STAT3 Dysregulation in Mature T and NK Cell Lymphomas

 , , ,
, , ,  ,
, 
Abstract
:1. Introduction
2. Overview of STAT3 in T Cell Lymphomas
3. Mechanism of Aberrant STAT3 Activation
3.1. Activating Mutations in JAK/STAT
3.2. Activating Kinase Mutations
3.3. Positive and Negative Regulators of STAT3
3.4. miRNA Regulation of STAT3
3.5. Disruptions in the Tumor Microenvironment
4. Targets of STAT3
4.1. BCL-2 Family Members
4.2. Micro-RNAs
4.3. PD-1/PD-L1 Axis
4.4. IRF4
4.5. IL-17 Cytokine
4.6. SOCS3 Protein
5. Therapeutic Targeting of STAT3
5.1. Upstream STAT3 Inhibition
5.2. Direct STAT3 Inhibition
6. Conclusions
Funding
Conflicts of Interest
References
- Vose, J.; Armitage, J.; Weisenburger, D. International peripheral T-cell and natural killer/T-cell lymphoma study: Pathology findings and clinical outcomes. J. Clin. Oncol. 2008, 26, 4124–4130. [Google Scholar]
- Groves, F.D.; Linet, M.S.; Travis, L.B.; Devesa, S.S. Cancer Surveillance Series: Non-Hodgkin’s Lymphoma Incidence by Histologic Subtype in the United States From 1978 Through 1995. J. Nat. Cancer Inst. 2000, 92, 1240–1251. [Google Scholar] [CrossRef] [PubMed]
- Van Arnam, J.S.; Lim, M.S.; Elenitoba-Johnson, K.S.J. Novel insights into the pathogenesis of T-cell lymphomas. Blood 2018, 131, 2320–2330. [Google Scholar] [CrossRef] [PubMed]
- Darnell, J.E., Jr. STATs and gene regulation. Science 1997, 277, 1630–1635. [Google Scholar] [CrossRef] [PubMed]
- Calò, V.; Migliavacca, M.; Bazan, V.; Macaluso, M.; Buscemi, M.; Gebbia, N.; Russo, A. STAT proteins: From normal control of cellular events to tumorigenesis. J. Cell Physiol. 2003, 197, 157–168. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; McBride, K.M.; Reich, N.C. STAT3 nuclear import is independent of tyrosine phosphorylation and mediated by importin-α3. Proc. Natl. Acad. Sci. USA 2005, 102, 8150–8155. [Google Scholar] [CrossRef]
- Darnell, J.E.; Kerr, I.M.; Stark, G.R. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science 1994, 264, 1415–1421. [Google Scholar] [CrossRef]
- Pranada, A.L.; Metz, S.; Herrmann, A.; Heinrich, P.C.; Muller-Newen, G. Real Time Analysis of STAT3 Nucleocytoplasmic Shuttling. J. Biol. Chem. 2003, 279, 15114–15123. [Google Scholar] [CrossRef]
- Meyer, T.; Gavenis, K.; Vinkemeier, U. Cell Type-Specific and Tyrosine Phosphorylation-Independent Nuclear Presence of STAT1 and STAT3. Exp. Cell Res. 2002, 272, 45–55. [Google Scholar] [CrossRef]
- Braunstein, J.; Brutsaert, S.; Olson, R.; Schindler, C. STATs Dimerize in the Absence of Phosphorylation. J. Biol. Chem. 2003, 278, 34133–34140. [Google Scholar] [CrossRef] [Green Version]
- Naito, T.; Tanaka, H.; Naoe, Y.; Taniuchi, I. Transcriptional control of T-cell development. Int. Immunol. 2011, 23, 661–668. [Google Scholar] [CrossRef] [PubMed]
- Vainchenker, W.; Constantinescu, S.N. The role of JAK-STAT signaling in haematological malignancies. Oncogene 2013, 32, 2601–2613. [Google Scholar] [CrossRef] [PubMed]
- Al Zaid Siddiquee, K.; Turkson, J. STAT3 as a target for inducing apoptosis in solid and hematological tumors. Cell Res. 2008, 18, 254–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waldmann, T.A.; Chen, J. Disorders of the JAK/STAT Pathway in T Cell Lymphoma Pathogenesis: Implications for Immunotherapy. Annu. Rev. Immunol. 2017, 35, 533–550. [Google Scholar] [CrossRef] [PubMed]
