Comprehensive Outline of Whole Exome Sequencing Data Analysis Tools Available in Clinical Oncology


Abstract
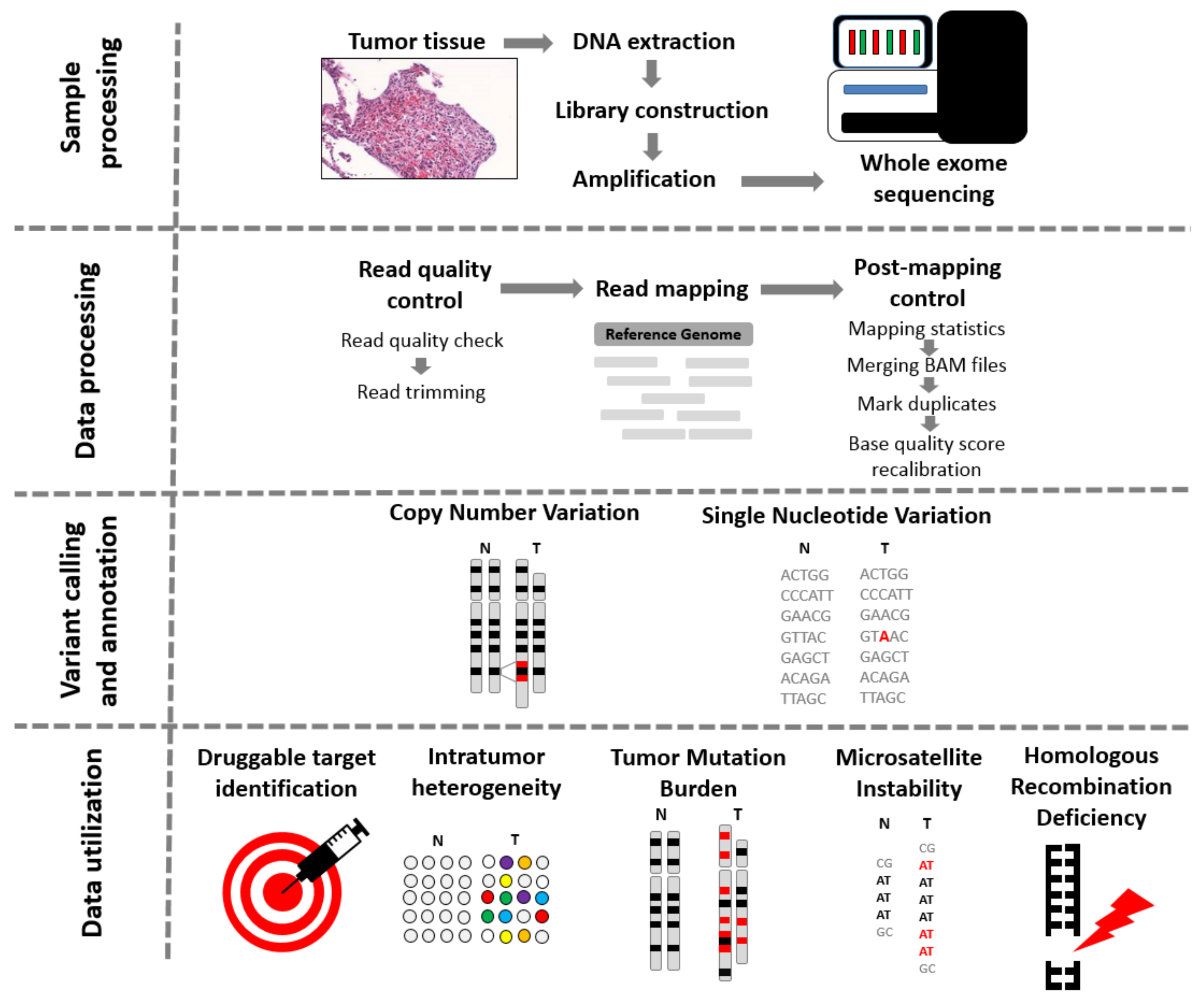
1. Introduction
2. First Steps of Whole Exome Sequencing
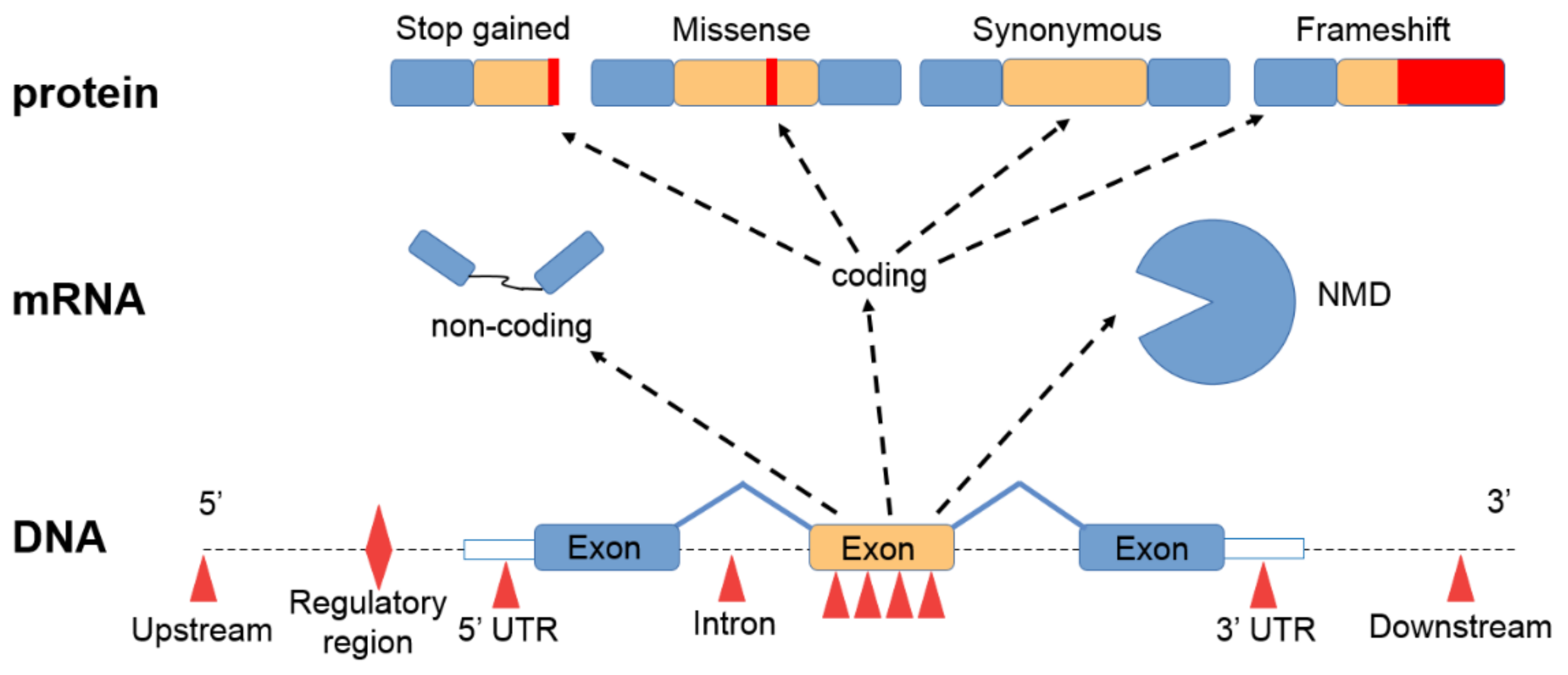
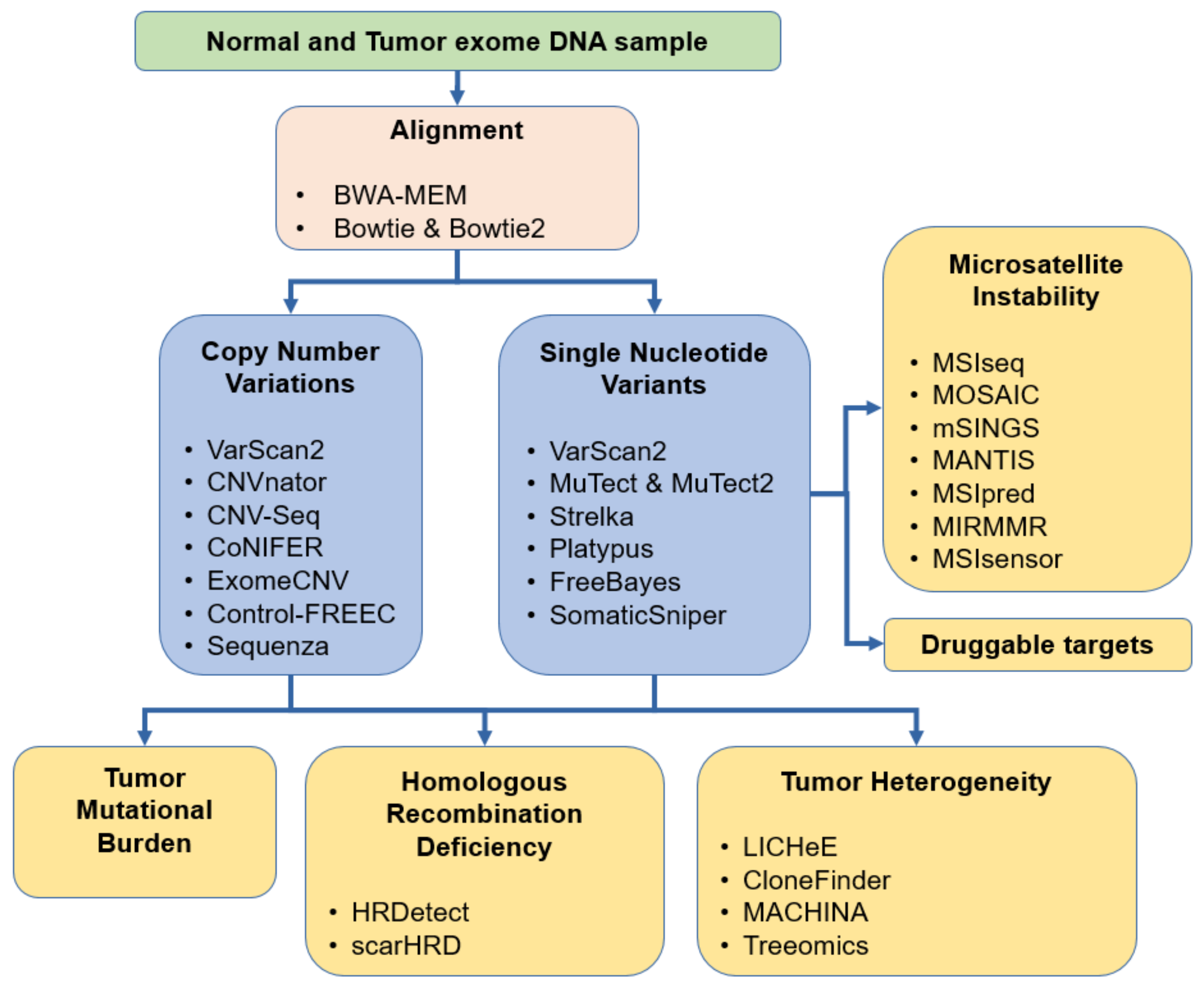
3. Short Nucleotide Variants
4. Integrated Tools
5. Galaxy—An Open Source, Web-Based Platform
6. Copy Number Variations
7. Homologous Recombination Deficiency
8. Response to Immunotherapy
9. Tumor Heterogeneity
10. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Menyhart, O.; Harami-Papp, H.; Sukumar, S.; Schafer, R.; Magnani, L.; de Barrios, O.; Gyorffy, B. Guidelines for the selection of functional assays to evaluate the hallmarks of cancer. Biochim. Biophys. Acta 2016, 1866, 300–319. [Google Scholar] [CrossRef] [PubMed]
- Schaub, M.A.; Boyle, A.P.; Kundaje, A.; Batzoglou, S.; Snyder, M. Linking disease associations with regulatory information in the human genome. Genome Res. 2012, 22, 1748–1759. [Google Scholar] [CrossRef]
- Li, G.; Pan, T.; Guo, D.; Li, L.C. Regulatory Variants and Disease: The E-Cadherin -160C/A SNP as an Example. Mol. Biol. Int. 2014, 2014, 967565. [Google Scholar] [CrossRef]
- Minde, D.P.; Anvarian, Z.; Rudiger, S.G.; Maurice, M.M. Messing up disorder: How do missense mutations in the tumor suppressor protein APC lead to cancer? Mol. Cancer 2011, 10, 101. [Google Scholar] [CrossRef] [PubMed]
- Gnarra, J.R.; Tory, K.; Weng, Y.; Schmidt, L.; Wei, M.H.; Li, H.; Latif, F.; Liu, S.; Chen, F.; Duh, F.M.; et al. Mutations of the VHL tumour suppressor gene in renal carcinoma. Nat. Genet. 1994, 7, 85–90. [Google Scholar] [CrossRef] [PubMed]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef]
- Shlien, A.; Malkin, D. Copy number variations and cancer. Genome Med. 2009, 1, 62. [Google Scholar] [CrossRef]
- Torgovnick, A.; Schumacher, B. DNA repair mechanisms in cancer development and therapy. Front. Genet. 2015, 6, 157. [Google Scholar] [CrossRef]
- Luchini, C.; Bibeau, F.; Ligtenberg, M.J.L.; Singh, N.; Nottegar, A.; Bosse, T.; Miller, R.; Riaz, N.; Douillard, J.Y.; Andre, F.; et al. ESMO recommendations on microsatellite instability testing for immunotherapy in cancer, and its relationship with PD-1/PD-L1 expression and tumour mutational burden: A systematic review-based approach. Ann. Oncol. Off. J. Eur. Soc. Med Oncol. 2019. [Google Scholar] [CrossRef]
- Morlote, D.; Janowski, K.M.; Siniard, R.C.; Guo, R.J.; Winokur, T.; DeFrank, G.; Harada, S. Effects of Improved DNA Integrity by Punch from Tissue Blocks as Compared to Pinpoint Extraction from Unstained Slides on Next-Generation Sequencing Quality Metrics. Am. J. Clin. Pathol. 2019, 152, 27–35. [Google Scholar] [CrossRef] [PubMed]
- McDonough, S.J.; Bhagwate, A.; Sun, Z.; Wang, C.; Zschunke, M.; Gorman, J.A.; Kopp, K.J.; Cunningham, J.M. Use of FFPE-derived DNA in next generation sequencing: DNA extraction methods. PLoS ONE 2019, 14, e0211400. [Google Scholar] [CrossRef] [PubMed]
- Warr, A.; Robert, C.; Hume, D.; Archibald, A.; Deeb, N.; Watson, M. Exome Sequencing: Current and Future Perspectives. G3 Genes Genomes Genet. 2015, 5, 1543–1550. [Google Scholar] [CrossRef] [PubMed]
- Chilamakuri, C.S.; Lorenz, S.; Madoui, M.A.; Vodak, D.; Sun, J.; Hovig, E.; Myklebost, O.; Meza-Zepeda, L.A. Performance comparison of four exome capture systems for deep sequencing. BMC Genom. 2014, 15, 449. [Google Scholar] [CrossRef] [PubMed]
- Pongor, L.; Kormos, M.; Hatzis, C.; Pusztai, L.; Szabo, A.; Gyorffy, B. A genome-wide approach to link genotype to clinical outcome by utilizing next generation sequencing and gene chip data of 6697 breast cancer patients. Genome Med. 2015, 7, 104. [Google Scholar] [CrossRef]
- Nagy, A.; Pongor, L.S.; Szabo, A.; Santarpia, M.; Gyorffy, B. KRAS driven expression signature has prognostic power superior to mutation status in non-small cell lung cancer. Int. J. Cancer 2017, 140, 930–937. [Google Scholar] [CrossRef]
- Gyorffy, B.; Pongor, L.; Bottai, G.; Li, X.; Budczies, J.; Szabo, A.; Hatzis, C.; Pusztai, L.; Santarpia, L. An integrative bioinformatics approach reveals coding and non-coding gene variants associated with gene expression profiles and outcome in breast cancer molecular subtypes. Br. J. Cancer 2018, 118, 1107–1114. [Google Scholar] [CrossRef]
- Menyhart, O.; Kakisaka, T.; Pongor, L.S.; Uetake, H.; Goel, A.; Gyorffy, B. Uncovering Potential Therapeutic Targets in Colorectal Cancer by Deciphering Mutational Status and Expression of Druggable Oncogenes. Cancers 2019, 11, 983. [Google Scholar] [CrossRef]
- Menyhart, O.; Pongor, L.S.; Gyorffy, B. Mutations Defining Patient Cohorts with Elevated PD-L1 Expression in Gastric Cancer. Front. Pharmacol. 2018, 9, 1522. [Google Scholar] [CrossRef]
- Cibulskis, K.; Lawrence, M.S.; Carter, S.L.; Sivachenko, A.; Jaffe, D.; Sougnez, C.; Gabriel, S.; Meyerson, M.; Lander, E.S.; Getz, G. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat. Biotechnol. 2013, 31, 213–219. [Google Scholar] [CrossRef]
- Koboldt, D.C.; Zhang, Q.; Larson, D.E.; Shen, D.; McLellan, M.D.; Lin, L.; Miller, C.A.; Mardis, E.R.; Ding, L.; Wilson, R.K. VarScan 2: Somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012, 22, 568–576. [Google Scholar] [CrossRef] [PubMed]
