinger, L.; Huuhtanen, J.; Lagger, S.; Hennighausen, L.; Valent, P.; et al. Twins with different personalities: STAT5B—but not STAT5A—has a key role in BCR/ABL-induced leukemia. Leukemia 2019. [Google Scholar] [CrossRef]
- Joliot, V.; Cormier, F.; Medyouf, H.; Alcalde, H.; Ghysdael, J. Constitutive STAT5 activation specifically cooperates with the loss of p53 function in B-cell lymphomagenesis. Oncogene 2006, 25, 4573–4584. [Google Scholar] [CrossRef][Green Version]
- Orlova, A.; Wingelhofer, B.; Neubauer, H.A.; Maurer, B.; Berger-Becvar, A.; Keserű, G.M.; Gunning, P.T.; Valent, P.; Moriggl, R. Emerging therapeutic targets in myeloproliferative neoplasms and peripheral T-cell leukemia and lymphomas. Expert Opin. Ther. Targets 2018, 22, 45–57. [Google Scholar] [CrossRef]
- Yu, H.; Jove, R. The STATs of cancer—New molecular targets come of age. Nat. Rev. Cancer 2004, 4, 97. [Google Scholar] [CrossRef]
- Ferbeyre, G.; Moriggl, R. The role of Stat5 transcription factors as tumor suppressors or oncogenes. Biochim. Biophys. Acta 2011, 1815, 104–114. [Google Scholar] [CrossRef]
- Dagvadorj, A.; Kirken, R.A.; Leiby, B.; Karras, J.; Nevalainen, M.T. Transcription Factor Signal Transducer and Activator of Transcription 5 Promotes Growth of Human Prostate Cancer Cells In vivo. Clin. Cancer Res. 2008, 14, 1317–1324. [Google Scholar] [CrossRef] [PubMed]
- David, M.; Wong, L.; Flavell, R.; Thompson, S.A.; Wells, A.; Larner, A.C.; Johnson, G.R. STAT Activation by Epidermal Growth Factor (EGF) and Amphiregulin: REQUIREMENT FOR THE EGF RECEPTOR KINASE BUT NOT FOR TYROSINE PHOSPHORYLATION SITES OR JAK1. J. Biol. Chem. 1996, 271, 9185–9188. [Google Scholar] [CrossRef] [PubMed]
- Blaas, L.; Kornfeld, J.W.; Schramek, D.; Musteanu, M.; Zollner, G.; Gumhold, J.; van Zijl, F.; Schneller, D.; Esterbauer, H.; Egger, G.; et al. Disruption of the growth hormone--signal transducer and activator of transcription 5--insulinlike growth factor 1 axis severely aggravates liver fibrosis in a mouse model of cholestasis. Hepatology 2010, 51, 1319–1326. [Google Scholar] [CrossRef] [PubMed]
- Sonkin, D.; Regnier, C.; Rong, X.; Fanton, C.; Palmer, M.; Holash, J.; Squires, M.; Sirulnik, L.A.; Radimerski, T.; Schlegel, R.; et al. Identification of pSTAT5 gene signature in hematologic malignancy. J. Clin. Oncol. 2013, 31, 7111. [Google Scholar] [CrossRef]
- De Araujo, E.D.; Erdogan, F.; Neubauer, H.A.; Meneksedag-Erol, D.; Manaswiyoungkul, P.; Eram, M.S.; Seo, H.S.; Qadree, A.K.; Israelian, J.; Orlova, A.; et al. Structural and functional consequences of the STAT5B(N642H) driver mutation. Nat. Commun. 2019, 10, 2517. [Google Scholar] [CrossRef]
- COSMIC. STAT5B Mutations Gene View. Available online: https://cancer.sanger.ac.uk/cosmic/gene/analysis?all_data=&coords=AA%3AAA&dr=&end=788&gd=&id=5369&ln=STAT5B&seqlen=788&sn=haematopoietic_and_lymphoid_tissue&start=1#ts (accessed on 19 September 2019).