- Pencik, J.; Pham, H.T.; Schmoellerl, J.; Javaheri, T.; Schlederer, M.; Culig, Z.; Merkel, O.; Moriggl, R.; Grebien, F.; Kenner, L. JAK-STAT signaling in cancer: From cytokines to non-coding genome. Cytokine 2016, 87, 26–36. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.O.; Pappu, B.P.; Nurieva, R.; Akimzhanov, A.; Kang, H.S.; Chung, Y.; Ma, L.; Shah, B.; Panopoulos, A.D.; Schluns, K.S.; et al. T helper 17 lineage differentiation is programmed by orphan nuclear receptors ROR alpha and ROR gamma. Immunity 2008, 28, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.O.; Panopoulos, A.D.; Nurieva, R.; Chang, S.H.; Wang, D.; Watowich, S.S.; Dong, C. STAT3 regulates cytokine-mediated generation of inflammatory helper T cells. J. Biol. Chem. 2007, 282, 9358–9363. [Google Scholar] [CrossRef]
- Egwuagu, C.E. STAT3 in CD4+ T helper cell differentiation and inflammatory diseases. Cytokine 2009, 47, 149–156. [Google Scholar] [CrossRef] [Green Version]
- Wilson, R.P.; Ives, M.L.; Lau, A.; Payne, K.; Kobayashi, M.; Arkwright, P.D.; Peake, J.; Wong, M.; Adelstein, S.; Smart, J.M.; et al. STAT3 is a critical cell-intrinsic regulator of human unconventional T cell numbers and function. J. Exp. Med. 2015, 212, 855–864. [Google Scholar] [CrossRef]
- Olson, J.A.; Jameson, S.C. Keeping STATs on memory CD8+ T cells. Immunity 2011, 35, 663–665. [Google Scholar] [CrossRef]
- Durant, L.; Watford, W.T.; Ramos, H.L.; Laurence, A.; Vahedi, G.; Wei, L.; Takahashi, H.; Sun, H.W.; Kanno, Y.; Powrie, F.; et al. Diverse targets of the transcription factor STAT3 contribute to T cell pathogenicity and homeostasis. Immunity 2010, 32, 605–615. [Google Scholar] [CrossRef] [PubMed]
- Crescenzo, R.; Abate, F.; Lasorsa, E.; Tabbo, F.; Gaudiano, M.; Chiesa, N.; Di Giacomo, F.; Spaccarotella, E.; Barbarossa, L.; Ercole, E.; et al. European T-Cell Lymphoma Study Group, T-Cell Project: Prospective Collection of Data in Patients with Peripheral T-Cell Lymphoma and the AIRC 5xMille Consortium “Genetics-Driven Targeted Management of Lymphoid Malignancies”. Convergent mutations and kinase fusions lead to oncogenic STAT3 activation in anaplastic large cell lymphoma. Cancer Cell 2015, 27, 516–532. [Google Scholar] [PubMed]
- Han, J.J.; O’byrne, M.; Stenson, M.J.; Maurer, M.J.; Wellik, L.E.; Feldman, A.L.; McPhail, E.D.; Witzig, T.E.; Gupta, M. Prognostic and therapeutic significance of phosphorylated STAT3 and protein tyrosine phosphatase-6 in peripheral-T cell lymphoma. Blood Cancer J. 2018, 8, 110. [Google Scholar] [CrossRef] [PubMed]
- Morris, S.W.; Kirstein, M.N.; Valentine, M.B.; Dittmer, K.G.; Shapiro, D.N.; Saltman, D.L.; Look, A.T. Fusion of a Kinase Gene, ALK, to a Nucleolar Protein Gene, NMP, in Non-Hodgkin’s Lymphoma. Science 1994, 263, 1281–1284. [Google Scholar] [CrossRef]
- Zhang, Q.; Raghunath, P.N.; Xue, L.; Majewski, M.; Carpentieri, D.F.; Odum, N.; Morris, S.; Skorski, T.; Wasik, M.A. Multilevel Dysregulation of STAT3 Activation in Anaplastic Lymphoma Kinase-Positive T/Null-Cell Lymphoma. J. Immunol. 2002, 168, 466–474. [Google Scholar] [CrossRef] [Green Version]
- Amin, H.M.; Lai, R. Pathobiology of ALK+ anaplastic large-cell lymphoma. Blood 2007, 110, 2259–2267. [Google Scholar] [CrossRef] [Green Version]