- Larson, D.E.; Harris, C.C.; Chen, K.; Koboldt, D.C.; Abbott, T.E.; Dooling, D.J.; Ley, T.J.; Mardis, E.R.; Wilson, R.K.; Ding, L. SomaticSniper: Identification of somatic point mutations in whole genome sequencing data. Bioinformatics 2012, 28, 311–317. [Google Scholar] [CrossRef] [PubMed]
- Saunders, C.T.; Wong, W.S.; Swamy, S.; Becq, J.; Murray, L.J.; Cheetham, R.K. Strelka: Accurate somatic small-variant calling from sequenced tumor-normal sample pairs. Bioinformatics 2012, 28, 1811–1817. [Google Scholar] [CrossRef]
- Erik Garrison, G.M. Haplotype-based variant detection from short-read sequencing. arXiv 2012, arXiv:1207.3907. [Google Scholar]
- Carbone, D.P.; Reck, M.; Paz-Ares, L.; Creelan, B.; Horn, L.; Steins, M.; Felip, E.; van den Heuvel, M.M.; Ciuleanu, T.E.; Badin, F.; et al. First-Line Nivolumab in Stage IV or Recurrent Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2017, 376, 2415–2426. [Google Scholar] [CrossRef] [PubMed]
- Cristescu, R.; Mogg, R.; Ayers, M.; Albright, A.; Murphy, E.; Yearley, J.; Sher, X.; Liu, X.Q.; Lu, H.; Nebozhyn, M.; et al. Pan-tumor genomic biomarkers for PD-1 checkpoint blockade-based immunotherapy. Science 2018, 362. [Google Scholar] [CrossRef]
- Hellmann, M.D.; Nathanson, T.; Rizvi, H.; Creelan, B.C.; Sanchez-Vega, F.; Ahuja, A.; Ni, A.; Novik, J.B.; Mangarin, L.M.B.; Abu-Akeel, M.; et al. Genomic Features of Response to Combination Immunotherapy in Patients with Advanced Non-Small-Cell Lung Cancer. Cancer Cell 2018, 33, 843–852.e4. [Google Scholar] [CrossRef]
- Hugo, W.; Zaretsky, J.M.; Sun, L.; Song, C.; Moreno, B.H.; Hu-Lieskovan, S.; Berent-Maoz, B.; Pang, J.; Chmielowski, B.; Cherry, G.; et al. Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell 2016, 165, 35–44. [Google Scholar] [CrossRef]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef]
- Riaz, N.; Havel, J.J.; Makarov, V.; Desrichard, A.; Urba, W.J.; Sims, J.S.; Hodi, F.S.; Martin-Algarra, S.; Mandal, R.; Sharfman, W.H.; et al. Tumor and Microenvironment Evolution during Immunotherapy with Nivolumab. Cell 2017, 171, 934–949.e16. [Google Scholar] [CrossRef]
- Rizvi, H.; Sanchez-Vega, F.; La, K.; Chatila, W.; Jonsson, P.; Halpenny, D.; Plodkowski, A.; Long, N.; Sauter, J.L.; Rekhtman, N.; et al. Molecular Determinants of Response to Anti-Programmed Cell Death (PD)-1 and Anti-Programmed Death-Ligand 1 (PD-L1) Blockade in Patients With Non-Small-Cell Lung Cancer Profiled With Targeted Next-Generation Sequencing. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2018, 36, 633–641. [Google Scholar] [CrossRef] [PubMed]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef] [PubMed]
- Snyder, A.; Makarov, V.; Merghoub, T.; Yuan, J.; Zaretsky, J.M.; Desrichard, A.; Walsh, L.A.; Postow, M.A.; Wong, P.; Ho, T.S.; et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N. Engl. J. Med. 2014, 371, 2189–2199. [Google Scholar] [CrossRef] [PubMed]
- Snyder, A.; Nathanson, T.; Funt, S.A.; Ahuja, A.; Buros Novik, J.; Hellmann, M.D.; Chang, E.; Aksoy, B.A.; Al-Ahmadie, H.; Yusko, E.; et al. Contribution of systemic and somatic factors to clinical response and resistance to PD-L1 blockade in urothelial cancer: An exploratory multi-omic analysis. PLoS Med. 2017, 14, e1002309. [Google Scholar] [CrossRef] [PubMed]
- Van Allen, E.M.; Miao, D.; Schilling, B.; Shukla, S.A.; Blank, C.; Zimmer, L.; Sucker, A.; Hillen, U.; Foppen, M.H.G.; Goldinger, S.M.; et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science 2015, 350, 207–211. [Google Scholar] [CrossRef]
- Rimmer, A.; Phan, H.; Mathieson, I.; Iqbal, Z.; Twigg, S.R.F.; Consortium, W.G.S.; Wilkie, A.O.M.; McVean, G.; Lunter, G. Integrating mapping-, assembly- and haplotype-based approaches for calling variants in clinical sequencing applications. Nat. Genet. 2014, 46, 912–918. [Google Scholar] [CrossRef]
- Wilm, A.; Aw, P.P.; Bertrand, D.; Yeo, G.H.; Ong, S.H.; Wong, C.H.; Khor, C.C.; Petric, R.; Hibberd, M.L.; Nagarajan, N. LoFreq: A sequence-quality aware, ultra-sensitive variant caller for uncovering cell-population heterogeneity from high-throughput sequencing datasets. Nucleic Acids Res. 2012, 40, 11189–11201. [Google Scholar] [CrossRef]
- Lai, Z.; Markovets, A.; Ahdesmaki, M.; Chapman, B.; Hofmann, O.; McEwen, R.; Johnson, J.; Dougherty, B.; Barrett, J.C.; Dry, J.R. VarDict: A novel and versatile variant caller for next-generation sequencing in cancer research. Nucleic Acids Res. 2016, 44, e108. [Google Scholar] [CrossRef]
- Roth, A.; Ding, J.; Morin, R.; Crisan, A.; Ha, G.; Giuliany, R.; Bashashati, A.; Hirst, M.; Turashvili, G.; Oloumi, A.; et al. JointSNVMix: A probabilistic model for accurate detection of somatic mutations in normal/tumour paired next-generation sequencing data. Bioinformatics 2012, 28, 907–913. [Google Scholar] [CrossRef]
- Ding, J.; Bashashati, A.; Roth, A.; Oloumi, A.; Tse, K.; Zeng, T.; Haffari, G.; Hirst, M.; Marra, M.A.; Condon, A.; et al. Feature-based classifiers for somatic mutation detection in tumour-normal paired sequencing data. Bioinformatics 2012, 28, 167–175. [Google Scholar] [CrossRef]