- Bandapalli, O.R.; Schuessele, S.; Kunz, J.B.; Rausch, T.; Stütz, A.M.; Tal, N.; Geron, I.; Gershman, N.; Izraeli, S.; Eilers, J.; et al. The activating STAT5B N642H mutation is a common abnormality in pediatric T-cell acute lymphoblastic leukemia and confers a higher risk of relapse. Haematologica 2014, 99, e188–e192. [Google Scholar] [CrossRef]
- Kontro, M.; Kuusanmaki, H.; Eldfors, S.; Burmeister, T.; Andersson, E.I.; Bruserud, O.; Brummendorf, T.H.; Edgren, H.; Gjertsen, B.T.; Itala-Remes, M.; et al. Novel activating STAT5B mutations as putative drivers of T-cell acute lymphoblastic leukemia. Leukemia 2014, 28, 1738–1742. [Google Scholar] [CrossRef]
- Schrader, A.; Crispatzu, G.; Oberbeck, S.; Mayer, P.; Pützer, S.; von Jan, J.; Vasyutina, E.; Warner, K.; Weit, N.; Pflug, N.; et al. Actionable perturbations of damage responses by TCL1/ATM and epigenetic lesions form the basis of T-PLL. Nat. Commun. 2018, 9, 697. [Google Scholar] [CrossRef]
- McKinney, M.; Moffitt, A.B.; Gaulard, P.; Travert, M.; De Leval, L.; Nicolae, A.; Raffeld, M.; Jaffe, E.S.; Pittaluga, S.; Xi, L.; et al. The Genetic Basis of Hepatosplenic T Cell Lymphoma. Cancer Discov. 2017. [Google Scholar] [CrossRef]
- Küçük, C.; Jiang, B.; Hu, X.; Zhang, W.; Chan, J.K.C.; Xiao, W.; Lack, N.; Alkan, C.; Williams, J.C.; Avery, K.N.; et al. Activating mutations of STAT5B and STAT3 in lymphomas derived from γδ-T or NK cells. Nat. Commun. 2015, 6. [Google Scholar] [CrossRef]
- Rajala, H.L.M.; Eldfors, S.; Kuusanmäki, H.; van Adrichem, A.J.; Olson, T.; Lagström, S.; Andersson, E.I.; Jerez, A.; Clemente, M.J.; Yan, Y.; et al. Discovery of somatic STAT5b mutations in large granular lymphocytic leukemia. Blood 2013, 121, 4541–4550. [Google Scholar] [CrossRef] [PubMed]
- Kiel, M.J.; Velusamy, T.; Rolland, D.; Sahasrabuddhe, A.A.; Chung, F.; Bailey, N.G.; Schrader, A.; Li, B.; Li, J.Z.; Ozel, A.B.; et al. Integrated genomic sequencing reveals mutational landscape of T-cell prolymphocytic leukemia. Blood 2014, 124, 1460–1472. [Google Scholar] [CrossRef]
- Dufva, O.; Kankainen, M.; Kelkka, T.; Sekiguchi, N.; Awad, S.A.; Eldfors, S.; Yadav, B.; Kuusanmäki, H.; Malani, D.; Andersson, E.I.; et al. Aggressive natural killer-cell leukemia mutational landscape and drug profiling highlight JAK-STAT signaling as therapeutic target. Nat. Commun. 2018, 9, 1567. [Google Scholar] [CrossRef] [PubMed]
- Pham, H.T.T.; Hengstschläger, M.; Moriggl, R. A haunted beast: Targeting STAT5B(N642H) in T-Cell Neoplasia. Mol. Cell. Oncol. 2018, 5, e1435181. [Google Scholar] [CrossRef]
- Nicolae, A.; Xi, L.; Pittaluga, S.; Abdullaev, Z.; Pack, S.D.; Chen, J.; Waldmann, T.A.; Jaffe, E.S.; Raffeld, M. Frequent STAT5B mutations in [gamma][delta] hepatosplenic T-cell lymphomas. Leukemia 2014, 28, 2244–2248. [Google Scholar] [CrossRef] [PubMed]
- Cross, N.C.P.; Hoade, Y.; Tapper, W.J.; Carreno-Tarragona, G.; Fanelli, T.; Jawhar, M.; Naumann, N.; Pieniak, I.; Lübke, J.; Ali, S.; et al. Recurrent activating STAT5B N642H mutation in myeloid neoplasms with eosinophilia. Leukemia 2018. [Google Scholar] [CrossRef]