- Matutes, E. The 2017 WHO update on mature T- and natural killer (NK) cell neoplasms. Int. J. Lab. Hematol. 2017, 40, 97–103. [Google Scholar] [CrossRef]
- Oishi, N.; Brody, G.S.; Ketterling, R.P.; Viswanatha, D.S.; He, R.; Dasari, S.; Mai, M.; Benson, H.K.; Sattler, C.A.; Boddicker, R.L.; et al. Genetic subtyping of breast implant-associated anaplastic large cell lymphoma. Blood 2018, 132, 544–547. [Google Scholar] [CrossRef]
- Morichika, K.; Karube, K.; Kayo, H.; Uchino, S.; Nishi, Y.; Nakachi, S.; Okamoto, S.; Morishima, S.; Ohshiro, K.; Nakazato, I.; et al. Phosphorylated STAT3 expression predicts better prognosis in smoldering type of adult T-cell leukemia/lymphoma. Cancer Sci. 2019, 110, 2982–2991. [Google Scholar] [CrossRef]
- Koskela, H.L.; Eldfors, S.; Ellonen, P.; van Adrichem, A.J.; Kuusanmäki, H.; Andersson, E.I.; Lagström, S.; Clemente, M.J.; Olson, T.; Jalkanen, S.E.; et al. Somatic STAT3 mutations in large granular lymphocytic leukemia. N. Engl. J. Med. 2012, 366, 1905–1913. [Google Scholar] [CrossRef]
- Coppe, A.; Andersson, E.; Binatti, A.; Gasparini, V.R.; Bortoluzzi, S.; Clemente, M.; Herling, M.; Maciejewski, J.; Mustjoki, S.; Bortoluzzi, S. Genomic landscape characterization of large granular lymphocyte leukemia with a systems genetics approach. Leukemia 2017, 31, 1243–1246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jerez, A.; Clemente, M.; Makishima, H.; Koskela, H.; Leblanc, F.; Peng Ng, K.; Olson, T.; Przychodzen, B.; Afable, M.; Gomez-Segui, I.; et al. STAT3 mutations unify the pathogenesis of chronic lymphoproliferative disorders of NK cells and T-cell large granular lymphocyte Leukemia. Blood 2012, 120, 3048–3057. [Google Scholar] [CrossRef] [PubMed]
- McKinney, M.; Moffitt, A.; Gaulard, P.; Travert, M.; De Leval, L.; Nicolae, A.; Raffeld, M.; Jaffe, E.S.; Pittaluga, S.; Xi, L.; et al. The Genetic Basis of Hepatosplenic T-cell Lymphoma. Cancer Discov. 2017, 7, 369–379. [Google Scholar] [CrossRef] [PubMed]
- Küçük, C.; Jiang, B.; Hu, X.; Zhang, W.; Chan, J.K.; Xiao, W.; Lack, N.; Alkan, C.; Williams, J.C.; Avery, K.N.; et al. Activating mutations of STAT5B and STAT3 in lymphomas derived from γδ-T or NK cells. Nat. Commun. 2015, 6, 6025. [Google Scholar] [CrossRef]
- Nicolae, A.; Xi, L.; Pittaluga, S.; Abdullaev, Z.; Pack, S.D.; Chen, J.; Waldmann, T.A.; Jaffe, E.S.; Raffeld, M. Frequent STAT5B mutations in γδ hepatosplenic T-cell lymphomas. Leukemia 2014, 28, 2244–2248. [Google Scholar] [CrossRef] [PubMed]
- Coppo, P.; Gouilleux-Gruart, V.; Huang, Y.; Bouhlal, H.; Bouamar, H.; Bouchet, S.; Perrot, C.; Vieillard, V.; Dartigues, P.; Gaulard, P.; et al. STAT3 transcription factor is constitutively activated and is oncogenic in nasal-type NK/T-cell lymphoma. Leukemia 2009, 23, 1667–1678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moffitt, A.B.; Ondrejka, S.L.; McKinney, M.; Rempel, R.E.; Goodlad, J.R.; Teh, C.H.; Leppa, S.; Mannisto, S.; Kovanen, P.E.; Tse, E.; et al. Enteropathy-associated T cell lymphoma subtypes are characterized by loss of function of SETD2. J. Exp. Med. 2017, 214, 1371–1386. [Google Scholar] [CrossRef] [PubMed]
- Hwang, S.T.; Janik, J.E.; Jaffe, E.S.; Wilson, W.H. Mycosis fungoides and Sézary syndrome. Lancet 2008, 371, 945–957. [Google Scholar] [CrossRef]