- Shiraishi, Y.; Sato, Y.; Chiba, K.; Okuno, Y.; Nagata, Y.; Yoshida, K.; Shiba, N.; Hayashi, Y.; Kume, H.; Homma, Y.; et al. An empirical Bayesian framework for somatic mutation detection from cancer genome sequencing data. Nucleic Acids Res. 2013, 41, e89. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Xi, L.; Hughes, D.S.; Zhang, J.; Zhang, J.; Futreal, P.A.; Wheeler, D.A.; Wang, W. MuSE: Accounting for tumor heterogeneity using a sample-specific error model improves sensitivity and specificity in mutation calling from sequencing data. Genome Biol. 2016, 17, 178. [Google Scholar] [CrossRef] [PubMed]
- Radenbaugh, A.J.; Ma, S.; Ewing, A.; Stuart, J.M.; Collisson, E.A.; Zhu, J.; Haussler, D. RADIA: RNA and DNA integrated analysis for somatic mutation detection. PLoS ONE 2014, 9, e111516. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Jeong, K.; Bhutani, K.; Lee, J.; Patel, A.; Scott, E.; Nam, H.; Lee, H.; Gleeson, J.G.; Bafna, V. Virmid: Accurate detection of somatic mutations with sample impurity inference. Genome Biol. 2013, 14, R90. [Google Scholar] [CrossRef] [PubMed]
- Gerstung, M.; Papaemmanuil, E.; Campbell, P.J. Subclonal variant calling with multiple samples and prior knowledge. Bioinformatics 2014, 30, 1198–1204. [Google Scholar] [CrossRef]
- Hansen, N.F.; Gartner, J.J.; Mei, L.; Samuels, Y.; Mullikin, J.C. Shimmer: Detection of genetic alterations in tumors using next-generation sequence data. Bioinformatics 2013, 29, 1498–1503. [Google Scholar] [CrossRef]
- Kassahn, K.S.; Holmes, O.; Nones, K.; Patch, A.M.; Miller, D.K.; Christ, A.N.; Harliwong, I.; Bruxner, T.J.; Xu, Q.; Anderson, M.; et al. Somatic point mutation calling in low cellularity tumors. PLoS ONE 2013, 8, e74380. [Google Scholar] [CrossRef]
- Cantarel, B.L.; Weaver, D.; McNeill, N.; Zhang, J.; Mackey, A.J.; Reese, J. BAYSIC: A Bayesian method for combining sets of genome variants with improved specificity and sensitivity. BMC Bioinform. 2014, 15, 104. [Google Scholar] [CrossRef]
- Fang, L.T.; Afshar, P.T.; Chhibber, A.; Mohiyuddin, M.; Fan, Y.; Mu, J.C.; Gibeling, G.; Barr, S.; Asadi, N.B.; Gerstein, M.B.; et al. An ensemble approach to accurately detect somatic mutations using SomaticSeq. Genome Biol. 2015, 16, 197. [Google Scholar] [CrossRef]
- Jones, D.; Raine, K.M.; Davies, H.; Tarpey, P.S.; Butler, A.P.; Teague, J.W.; Nik-Zainal, S.; Campbell, P.J. cgpCaVEManWrapper: Simple Execution of CaVEMan in Order to Detect Somatic Single Nucleotide Variants in NGS Data. Curr. Protoc. Bioinform. 2016, 56, 15. [Google Scholar] [CrossRef]
- Spinella, J.F.; Mehanna, P.; Vidal, R.; Saillour, V.; Cassart, P.; Richer, C.; Ouimet, M.; Healy, J.; Sinnett, D. SNooPer: A machine learning-based method for somatic variant identification from low-pass next-generation sequencing. BMC Genom. 2016, 17, 912. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Loewer, M.; Aluru, S.; Schmidt, B. SNVSniffer: An integrated caller for germline and somatic single-nucleotide and indel mutations. BMC Syst. Biol. 2016, 10 (Suppl. 2), 47. [Google Scholar] [CrossRef]
- Usuyama, N.; Shiraishi, Y.; Sato, Y.; Kume, H.; Homma, Y.; Ogawa, S.; Miyano, S.; Imoto, S. HapMuC: Somatic mutation calling using heterozygous germ line variants near candidate mutations. Bioinformatics 2014, 30, 3302–3309. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Wang, P.; Xu, F.; Luo, R.; Wong, M.P.; Lam, T.W.; Wang, J. FaSD-somatic: A fast and accurate somatic SNV detection algorithm for cancer genome sequencing data. Bioinformatics 2014, 30, 2498–2500. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sengupta, S.; Gulukota, K.; Zhu, Y.; Ober, C.; Naughton, K.; Wentworth-Sheilds, W.; Ji, Y. Ultra-fast local-haplotype variant calling using paired-end DNA-sequencing data reveals somatic mosaicism in tumor and normal blood samples. Nucleic Acids Res. 2016, 44, e25. [Google Scholar] [CrossRef] [PubMed]
- Carrot-Zhang, J.; Majewski, J. LoLoPicker: Detecting low allelic-fraction variants from low-quality cancer samples. Oncotarget 2017, 8, 37032–37040. [Google Scholar] [CrossRef] [PubMed]
- Xu, C. A review of somatic single nucleotide variant calling algorithms for next-generation sequencing data. Comput. Struct. Biotechnol. J. 2018, 16, 15–24. [Google Scholar] [CrossRef]
- Liu, Z.K.; Shang, Y.K.; Chen, Z.N.; Bian, H. A three-caller pipeline for variant analysis of cancer whole-exome sequencing data. Mol. Med. Rep. 2017, 15, 2489–2494. [Google Scholar] [CrossRef]
- Kroigard, A.B.; Thomassen, M.; Laenkholm, A.V.; Kruse, T.A.; Larsen, M.J. Evaluation of Nine Somatic Variant Callers for Detection of Somatic Mutations in Exome and Targeted Deep Sequencing Data. PLoS ONE 2016, 11, e0151664. [Google Scholar] [CrossRef]
- Cai, L.; Yuan, W.; Zhang, Z.; He, L.; Chou, K.C. In-depth comparison of somatic point mutation callers based on different tumor next-generation sequencing depth data. Sci. Rep. 2016, 6, 36540. [Google Scholar] [CrossRef]
- Kumaran, M.; Subramanian, U.; Devarajan, B. Performance assessment of variant calling pipelines using human whole exome sequencing and simulated data. BMC Bioinform. 2019, 20, 342. [Google Scholar] [CrossRef] [PubMed]