- Luo, Q.; Shen, J.; Yang, Y.; Tang, H.; Shi, M.; Liu, J.; Liu, Z.; Shi, X.; Yi, Y. CSF3R T618I, ASXL1 G942 fs and STAT5B N642H trimutation co-contribute to a rare chronic neutrophilic leukaemia manifested by rapidly progressive leucocytosis, severe infections, persistent fever and deep venous thrombosis. Br. J. Haematol. 2018, 180, 892–894. [Google Scholar] [CrossRef]
- Waldmann, T.A.; Chen, J. Disorders of the JAK/STAT Pathway in T Cell Lymphoma Pathogenesis: Implications for Immunotherapy. Annu. Rev. Immunol. 2017, 35, 533–550. [Google Scholar] [CrossRef]
- Klein, K.; Witalisz-Siepracka, A.; Maurer, B.; Prinz, D.; Heller, G.; Leidenfrost, N.; Prchal-Murphy, M.; Suske, T.; Moriggl, R.; Sexl, V. STAT5BN642H drives transformation of NKT cells: A novel mouse model for CD56+ T-LGL leukemia. Leukemia 2019. [Google Scholar] [CrossRef]
- Kelly, J.A.; Spolski, R.; Kovanen, P.E.; Suzuki, T.; Bollenbacher, J.; Pise-Masison, C.A.; Radonovich, M.F.; Lee, S.; Jenkins, N.A.; Copeland, N.G.; et al. Stat5 Synergizes with T Cell Receptor/Antigen Stimulation in the Development of Lymphoblastic Lymphoma. J. Exp. Med. 2003, 198, 79–89. [Google Scholar] [CrossRef]
- Chen, B.; Yi, B.; Mao, R.; Liu, H.; Wang, J.; Sharma, A.; Peiper, S.; Leonard, W.J.; She, J.X. Enhanced T Cell Lymphoma in NOD.Stat5b Transgenic Mice Is Caused by Hyperactivation of Stat5b in CD8(+) Thymocytes. PLoS ONE 2013, 8, e56600. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.C.; Schmidt, J.W.; Creamer, B.A.; Triplett, A.A.; Wagner, K.U. Gain-of-Function of Stat5 Leads to Excessive Granulopoiesis and Lethal Extravasation of Granulocytes to the Lung. PLoS ONE 2013, 8, e60902. [Google Scholar] [CrossRef] [PubMed]
- Kadekar, D.; Agerholm, R.; Rizk, J.; Neubauer, H.; Suske, T.; Maurer, B.; Viñals, M.T.; Comelli, E.; Taibi, A.; Moriggl, R.; et al. The neonatal microenvironment programs conventional and intestinal Tbet+ γδT17 cells through the transcription factor STAT5. bioRxiv 2019, 658542. [Google Scholar] [CrossRef]
- De Bock, C.E.; Demeyer, S.; Degryse, S.; Verbeke, D.; Sweron, B.; Gielen, O.; Vandepoel, R.; Vicente, C.; Vanden Bempt, M.; Dagklis, A.; et al. HOXA9 Cooperates with Activated JAK/STAT Signaling to Drive Leukemia Development. Cancer Discov. 2018, 8, 616–631. [Google Scholar] [CrossRef] [PubMed]
- Rossari, F.; Minutolo, F.; Orciuolo, E. Past, present, and future of Bcr-Abl inhibitors: From chemical development to clinical efficacy. J. Hematol. Oncol. 2018, 11, 84. [Google Scholar] [CrossRef] [PubMed]
- Hoelbl, A.; Schuster, C.; Kovacic, B.; Zhu, B.; Wickre, M.; Hoelzl, M.A.; Fajmann, S.; Grebien, F.; Warsch, W.; Stengl, G.; et al. Stat5 is indispensable for the maintenance of bcr/abl-positive leukaemia. EMBO Mol. Med. 2010, 2, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Walz, C.; Ahmed, W.; Lazarides, K.; Betancur, M.; Patel, N.; Hennighausen, L.; Zaleskas, V.M.; Van Etten, R.A. Essential role for Stat5a/b in myeloproliferative neoplasms induced by BCR-ABL1 and JAK2V617F in mice. Blood 2012, 119, 3550–3560. [Google Scholar] [CrossRef]