- Foss, F.M.; Sausville, E.A. Prognosis and staging of cutaneous T-cell lymphoma. Hematol. Oncol. Clin. N. Am. 1995, 9, 1011–1019. [Google Scholar] [CrossRef]
- Kim, Y.H.; Liu, H.; Mraz-Gernhard, S.; Varghese, A.; Hoppe, R.T. Long-term outcome of 525 patients with mycosis fungoides and Sezary syndrome: Clinical prognostic factors and risk for disease progression. Arch. Dermatol. 2003, 139, 857–866. [Google Scholar] [CrossRef]
- Fujii, K. New Therapies and Immunological Findings in Cutaneous T-Cell Lymphoma. Front. Oncol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, M.; Kaltoft, K.; Nordahl, M.; Röpke, C.; Geisler, C.; Mustelin, T.; Dobson, P.; Svejgaard, A.; Odum, N. Constitutive activation of a slowly migrating isoform of Stat3 in mycosis fungoides: Tyrphostin AG490 inhibits Stat3 activation and growth of mycosis fungoides tumor cell lines. Proc. Natl. Acad. Sci. USA 1997, 94, 6764–6769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sommer, V.H.; Clemmensen, O.; Nielsen, O.; Wasik, M.; Lovato, P.; Brender, C.; Eriksen, K.W.; Woetmann, A.; Kaestel, C.G.; Nissen, M.H.; et al. In vivo activation of STAT3 in cutaneous T-cell lymphoma. Evidence for an antiapoptotic function of STAT3. Leukemia 2004, 18, 1288–1295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Kester, M.S.; Out-Luiting, J.J.; von dem Borne, P.A.; Willemze, R.; Tensen, C.P.; Vermeer, M.H. Cucurbitacin I inhibits Stat3 and induces apoptosis in Sezary cells. J. Investig. Dermatol. 2008, 128, 1691–1695. [Google Scholar] [CrossRef]
- Eriksen, K.W.; Kaltoft, K.; Mikkelsen, G.; Nielsen, M.; Zhang, Q.; Geisler, C.; Nissen, M.H.; Ropke, C.; Waskik, M.A.; Odum, N. Constitutive STAT3-activation in Sezary syndrome: Tyrphostin AG490 inhibits STAT3-activation, interleukin-2 receptor expression and growth of leukemic Sezary cells. Leukemia 2001, 15, 787–793. [Google Scholar] [CrossRef]
- Greenplate, A.; Wang, K.; Tripathi, R.M.; Palma, N.; Ali, S.M.; Stephens, P.J.; Miller, V.A.; Shyr, Y.; Guo, Y.; Reddy, N.M.; et al. Genomic Profiling of T-Cell Neoplasms Reveals Frequent JAK1 and JAK3 Mutations with Clonal Evasion from Targeted Therapies. JCO Precis. Oncol. 2018, 2, 1–16. [Google Scholar]
- Zamo, A.; Chiarle, R.; Piva, R.; Howes, J.; Fan, Y.; Chilosi, M.; Levy, D.E.; Inghirami, G. Anaplastic lymphoma kinase (ALK) activates Stat3 and protects hematopoietic cells from cell death. Oncogene 2002, 21, 1038–1047. [Google Scholar] [CrossRef] [Green Version]
- Aggarwal, B.B.; Kunnumakkara, A.B.; Harikumar, K.B.; Gupta, S.R.; Tharakan, S.T.; Koca, C.; Dey, S.; Sung, B. Signal transducer and activator of transcription-3, inflammation, and cancer: How intimate is the relationship? Ann. N. Y. Acad. Sci. 2009, 1171, 59–76. [Google Scholar] [CrossRef]
- Chiarle, R.; Gong, J.Z.; Guasparri, I.; Pesci, A.; Cai, J.; Liu, J.; Simmons, W.J.; Dhall, G.; Howes, J.; Piva, R.; et al. NPM-ALK transgenic mice spontaneously develop T-cell lymphomas and plasma cell tumors. Blood 2003, 101, 1919–1927. [Google Scholar] [CrossRef] [Green Version]
- Chiarle, R.; Simmons, W.; Cai, H.; Dhall, G.; Zamo, A.; Raz, R.; Karras, J.G.; Levy, D.E.; Inghirami, G. Stat3 is required for ALK-mediated lymphomagenesis and provides a possible therapeutic target. Nat. Med. 2005, 11, 623–629. [Google Scholar] [CrossRef]