- Hwang, S.; Kim, E.; Lee, I.; Marcotte, E.M. Systematic comparison of variant calling pipelines using gold standard personal exome variants. Sci. Rep. 2015, 5, 17875. [Google Scholar] [CrossRef] [PubMed]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef] [PubMed]
- Singer, J.; Ruscheweyh, H.J.; Hofmann, A.L.; Thurnherr, T.; Singer, F.; Toussaint, N.C.; Ng, C.K.Y.; Piscuoglio, S.; Beisel, C.; Christofori, G.; et al. NGS-pipe: A flexible, easily extendable and highly configurable framework for NGS analysis. Bioinformatics 2018, 34, 107–108. [Google Scholar] [CrossRef]
- Lawrence, M.; Gentleman, R. VariantTools: An extensible framework for developing and testing variant callers. Bioinformatics 2017, 33, 3311–3313. [Google Scholar] [CrossRef]
- Knaus, B.J.; Grunwald, N.J. vcfr: A package to manipulate and visualize variant call format data in R. Mol. Ecol. Resour. 2017, 17, 44–53. [Google Scholar] [CrossRef]
- Pietrelli, A.; Valenti, L. myVCF: A desktop application for high-throughput mutations data management. Bioinformatics 2017, 33, 3676–3678. [Google Scholar] [CrossRef]
- Rashid, M.; Robles-Espinoza, C.D.; Rust, A.G.; Adams, D.J. Cake: A bioinformatics pipeline for the integrated analysis of somatic variants in cancer genomes. Bioinformatics 2013, 29, 2208–2210. [Google Scholar] [CrossRef]
- Di Nanni, N.; Moscatelli, M.; Gnocchi, M.; Milanesi, L.; Mosca, E. isma: An R package for the integrative analysis of mutations detected by multiple pipelines. BMC Bioinform. 2019, 20, 107. [Google Scholar] [CrossRef]
- Huang, W.; Guo, Y.A.; Muthukumar, K.; Baruah, P.; Chang, M.M.; Skanderup, A.J. SMuRF: Portable and accurate ensemble prediction of somatic mutations. Bioinformatics 2019. [Google Scholar] [CrossRef]
- Anzar, I.; Sverchkova, A.; Stratford, R.; Clancy, T. NeoMutate: An ensemble machine learning framework for the prediction of somatic mutations in cancer. BMC Med. Genom. 2019, 12, 63. [Google Scholar] [CrossRef] [PubMed]
- Wolstencroft, K.; Haines, R.; Fellows, D.; Williams, A.; Withers, D.; Owen, S.; Soiland-Reyes, S.; Dunlop, I.; Nenadic, A.; Fisher, P.; et al. The Taverna workflow suite: Designing and executing workflows of Web Services on the desktop, web or in the cloud. Nucleic Acids Res. 2013, 41, W557–W561. [Google Scholar] [CrossRef] [PubMed]
- Berthold, M.R.; Cebron, N.; Dill, F.; Gabriel, T.R.; Kötter, T.; Meinl, T.; Ohl, P.; Sieb, C.; Thiel, K.; Wiswedel, B. KNIME: The Konstanz Information Miner; Springer: Berlin/Heidelberg, Germany, 2008; pp. 319–326. [Google Scholar]
- Langmead, B.; Nellore, A. Cloud computing for genomic data analysis and collaboration. Nat. Rev. Genet. 2018, 19, 208–219. [Google Scholar] [CrossRef] [PubMed]
- Lau, J.W.; Lehnert, E.; Sethi, A.; Malhotra, R.; Kaushik, G.; Onder, Z.; Groves-Kirkby, N.; Mihajlovic, A.; DiGiovanna, J.; Srdic, M.; et al. The Cancer Genomics Cloud: Collaborative, Reproducible, and Democratized-A New Paradigm in Large-Scale Computational Research. Cancer Res. 2017, 77, e3–e6. [Google Scholar] [CrossRef] [PubMed]
- Reich, M.; Liefeld, T.; Gould, J.; Lerner, J.; Tamayo, P.; Mesirov, J.P. GenePattern 2.0. Nat. Genet. 2006, 38, 500–501. [Google Scholar] [CrossRef]
- Giardine, B.; Riemer, C.; Hardison, R.C.; Burhans, R.; Elnitski, L.; Shah, P.; Zhang, Y.; Blankenberg, D.; Albert, I.; Taylor, J.; et al. Galaxy: A platform for interactive large-scale genome analysis. Genome Res. 2005, 15, 1451–1455. [Google Scholar] [CrossRef]
- Okonechnikov, K.; Golosova, O.; Fursov, M.; Ugene Team. Unipro UGENE: A unified bioinformatics toolkit. Bioinformatics 2012, 28, 1166–1167. [Google Scholar] [CrossRef]
- Wang, L.; Lu, Z.; Van Buren, P.; Ware, D. SciApps: A cloud-based platform for reproducible bioinformatics workflows. Bioinformatics 2018, 34, 3917–3920. [Google Scholar] [CrossRef]
- Afgan, E.; Baker, D.; Batut, B.; van den Beek, M.; Bouvier, D.; Cech, M.; Chilton, J.; Clements, D.; Coraor, N.; Gruning, B.A.; et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res. 2018, 46, W537–W544. [Google Scholar] [CrossRef]
- Conrad, D.F.; Pinto, D.; Redon, R.; Feuk, L.; Gokcumen, O.; Zhang, Y.; Aerts, J.; Andrews, T.D.; Barnes, C.; Campbell, P.; et al. Origins and functional impact of copy number variation in the human genome. Nature 2010, 464, 704–712. [Google Scholar] [CrossRef]
- Handsaker, R.E.; Van Doren, V.; Berman, J.R.; Genovese, G.; Kashin, S.; Boettger, L.M.; McCarroll, S.A. Large multiallelic copy number variations in humans. Nat. Genet. 2015, 47, 296–303. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Ho, S.S.; Zhang, X.; Pattni, R.; Haraksingh, R.R.; Urban, A.E. Whole-genome sequencing analysis of CNV using low-coverage and paired-end strategies is efficient and outperforms array-based CNV analysis. J. Med Genet. 2018, 55, 735–743. [Google Scholar] [CrossRef] [PubMed]
- Tan, R.; Wang, Y.; Kleinstein, S.E.; Liu, Y.; Zhu, X.; Guo, H.; Jiang, Q.; Allen, A.S.; Zhu, M. An evaluation of copy number variation detection tools from whole-exome sequencing data. Hum. Mutat. 2014, 35, 899–907. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Morrison, C.D.; Johnson, C.S.; Trump, D.L.; Qin, M.; Conroy, J.C.; Wang, J.; Liu, S. Computational methods for detecting copy number variations in cancer genome using next generation sequencing: Principles and challenges. Oncotarget 2013, 4, 1868–1881. [Google Scholar] [CrossRef]