- Hantschel, O.; Warsch, W.; Eckelhart, E.; Kaupe, I.; Grebien, F.; Wagner, K.U.; Superti-Furga, G.; Sexl, V. BCR-ABL uncouples canonical JAK2-STAT5 signaling in chronic myeloid leukemia. Nat. Chem. Biol. 2012, 8, 285. [Google Scholar] [CrossRef]
- Warsch, W.; Kollmann, K.; Eckelhart, E.; Fajmann, S.; Cerny-Reiterer, S.; Hölbl, A.; Gleixner, K.V.; Dworzak, M.; Mayerhofer, M.; Hoermann, G.; et al. High STAT5 levels mediate imatinib resistance and indicate disease progression in chronic myeloid leukemia. Blood 2011, 117, 3409–3420. [Google Scholar] [CrossRef]
- Ma, L.; Pak, M.L.; Ou, J.; Yu, J.; St. Louis, P.; Shan, Y.; Hutchinson, L.; Li, S.; Brehm, M.A.; Zhu, L.J.; et al. Prosurvival kinase PIM2 is a therapeutic target for eradication of chronic myeloid leukemia stem cells. Proc. Natl. Acad. Sci. USA 2019, 116, 10482–10487. [Google Scholar] [CrossRef]
- Minieri, V.; De Dominici, M.; Porazzi, P.; Mariani, S.A.; Spinelli, O.; Rambaldi, A.; Peterson, L.F.; Porcu, P.; Nevalainen, M.T.; Calabretta, B. Targeting STAT5 or STAT5-regulated pathways suppresses leukemogenesis of Ph+ acute lymphoblastic leukemia. Cancer Res. 2018. [Google Scholar] [CrossRef] [PubMed]
- Najjar, I.; Grimont, A.; Martin, N.; Martin-Lanneree, S.; Bonnet, M.L.; Guilhot, F.; Chomel, J.C.; Lauret, E.; Turhan, A.G.; Dusanter-Fourt, I. Distinct Functions of Stat5A and Stat5B in Chronic Myeloid Leukemia (CML): Stat5B Is Implicated in Survival and Self-Renewal and Stat5A in Imatinib Resistance. Blood 2010, 116, 1214. [Google Scholar] [CrossRef]
- Casetti, L.; Martin-Lannerée, S.; Najjar, I.; Plo, I.; Augé, S.; Roy, L.; Chomel, J.C.; Lauret, E.; Turhan, A.G.; Dusanter-Fourt, I. Differential Contributions of STAT5A and STAT5B to Stress Protection and Tyrosine Kinase Inhibitor Resistance of Chronic Myeloid Leukemia Stem/Progenitor Cells. Cancer Res. 2013, 73, 2052–2058. [Google Scholar] [CrossRef] [PubMed]
- Ducray, S.P.; Natarajan, K.; Garland, G.D.; Turner, S.D.; Egger, G. The Transcriptional Roles of ALK Fusion Proteins in Tumorigenesis. Cancers 2019, 11, 1074. [Google Scholar] [CrossRef]
- Werner, M.T.; Zhao, C.; Zhang, Q.; Wasik, M.A. Nucleophosmin-anaplastic lymphoma kinase: The ultimate oncogene and therapeutic target. Blood 2017, 129, 823–831. [Google Scholar] [CrossRef]
- Zhang, Q.; Wang, H.Y.; Liu, X.; Wasik, M.A. STAT5A is epigenetically silenced by the tyrosine kinase NPM1-ALK and acts as a tumor suppressor by reciprocally inhibiting NPM1-ALK expression. Nat. Med. 2007, 13, 1341. [Google Scholar] [CrossRef]
- Recio, C.; Guerra, B.; Guerra-Rodríguez, M.; Aranda-Tavío, H.; Martín-Rodríguez, P.; de Mirecki-Garrido, M.; Brito-Casillas, Y.; García-Castellano, J.M.; Estévez-Braun, A.; Fernández-Pérez, L. Signal transducer and activator of transcription (STAT)-5: An opportunity for drug development in oncohematology. Oncogene 2019, 38, 4657–4668. [Google Scholar] [CrossRef]
- Gadina, M.; Johnson, C.; Schwartz, D.; Bonelli, M.; Hasni, S.; Kanno, Y.; Changelian, P.; Laurence, A.; O’Shea, J.J. Translational and clinical advances in JAK-STAT biology: The present and future of jakinibs. J. Leukoc. Biol. 2018, 104, 499–514. [Google Scholar] [CrossRef]
- Senkevitch, E.; Durum, S. The promise of Janus kinase inhibitors in the treatment of hematological malignancies. Cytokine 2017, 98, 33–41. [Google Scholar] [CrossRef]
- Cumaraswamy, A.A.; Lewis, A.M.; Geletu, M.; Todic, A.; Diaz, D.B.; Cheng, X.R.; Brown, C.E.; Laister, R.C.; Muench, D.; Kerman, K.; et al. Nanomolar-Potency Small Molecule Inhibitor of STAT5 Protein. ACS Med. Chem. Lett. 2014, 5, 1202–1206. [Google Scholar] [CrossRef]
- Gleixner, K.V.; Schneeweiss, M.; Eisenwort, G.; Berger, D.; Herrmann, H.; Blatt, K.; Greiner, G.; Byrgazov, K.; Hoermann, G.; Konopleva, M.; et al. Combined targeting of STAT3 and STAT5: A novel approach to overcome drug resistance in chronic myeloid leukemia. Haematologica 2017, 102, 1519–1529. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wingelhofer, B.; Maurer, B.; Heyes, E.C.; Cumaraswamy, A.A.; Berger-Becvar, A.; de Araujo, E.D.; Orlova, A.; Freund, P.; Ruge, F.; Park, J.; et al. Pharmacologic inhibition of STAT5 in acute myeloid leukemia. Leukemia 2018, 32, 1135–1146. [Google Scholar] [CrossRef] [PubMed]
- Liao, Z.; Gu, L.; Vergalli, J.; Mariani, S.A.; De Dominici, M.; Lokareddy, R.K.; Dagvadorj, A.; Purushottamachar, P.; McCue, P.A.; Trabulsi, E.; et al. Structure-Based Screen Identifies a Potent Small Molecule Inhibitor of Stat5a/b with Therapeutic Potential for Prostate Cancer and Chronic Myeloid Leukemia. Mol. Cancer Ther. 2015, 14, 1777–1793. [Google Scholar] [CrossRef] [PubMed]
- Natarajan, K.; Müller-Klieser, D.; Rubner, S.; Berg, T. Stafia-1: A STAT5a-selective inhibitor developed via docking-based screening of in silico O-phosphorylated fragments. Chem. A Eur. J. 2019. [Google Scholar] [CrossRef]
- Elumalai, N.; Berg, A.; Rubner, S.; Berg, T. Phosphorylation of Capsaicinoid Derivatives Provides Highly Potent and Selective Inhibitors of the Transcription Factor STAT5b. ACS Chem. Biol. 2015, 10, 2884–2890. [Google Scholar] [CrossRef] [PubMed]
- Elumalai, N.; Berg, A.; Natarajan, K.; Scharow, A.; Berg, T. Nanomolar inhibitors of the transcription factor STAT5b with high selectivity over STAT5a. Angew. Chem. Int. Ed. 2015, 54, 4758–4763. [Google Scholar] [CrossRef]
- Elumalai, N.; Berg, A.; Rubner, S.; Blechschmidt, L.; Song, C.; Natarajan, K.; Matysik, J.; Berg, T. Rational development of Stafib-2: A selective, nanomolar inhibitor of the transcription factor STAT5b. Sci. Rep. 2017, 7, 819. [Google Scholar] [CrossRef]
- Gräb, J.; Berg, A.; Blechschmidt, L.; Klüver, B.; Rubner, S.; Fu, D.Y.; Meiler, J.; Gräber, M.; Berg, T. The STAT5b Linker Domain Mediates the Selectivity of Catechol Bisphosphates for STAT5b over STAT5a. ACS Chem. Biol. 2019, 14, 796–805. [Google Scholar] [CrossRef]
- Behbod, F.; Nagy, Z.S.; Stepkowski, S.M.; Karras, J.; Johnson, C.R.; Jarvis, W.D.; Kirken, R.A. Specific Inhibition of Stat5a/b Promotes Apoptosis of IL-2-Responsive Primary and Tumor-Derived Lymphoid Cells. J. Immunol. 2003, 171, 3919–3927. [Google Scholar] [CrossRef]