- Abyzov, A.; Urban, A.E.; Snyder, M.; Gerstein, M. CNVnator: An approach to discover, genotype, and characterize typical and atypical CNVs from family and population genome sequencing. Genome Res. 2011, 21, 974–984. [Google Scholar] [CrossRef]
- Xie, C.; Tammi, M.T. CNV-seq, a new method to detect copy number variation using high-throughput sequencing. BMC Bioinform. 2009, 10, 80. [Google Scholar] [CrossRef]
- Krumm, N.; Sudmant, P.H.; Ko, A.; O’Roak, B.J.; Malig, M.; Coe, B.P.; Project, N.E.S.; Quinlan, A.R.; Nickerson, D.A.; Eichler, E.E. Copy number variation detection and genotyping from exome sequence data. Genome Res. 2012, 22, 1525–1532. [Google Scholar] [CrossRef]
- Boeva, V.; Popova, T.; Bleakley, K.; Chiche, P.; Cappo, J.; Schleiermacher, G.; Janoueix-Lerosey, I.; Delattre, O.; Barillot, E. Control-FREEC: A tool for assessing copy number and allelic content using next-generation sequencing data. Bioinformatics 2012, 28, 423–425. [Google Scholar] [CrossRef]
- Sathirapongsasuti, J.F.; Lee, H.; Horst, B.A.; Brunner, G.; Cochran, A.J.; Binder, S.; Quackenbush, J.; Nelson, S.F. Exome sequencing-based copy-number variation and loss of heterozygosity detection: ExomeCNV. Bioinformatics 2011, 27, 2648–2654. [Google Scholar] [CrossRef]
- Fromer, M.; Moran, J.L.; Chambert, K.; Banks, E.; Bergen, S.E.; Ruderfer, D.M.; Handsaker, R.E.; McCarroll, S.A.; O’Donovan, M.C.; Owen, M.J.; et al. Discovery and statistical genotyping of copy-number variation from whole-exome sequencing depth. Am. J. Hum. Genet. 2012, 91, 597–607. [Google Scholar] [CrossRef]
- Plagnol, V.; Curtis, J.; Epstein, M.; Mok, K.Y.; Stebbings, E.; Grigoriadou, S.; Wood, N.W.; Hambleton, S.; Burns, S.O.; Thrasher, A.J.; et al. A robust model for read count data in exome sequencing experiments and implications for copy number variant calling. Bioinformatics 2012, 28, 2747–2754. [Google Scholar] [CrossRef] [PubMed]
- Klambauer, G.; Schwarzbauer, K.; Mayr, A.; Clevert, D.A.; Mitterecker, A.; Bodenhofer, U.; Hochreiter, S. cn.MOPS: Mixture of Poissons for discovering copy number variations in next-generation sequencing data with a low false discovery rate. Nucleic Acids Res. 2012, 40, e69. [Google Scholar] [CrossRef] [PubMed]
- Talevich, E.; Shain, A.H.; Botton, T.; Bastian, B.C. CNVkit: Genome-Wide Copy Number Detection and Visualization from Targeted DNA Sequencing. PLoS Comput. Biol. 2016, 12, e1004873. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Lupat, R.; Amarasinghe, K.C.; Thompson, E.R.; Doyle, M.A.; Ryland, G.L.; Tothill, R.W.; Halgamuge, S.K.; Campbell, I.G.; Gorringe, K.L. CONTRA: Copy number analysis for targeted resequencing. Bioinformatics 2012, 28, 1307–1313. [Google Scholar] [CrossRef] [PubMed]
- Favero, F.; Joshi, T.; Marquard, A.M.; Birkbak, N.J.; Krzystanek, M.; Li, Q.; Szallasi, Z.; Eklund, A.C. Sequenza: Allele-specific copy number and mutation profiles from tumor sequencing data. Ann. Oncol. Off. J. Eur. Soc. Med Oncol. 2015, 26, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Magi, A.; Tattini, L.; Cifola, I.; D’Aurizio, R.; Benelli, M.; Mangano, E.; Battaglia, C.; Bonora, E.; Kurg, A.; Seri, M.; et al. EXCAVATOR: Detecting copy number variants from whole-exome sequencing data. Genome Biol. 2013, 14, R120. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Oldridge, D.A.; Diskin, S.J.; Zhang, N.R. CODEX: A normalization and copy number variation detection method for whole exome sequencing. Nucleic Acids Res. 2015, 43, e39. [Google Scholar] [CrossRef]
- Amarasinghe, K.C.; Li, J.; Hunter, S.M.; Ryland, G.L.; Cowin, P.A.; Campbell, I.G.; Halgamuge, S.K. Inferring copy number and genotype in tumour exome data. BMC Genom. 2014, 15, 732. [Google Scholar] [CrossRef]
- Deng, X. SeqGene: A comprehensive software solution for mining exome- and transcriptome- sequencing data. BMC Bioinform. 2011, 12, 267. [Google Scholar] [CrossRef]
- Shi, Y.; Majewski, J. FishingCNV: A graphical software package for detecting rare copy number variations in exome-sequencing data. Bioinformatics 2013, 29, 1461–1462. [Google Scholar] [CrossRef]
- Gambin, T.; Akdemir, Z.C.; Yuan, B.; Gu, S.; Chiang, T.; Carvalho, C.M.B.; Shaw, C.; Jhangiani, S.; Boone, P.M.; Eldomery, M.K.; et al. Homozygous and hemizygous CNV detection from exome sequencing data in a Mendelian disease cohort. Nucleic Acids Res. 2017, 45, 1633–1648. [Google Scholar] [CrossRef] [PubMed]
- Coin, L.J.; Cao, D.; Ren, J.; Zuo, X.; Sun, L.; Yang, S.; Zhang, X.; Cui, Y.; Li, Y.; Jin, X.; et al. An exome sequencing pipeline for identifying and genotyping common CNVs associated with disease with application to psoriasis. Bioinformatics 2012, 28, i370–i374. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Packer, J.S.; Maxwell, E.K.; O’Dushlaine, C.; Lopez, A.E.; Dewey, F.E.; Chernomorsky, R.; Baras, A.; Overton, J.D.; Habegger, L.; Reid, J.G. CLAMMS: A scalable algorithm for calling common and rare copy number variants from exome sequencing data. Bioinformatics 2016, 32, 133–135. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Bell, J.M.; Zavala, N.A.; Ji, H.P.; Zhang, N.R. Allele-specific copy number profiling by next-generation DNA sequencing. Nucleic Acids Res. 2015, 43, e23. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zhang, Z.; Hao, K. SAAS-CNV: A Joint Segmentation Approach on Aggregated and Allele Specific Signals for the Identification of Somatic Copy Number Alterations with Next-Generation Sequencing Data. PLoS Comput. Biol. 2015, 11, e1004618. [Google Scholar] [CrossRef]