- Sen, M.; Grandis, J.R. Nucleic acid-based approaches to STAT inhibition. JAK-STAT 2012, 1, 285–291. [Google Scholar] [CrossRef]
- Liu, L.J.; Wang, W.; Kang, T.S.; Liang, J.X.; Liu, C.; Kwong, D.W.J.; Wong, V.K.W.; Ma, D.L.; Leung, C.H. Antagonizing STAT5B dimerization with an osmium complex. Sci. Rep. 2016, 6, 36044. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.W.; Kim, J.H.; Park, M.; Yeom, J.H.; Go, H.; Kim, S.; Han, M.S.; Lee, K.; Bae, J. Modulation of biological processes in the nucleus by delivery of DNA oligonucleotides conjugated with gold nanoparticles. Biomaterials 2011, 32, 2593–2604. [Google Scholar] [CrossRef] [PubMed]
- Weber, A.; Borghouts, C.; Brendel, C.; Moriggl, R.; Delis, N.; Brill, B.; Vafaizadeh, V.; Groner, B. The inhibition of stat5 by a Peptide aptamer ligand specific for the DNA binding domain prevents target gene transactivation and the growth of breast and prostate tumor cells. Pharmaceuticals 2013, 6, 960–987. [Google Scholar] [CrossRef]
- Mullard, A. Protein–protein interaction inhibitors get into the groove. Nat. Rev. Drug Discov. 2012, 11, 173. [Google Scholar] [CrossRef]
- Berg, A.; Sperl, B.; Berg, T. ATP Inhibits the Transcription Factor STAT5b. ChemBioChem 2019. [Google Scholar] [CrossRef]
- Mandal, M.; Powers, S.E.; Maienschein-Cline, M.; Bartom, E.T.; Hamel, K.M.; Kee, B.L.; Dinner, A.R.; Clark, M.R. Epigenetic repression of the Igk locus by STAT5-mediated recruitment of the histone methyltransferase Ezh2. Nat. Immunol. 2011, 12, 1212. [Google Scholar] [CrossRef]
- Gall Trošelj, K.; Novak Kujundzic, R.; Ugarkovic, D. Polycomb repressive complex’s evolutionary conserved function: The role of EZH2 status and cellular background. Clin. Epigenetics 2016, 8, 55. [Google Scholar] [CrossRef]
- Goldenson, B.; Crispino, J.D. The aurora kinases in cell cycle and leukemia. Oncogene 2015, 34, 537–545. [Google Scholar] [CrossRef]
- Tang, A.; Gao, K.; Chu, L.; Zhang, R.; Yang, J.; Zheng, J. Aurora kinases: Novel therapy targets in cancers. Oncotarget 2017, 8, 23937–23954. [Google Scholar] [CrossRef]
- Bavetsias, V.; Linardopoulos, S. Aurora Kinase Inhibitors: Current Status and Outlook. Front. Oncol. 2015, 5, 278. [Google Scholar] [CrossRef]
- Porpaczy, E.; Tripolt, S.; Hoelbl-Kovacic, A.; Gisslinger, B.; Bago-Horvath, Z.; Casanova-Hevia, E.; Clappier, E.; Decker, T.; Fajmann, S.; Fux, D.A.; et al. Aggressive B-cell lymphomas in patients with myelofibrosis receiving JAK1/2 inhibitor therapy. Blood 2018, 132, 694–706. [Google Scholar] [CrossRef] [PubMed]
- Bottos, A.; Gotthardt, D.; Gill, J.W.; Gattelli, A.; Frei, A.; Tzankov, A.; Sexl, V.; Wodnar-Filipowicz, A.; Hynes, N.E. Decreased NK-cell tumour immunosurveillance consequent to JAK inhibition enhances metastasis in breast cancer models. Nat. Commun. 2016, 7, 12258. [Google Scholar] [CrossRef]