- Straver, R.; Weiss, M.M.; Waisfisz, Q.; Sistermans, E.A.; Reinders, M.J.T. WISExome: A within-sample comparison approach to detect copy number variations in whole exome sequencing data. Eur. J. Hum. Genet. 2017, 25, 1354–1363. [Google Scholar] [CrossRef]
- Zare, F.; Dow, M.; Monteleone, N.; Hosny, A.; Nabavi, S. An evaluation of copy number variation detection tools for cancer using whole exome sequencing data. BMC Bioinform. 2017, 18, 286. [Google Scholar] [CrossRef]
- Kim, H.Y.; Choi, J.W.; Lee, J.Y.; Kong, G. Gene-based comparative analysis of tools for estimating copy number alterations using whole-exome sequencing data. Oncotarget 2017, 8, 27277–27285. [Google Scholar] [CrossRef]
- Alkodsi, A.; Louhimo, R.; Hautaniemi, S. Comparative analysis of methods for identifying somatic copy number alterations from deep sequencing data. Brief. Bioinform. 2015, 16, 242–254. [Google Scholar] [CrossRef]
- Nam, J.Y.; Kim, N.K.; Kim, S.C.; Joung, J.G.; Xi, R.; Lee, S.; Park, P.J.; Park, W.Y. Evaluation of somatic copy number estimation tools for whole-exome sequencing data. Brief. Bioinform. 2016, 17, 185–192. [Google Scholar] [CrossRef]
- Gao, J.; Wan, C.; Zhang, H.; Li, A.; Zang, Q.; Ban, R.; Ali, A.; Yu, Z.; Shi, Q.; Jiang, X.; et al. Anaconda: AN automated pipeline for somatic COpy Number variation Detection and Annotation from tumor exome sequencing data. BMC Bioinform. 2017, 18, 436. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Wang, R.; Urrutia, E.; Anastopoulos, I.N.; Nathanson, K.L.; Zhang, N.R. CODEX2: Full-spectrum copy number variation detection by high-throughput DNA sequencing. Genome Biol. 2018, 19, 202. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, N.; Walker, G.C. Mechanisms of DNA damage, repair, and mutagenesis. Environ. Mol. Mutagen. 2017, 58, 235–263. [Google Scholar] [CrossRef] [PubMed]
- Shee, C.; Gibson, J.L.; Rosenberg, S.M. Two mechanisms produce mutation hotspots at DNA breaks in Escherichia coli. Cell Rep. 2012, 2, 714–721. [Google Scholar] [CrossRef] [PubMed]
- Hoppe, M.M.; Sundar, R.; Tan, D.S.P.; Jeyasekharan, A.D. Biomarkers for Homologous Recombination Deficiency in Cancer. J. Natl. Cancer Inst. 2018, 110, 704–713. [Google Scholar] [CrossRef]
- Davies, H.; Glodzik, D.; Morganella, S.; Yates, L.R.; Staaf, J.; Zou, X.; Ramakrishna, M.; Martin, S.; Boyault, S.; Sieuwerts, A.M.; et al. HRDetect is a predictor of BRCA1 and BRCA2 deficiency based on mutational signatures. Nat. Med. 2017, 23, 517–525. [Google Scholar] [CrossRef]
- Sztupinszki, Z.; Diossy, M.; Krzystanek, M.; Reiniger, L.; Csabai, I.; Favero, F.; Birkbak, N.J.; Eklund, A.C.; Syed, A.; Szallasi, Z. Migrating the SNP array-based homologous recombination deficiency measures to next generation sequencing data of breast cancer. NPJ Breast Cancer 2018, 4, 16. [Google Scholar] [CrossRef]
- Riaz, N.; Blecua, P.; Lim, R.S.; Shen, R.; Higginson, D.S.; Weinhold, N.; Norton, L.; Weigelt, B.; Powell, S.N.; Reis-Filho, J.S. Pan-cancer analysis of bi-allelic alterations in homologous recombination DNA repair genes. Nat. Commun. 2017, 8, 857. [Google Scholar] [CrossRef]
- Hargadon, K.M.; Johnson, C.E.; Williams, C.J. Immune checkpoint blockade therapy for cancer: An overview of FDA-approved immune checkpoint inhibitors. Int. Immunopharmacol. 2018, 62, 29–39. [Google Scholar] [CrossRef]
- Melendez, B.; Van Campenhout, C.; Rorive, S.; Remmelink, M.; Salmon, I.; D’Haene, N. Methods of measurement for tumor mutational burden in tumor tissue. Transl. Lung Cancer Res. 2018, 7, 661–667. [Google Scholar] [CrossRef]
- Buttner, R.; Longshore, J.W.; Lopez-Rios, F.; Merkelbach-Bruse, S.; Normanno, N.; Rouleau, E.; Penault-Llorca, F. Implementing TMB measurement in clinical practice: Considerations on assay requirements. ESMO Open 2019, 4, e000442. [Google Scholar] [CrossRef] [PubMed]
- FoCR. Friends of Cancer Research Announces Launch of Phase II TMB Harmonization Project; FoCR: Washington, DC, USA, 2018. [Google Scholar]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Borresen-Dale, A.L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, L.B.; Kim, J.; Haradhvala, N.J.; Huang, M.N.; Ng, A.W.; Wu, Y.; Boot, A.; Covington, K.R.; Gordenin, D.A.; Bergstrom, E.N.; et al. The Repertoire of Mutational Signatures in Human Cancer. bioRxiv 2019. [Google Scholar] [CrossRef]
- Rosenthal, R.; McGranahan, N.; Herrero, J.; Taylor, B.S.; Swanton, C. DeconstructSigs: Delineating mutational processes in single tumors distinguishes DNA repair deficiencies and patterns of carcinoma evolution. Genome Biol. 2016, 17, 31. [Google Scholar] [CrossRef] [PubMed]
- Blokzijl, F.; Janssen, R.; van Boxtel, R.; Cuppen, E. MutationalPatterns: Comprehensive genome-wide analysis of mutational processes. Genome Med. 2018, 10, 33. [Google Scholar] [CrossRef] [PubMed]
- Maura, F.; Degasperi, A.; Nadeu, F.; Leongamornlert, D.; Davies, H.; Moore, L.; Royo, R.; Ziccheddu, B.; Puente, X.S.; Avet-Loiseau, H.; et al. A practical guide for mutational signature analysis in hematological malignancies. Nat. Commun. 2019, 10, 2969. [Google Scholar] [CrossRef]
- Van Hoeck, A.; Tjoonk, N.H.; van Boxtel, R.; Cuppen, E. Portrait of a cancer: Mutational signature analyses for cancer diagnostics. BMC Cancer 2019, 19, 457. [Google Scholar] [CrossRef]
- Germano, G.; Lamba, S.; Rospo, G.; Barault, L.; Magri, A.; Maione, F.; Russo, M.; Crisafulli, G.; Bartolini, A.; Lerda, G.; et al. Inactivation of DNA repair triggers neoantigen generation and impairs tumour growth. Nature 2017, 552, 116–120. [Google Scholar] [CrossRef]
- Baudrin, L.G.; Deleuze, J.F.; How-Kit, A. Molecular and Computational Methods for the Detection of Microsatellite Instability in Cancer. Front. Oncol. 2018, 8, 621. [Google Scholar] [CrossRef]
- Kautto, E.A.; Bonneville, R.; Miya, J.; Yu, L.; Krook, M.A.; Reeser, J.W.; Roychowdhury, S. Performance evaluation for rapid detection of pan-cancer microsatellite instability with MANTIS. Oncotarget 2017, 8, 7452–7463. [Google Scholar] [CrossRef]
- Huang, M.N.; McPherson, J.R.; Cutcutache, I.; Teh, B.T.; Tan, P.; Rozen, S.G. MSIseq: Software for Assessing Microsatellite Instability from Catalogs of Somatic Mutations. Sci. Rep. 2015, 5, 13321. [Google Scholar] [CrossRef]
- Wang, C.; Liang, C. MSIpred: A python package for tumor microsatellite instability classification from tumor mutation annotation data using a support vector machine. Sci. Rep. 2018, 8, 17546. [Google Scholar] [CrossRef] [PubMed]
- Foltz, S.M.; Liang, W.W.; Xie, M.; Ding, L. MIRMMR: Binary classification of microsatellite instability using methylation and mutations. Bioinformatics 2017, 33, 3799–3801. [Google Scholar] [CrossRef] [PubMed]
- Hause, R.J.; Pritchard, C.C.; Shendure, J.; Salipante, S.J. Classification and characterization of microsatellite instability across 18 cancer types. Nat. Med. 2016, 22, 1342–1350. [Google Scholar] [CrossRef]
- Xia, J.; Chiu, L.Y.; Nehring, R.B.; Bravo Nunez, M.A.; Mei, Q.; Perez, M.; Zhai, Y.; Fitzgerald, D.M.; Pribis, J.P.; Wang, Y.; et al. Bacteria-to-Human Protein Networks Reveal Origins of Endogenous DNA Damage. Cell 2019, 176, 127–143.e24. [Google Scholar] [CrossRef] [PubMed]
- Santarpia, L.; Bottai, G.; Kelly, C.M.; Gyorffy, B.; Szekely, B.; Pusztai, L. Deciphering and Targeting Oncogenic Mutations and Pathways in Breast Cancer. Oncologist 2016, 21, 1063–1078. [Google Scholar] [CrossRef]
- Oh, B.Y.; Shin, H.T.; Yun, J.W.; Kim, K.T.; Kim, J.; Bae, J.S.; Cho, Y.B.; Lee, W.Y.; Yun, S.H.; Park, Y.A.; et al. Intratumor heterogeneity inferred from targeted deep sequencing as a prognostic indicator. Sci. Rep. 2019, 9, 4542. [Google Scholar] [CrossRef]
- Goh, G.; McGranahan, N.; Wilson, G.A. Computational Methods for Analysis of Tumor Clonality and Evolutionary History. Methods Mol. Biol. 2019, 1878, 217–226. [Google Scholar] [CrossRef]
- Miura, S.; Gomez, K.; Murillo, O.; Huuki, L.A.; Vu, T.; Buturla, T.; Kumar, S. Predicting clone genotypes from tumor bulk sequencing of multiple samples. Bioinformatics 2018, 34, 4017–4026. [Google Scholar] [CrossRef]
- Miura, S.; Vu, T.; Deng, J.; Buturla, T.; Choi, J.; Kumar, S. Power and pitfalls of computational methods for inferring clone phylogenies and mutation orders from bulk sequencing data. bioRxiv 2019. [Google Scholar] [CrossRef]
- Pongor, L.; Harami-Papp, H.; Mehes, E.; Czirok, A.; Gyorffy, B. Cell Dispersal Influences Tumor Heterogeneity and Introduces a Bias in NGS Data Interpretation. Sci. Rep. 2017, 7, 7358. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Lee, M.S.; Lu, H.; Oh, D.Y.; Kim, Y.J.; Park, D.; Park, G.; Ren, X.; Bristow, C.A.; Haseley, P.S.; et al. Analyzing Somatic Genome Rearrangements in Human Cancers by Using Whole-Exome Sequencing. Am. J. Hum. Genet. 2016, 98, 843–856. [Google Scholar] [CrossRef] [PubMed]
- D’Agaro, E. Artificial intelligence used in genome analysis studies. EuroBiotech J. 2018, 2, 78–88. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Name | Published | Cited in 2018 | Control Needed | InDel detection | Contamination Correction | Trained on Cancer Data | Environment | Ref |
|---|---|---|---|---|---|---|---|---|
| Varscan2 | 2012 | 2229 | + | + | − | + | Java, Perl, R, Galaxy | [21] |
| MuTect2 * | 2013 | 2005 | + | − | + | + | Java, R | [20] |
| FreeBayes | 2012 | 1121 | − | + | − | + | C, C++, Galaxy | [24] |
| Strelka * | 2012 | 759 | + | + | − | + | C++, Perl | [23] |
| Platypus * | 2014 | 462 | − | + | − | + | C, Cython, Python | [36] |
| SomaticSniper * | 2012 | 373 | + | − | − | + | C, Galaxy | [22] |
| LoFreq * | 2012 | 349 | − | + | + | + | Python | [37] |
| VarDict * | 2016 | 171 | − | + | − | + | Perl | [38] |
| JointSNVMix * | 2012 | 160 | + | − | − | + | C, C++, Python, Galaxy | [39] |
| MutationSeq * | 2012 | 108 | + | − | − | + | C++, Python | [40] |
| EBCall * | 2013 | 85 | + | + | − | + | C++, Perl, R, Shell | [41] |
| MuSE * | 2016 | 65 | + | − | + | + | C, C++ | [42] |
| RADIA | 2014 | 53 | + | − | + | + | Python | [43] |
| Virmid | 2013 | 49 | + | − | + | + | Java | [44] |