- Gadina, M.; Le, M.T.; Schwartz, D.M.; Silvennoinen, O.; Nakayamada, S.; Yamaoka, K.; O’Shea, J.J. Janus kinases to jakinibs: From basic insights to clinical practice. Rheumatology 2019, 58, i4–i16. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Okhovat, J.P.; Hong, E.K.; Kim, Y.H.; Wood, G.S. Preclinical studies support combined inhibition of BET family proteins and histone deacetylases as epigentic therapy for cutaneous T-cell lymphoma. Neoplasia 2019, 21, 82–92. [Google Scholar] [CrossRef] [PubMed]
- Pinz, S.; Unser, S.; Buob, D.; Fischer, P.; Jobst, B.; Rascle, A. Deacetylase inhibitors repress STAT5-mediated transcription by interfering with bromodomain and extra-terminal (BET) protein function. Nucleic Acids Res. 2015, 43, 3524–3545. [Google Scholar] [CrossRef]
- Liu, S.; Walker, S.R.; Nelson, E.A.; Cerulli, R.; Xiang, M.; Toniolo, P.A.; Qi, J.; Stone, R.M.; Wadleigh, M.; Bradner, J.E.; et al. Targeting STAT5 in hematologic maligancies through inhibition of the bromodomain and extra-terminal (BET) bromodomain BRD2. Mol. Cancer Ther. 2014, 13, 1194–1205. [Google Scholar] [CrossRef]
- Fiskus, W.; Sharma, S.; Qi, J.; Shah, B.; Devaraj, S.G.T.; Leveque, C.; Portier, B.P.; Iyer, S.; Bradner, J.E.; Bhalla, K.N. BET protein antagonist JQ1 is synergistically lethal with FLT3 tyrosine kinase inhibitor (KI) and overcomes resistance to FLT3-TKI in AML cells expressing FLT-ITD. Mol. Cancer Ther. 2014, 13, 2315–2327. [Google Scholar] [CrossRef]
- Nguyen, T.; Dai, Y.; Attkisson, E.; Kramer, L.; Jordan, N.; Nguyen, N.; Kolluri, N.; Muschen, M.; Grant, S. HDAC inhibitors potentiate the activity of the BCAR/ABL kinase inhibitor KW-2449 in imatinib-sensitive or -resistant BCR/ABL+ leukemia cells in vitro and in vivo. Clin. Cancer Res. 2011, 17, 3219–3232. [Google Scholar] [CrossRef]
- Rascle, A.; Johnston, J.A.; Amati, B. Deacetylase activity is required for recruitment of the basal transcription machinery and transactivation by STAT5. Mol. Cell Biol. 2003, 23, 4162–4173. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Deficiency | Phenotype | Reference | |
|---|---|---|---|
| STAT5A/B | Stat5a/bΔN | truncated N-termini of Stat5a and Stat5b forms dimers but no tetramers smaller, infertile, less CD25+CD4+ T cells | Teglund et al. 1998 [56] |
| Stat5a/b-/- | total body knockout perinatal lethal, 1–2% survivors dwarfism, anemia, reduced T- and NK cell numbers, block in pre–pro-B cell stage | Cui et al. 2004 [59] | |
| Stat5a/bDKI | N-termini mutations in Stat5a and Stat5b no tetramer formation less CD25+CD4+ and CD8+ T cells, less NK cells | Lin et al. 2012 [57] | |
| Stat5a/bfl/fl | loxP-sites spanning Stat5a and Stat5b for tissue-specific or inducible deletion—e.g., in the hematopoietic system: | Cui et al. 2004 [59] | |
| vav-Cre: Anemia, lymphopenia, reduced repopulation capacity (upon BM transplants) | Wang et al. 2015 [60] | ||
| Tie2-Cre: Anemia, lymphopenia, reduced repopulation capacity (upon BM transplants) | Zhu et al. 2008 [61] | ||
| Mx1-Cre: Reduced repopulation capacity (upon BM transplants) | Wang et al. 2009 [62] | ||
| STAT5A | Stat5aΔN | truncated N-termini of Stat5a forms dimers but no tetramers reduced prolactin signaling (mammary gland) | Teglund et al. 1998 [56] |
| Stat5a−/− | total body knockout of Stat5a failure in mammary gland formation and function | Liu et al. 1997 [9] | |
| Stat5aKI | N-termini mutation of Stat5a no STAT5A tetramers slight reduction of IL-2R expression on T cells | Lin et al. 2012 [57] | |
| STAT5B | Stat5bΔN | truncated N-termini of Stat5b forms dimers but no tetramers dwarfism, reduced IGF-1 levels | Teglund et al. 1998 [56] |
| Stat5b−/− | total body knockout of Stat5b dwarfism, failure in GH signaling, reduced numbers of NK and T cells | Udy et al. 1997 [63] | |
| Stat5bKI | N-termini mutations of Stat5b no STAT5B tetramers same, but more pronounced as in Stat5aKI | Lin et al. 2012 [57] | |
| Transgene | Promoter | Phenotype | Reference |
|---|---|---|---|
| Stat5b-tg | H-2Kb promoter and IgM enhancer (T, B, NK cells) | more CD8+ T cells ~12% thymic T cell lymphoblastic lymphoma (CD8+CD4+ or CD8+) | Kelly et al. 2003 [105,153] |
| NOD background | 75% CD8+ T cell lymphoblastic lymphoma | Chen et al. 2013 [154] | |
| Stat5b-CA-tg | Lck promoter and IgM enhancer | expansion of CD8+ and γδ T cells late emergence of clonal B cell lymphoma/leukemia (low incidence) | Burchill et al. 2003 [88] |
| pre-BCR pathway defects | increased B-ALL incidence | Katerndahl et al. 2017 [89] | |
| cS5F | Eµ enhancer (lymphoid specific) | increase of CD8+ T cells, late emergence of clonal B cell lymphoma/leukemia (low incidence) | Joliot et al. 2006 [129] |
| MMTV-tTA TetO-cS5F | cS5F-Tet-OFF under MMTV-LTR promoter | hematopoiesis unaffected cross to Stat5a/bΔN → expansion of granulocytes | Lin et al. 2013 [155] |
| hSTAT5B | vav promoter | mild expansion of CD8+ T cells no disease | Pham et al. 2018 [84] |
| hSTAT5BN642H | vav promoter | aggressive CD8+ T cell neoplasia enhanced numbers of HSCs and lymphoid progenitors | Pham et al. 2018 [84] |
| cS5F lo | vav promoter | mild expansion of CD8+ T cells no disease | Maurer et al. 2019 [83] |
| cS5F hi | vav promoter | CD8+ T cell neoplasia correlating to PTCL-NOS enhanced numbers of HSCs and lymphoid progenitors | Maurer et al. 2019 [83] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maurer, B.; Kollmann, S.; Pickem, J.; Hoelbl-Kovacic, A.; Sexl, V. STAT5A and STAT5B—Twins with Different Personalities in Hematopoiesis and Leukemia. Cancers 2019, 11, 1726. https://doi.org/10.3390/cancers11111726
Maurer B, Kollmann S, Pickem J, Hoelbl-Kovacic A, Sexl V. STAT5A and STAT5B—Twins with Different Personalities in Hematopoiesis and Leukemia. Cancers. 2019; 11(11):1726. https://doi.org/10.3390/cancers11111726
Chicago/Turabian StyleMaurer, Barbara, Sebastian Kollmann, Judith Pickem, Andrea Hoelbl-Kovacic, and Veronika Sexl. 2019. "STAT5A and STAT5B—Twins with Different Personalities in Hematopoiesis and Leukemia" Cancers 11, no. 11: 1726. https://doi.org/10.3390/cancers11111726
APA StyleMaurer, B., Kollmann, S., Pickem, J., Hoelbl-Kovacic, A., & Sexl, V. (2019). STAT5A and STAT5B—Twins with Different Personalities in Hematopoiesis and Leukemia. Cancers, 11(11), 1726. https://doi.org/10.3390/cancers11111726