| deepSNV * | 2014 | 47 | + | − | − | + | R | [45] |
| Shimmer * | 2013 | 45 | + | − | + | + | C, Perl, R | [46] |
| qSNP * | 2013 | 40 | + | − | + | − | Java | [47] |
| BAYSIC | 2014 | 39 | + | − | − | + | R | [48] |
| SomaticSeq * | 2015 | 38 | + | + | − | + | Python, R | [49] |
| CaVEMan * | 2016 | 31 | + | − | + | + | C | [50] |
| SNooPer * | 2016 | 26 | − | + | + | + | Perl | [51] |
| SNVSniffer * | 2016 | 17 | − | + | − | + | C++ | [52] |
| HapMuC | 2014 | 15 | − | + | − | + | C++, Python, Ruby | [53] |
| FaSD-somatic | 2014 | 13 | − | − | − | + | C, C++ | [54] |
| LocHap * | 2016 | 8 | + | + | + | + | g++ complier, GNU Make | [55] |
| LoLoPicker * | 2017 | 6 | + | − | + | + | Python | [56] |
| Name | Description | Year | Citation | License | System type | Ref. |
|---|---|---|---|---|---|---|
| Galaxy | Open-source web-platform with several analysis tools | 2005 | 1977 | free | cloud-based | [77] |
| GenePattern | Workflow management system, provides access to multiple genomic analysis tools | 2006 | 1573 | free | cloud-based | [76] |
| KNIME | Software enabling creation, analysis, and visualization of data | 2008 | 1476 | free | local installation needed | [73] |
| UGENE | Workflow management system installed on a local computer | 2012 | 876 | free | local installation needed | [78] |
| Taverna | Open source software tool for designing and executing workflows | 2013 | 643 | free | local installation needed | [72] |
| Cancer Genomics Cloud | Provides access to data, tools, and computing resources | 2017 | 32 | commercial | cloud-based | [75] |
| SciApps | Platform for building, running, and sharing scientific workflows | 2018 | 5 | free | cloud-based | [79] |
| Terra | Bioinformatic workspace, including a repository of public best practices, methods, and public data sets | − | − | commercial | cloud-based | − |
| Name | Published | Control Needed | Contamination Correction | GC-Content Correction | Trained on Cancer Data | Cited in 2018 | Environment | Ref. |
|---|---|---|---|---|---|---|---|---|
| Varscan2 | 2012 | + | − | − | + | 2229 | Java, Perl, R, Galaxy | [21] |
| CNVnator | 2011 | + | − | + | − | 767 | C++ | [86] |
| CNV-Seq | 2009 | + | − | − | − | 463 | Perl, R | [87] |
| CoNIFER | 2012 | − | + | − | − | 378 | Python | [88] |
| Control-FREEC * | 2012 | − | + | + | + | 342 | C, C++, R | [89] |
| ExomeCNV | 2011 | + | + | − | + | 338 | R | [90] |
| XHMM | 2012 | − | + | + | + | 322 | C++ | [91] |
| ExomeDepth | 2012 | + | − | + | − | 264 | R | [92] |
| cn.MOPS | 2012 | − | + | + | − | 249 | R | [93] |
| Cnvkit * | 2016 | + | + | + | + | 219 | Python, Galaxy | [94] |
| CONTRA | 2012 | − | − | + | − | 194 | Python, R | [95] |
| Sequenza * | 2015 | + | − | + | + | 167 | Python, R | [96] |
| EXCAVATOR | 2013 | + | + | + | + | 155 | Perl | [97] |
| CODEX | 2015 | − | + | + | + | 72 | R | [98] |
| ADTEx | 2014 | + | + | − | + | 57 | Python, R | [99] |
| Seqgene | 2011 | + | − | − | + | 43 | R | [100] |
| FishingCNV | 2013 | − | − | − | − | 41 | Java, R | [101] |
| HMZDelFinder | 2017 | − | − | − | − | 33 | R | [102] |
| ExoCNVTest | 2012 | + | − | − | − | 27 | Java, R | [103] |
| CLAMMS | 2016 | − | − | + | − | 23 | C | [104] |
| falcon | 2015 | + | + | − | + | 22 | C | [105] |
| saasCNV * | 2015 | + | + | − | + | 17 | R | [106] |
| WISExome | 2017 | − | − | − | − | 1 | C, C++ | [107] |
| Tradename | Description | Year | Target | Tumor | Utility |
|---|---|---|---|---|---|
| Illumina MiSeqDX platform | High throughput DNA sequence analyzer | 2013 | - | - | technology |
| FoundationFocus CDxBRCA | NGS oncology panel, somatic or germline variant detection system | 2016 | BRCA | ovarian | diagnosis |
| MSK-IMPACT | NGS-based tumor profiling test | 2017 | 468 genes | various | predisposition, diagnosis |
| FoundationOne CDx | NGS oncology panel, somatic or germline variant detection system | 2017 | 324 genes | various | predisposition, diagnosis |
| Oncomine Dx Target Test | NGS oncology panel, somatic or germline variant detection system | 2017 | 24 genes | lung | diagnosis |
| Praxis Extended RAS Panel | NGS oncology panel, somatic or germline variant detection system | 2017 | RAS | colon | diagnosis |
| Adaptive Biotechnologies clonoSEQ | DNA-based test for minimal residual disease for hematologic malignancies | 2018 | BCL1, BCL2 | leukemia, myeloma | follow-up |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bartha, Á.; Győrffy, B. Comprehensive Outline of Whole Exome Sequencing Data Analysis Tools Available in Clinical Oncology. Cancers 2019, 11, 1725. https://doi.org/10.3390/cancers11111725
Bartha Á, Győrffy B. Comprehensive Outline of Whole Exome Sequencing Data Analysis Tools Available in Clinical Oncology. Cancers. 2019; 11(11):1725. https://doi.org/10.3390/cancers11111725
Chicago/Turabian StyleBartha, Áron, and Balázs Győrffy. 2019. "Comprehensive Outline of Whole Exome Sequencing Data Analysis Tools Available in Clinical Oncology" Cancers 11, no. 11: 1725. https://doi.org/10.3390/cancers11111725
APA StyleBartha, Á., & Győrffy, B. (2019). Comprehensive Outline of Whole Exome Sequencing Data Analysis Tools Available in Clinical Oncology. Cancers, 11(11), 1725. https://doi.org/10.3390/cancers11111725