- Zhang, Q.; Raghunath, P.; Vonderheid, E.; Odum, N.; Wasik, M.A. Lack of Phosphotyrosine Phosphatase SHP-1 Expression in Malignant T-Cell Lymphoma Cells Results from Methylation of the SHP-1 Promoter. Am. J. Pathol. 2000, 157, 1137–1146. [Google Scholar] [CrossRef] [Green Version]
- Khoury, J.D.; Rassidakis, G.; Medeiros, L.J.; Amin, H.M.; Lai, R. Methylation of SHP1 gene and loss of SHP1 protein expression are frequent in systemic anaplastic large cell lymphoma. Blood 2004, 104, 1580–1581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, Y.; Amin, H.; Franko, B.; Frantz, C.; Shi, X.; Lai, R. Loss of SHP1 enhances JAK3/STAT3 signaling and decreases proteasome degradation of JAK3 and NPM-ALK in ALK. Blood 2006, 108, 2796–2803. [Google Scholar] [CrossRef] [PubMed]
- Nasr, M.R.; Laver, J.H.; Chang, M.; Hutchison, R.E. Expression of anaplastic lymphoma kinase, tyrosine-phosphorylated STAT3, and associated factors in pediatric anaplastic large cell lymphoma: A report from the children’s oncology group. Am. J. Clin. Pathol. 2007, 127, 770–778. [Google Scholar] [CrossRef] [PubMed]
- Desjobert, C.; Renalier, M.; Bergalet, J.; Dejean, E.; Joseph, N.; Kruczynski, A.; Soulier, J.; Espinos, E.; Meggetto, F.; Cavaillé, J.; et al. MiR-29a down-regulation in ALK-positive anaplastic large cell lymphomas contributes to apoptosis blockade through MCL-1 overexpression. Blood 2011, 117, 6627–6637. [Google Scholar] [CrossRef] [Green Version]
- Atsaves, V.; Tsesmetzis, N.; Chioureas, D.; Kis, L.; Leventaki, V.; Drakos, E.; Panaretakis, T.; Grander, D.; Medeiros, L.J.; Young, K.H.; et al. PD-L1 is commonly expressed and transcriptionally regulated by STAT3 and MYC in ALK-negative anaplastic large-cell lymphoma. Leukemia 2017, 31, 1633–1637. [Google Scholar] [CrossRef] [PubMed]
- Marzec, M.; Zhang, Q.; Goradia, A.; Raghunath, P.N.; Liu, X.; Paessler, M.; Wang, H.Y.; Wysocka, M.; Cheng, M.; Ruggeri, B.A.; et al. Oncogenic kinase NPM/ALK induces through STAT3 expression of immunosuppressive protein CD274 (PD-L1, B7-H1). Proc. Natl. Acad. Sci. USA 2008, 105, 20852–20857. [Google Scholar] [CrossRef] [Green Version]
- Bandini, C.; Pupuleku, A.; Spaccarotella, E.; Pellegrino, E.; Wang, R.; Vitale, N.; Duval, C.; Cantarella, D.; Rinaldi, A.; Provero, P.; et al. IRF4 Mediates the Oncogenic Effects of STAT3 in Anaplastic Large Cell Lymphomas. Cancers 2018, 10, 21. [Google Scholar] [CrossRef]
- Weilemann, A.; Grau, M.; Erdmann, T.; Merkel, O.; Sobhiafshar, U.; Anagnostopoulos, I.; Hummel, M.; Siegert, A.; Hayford, C.; Madle, H.; et al. Essential role of IRF4 and MYC signaling for survival of anaplastic large cell lymphoma. Blood 2015, 125, 124–132. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Zhang, Y.; Petrus, M.N.; Xiao, W.; Nicolae, A.; Raffeld, M.; Pittaluga, S.; Bamford, R.N.; Nakagawa, M.; Ouyang, S.T.; et al. Cytokine receptor signaling is required for the survival of ALK− anaplastic large cell lymphoma, even in the presence of JAK1/STAT3 mutations. Proc. Natl. Acad. Sci. USA 2017, 114, 3975–3980. [Google Scholar] [CrossRef]
- Merkel, O.; Hamacher, F.; Griessl, R.; Grabner, L.; Schiefer, A.I.; Prutsch, N.; Baer, C.; Egger, G.; Schlederer, M.; Krenn, P.W.; et al. Oncogenic role of miR-155 in anaplastic large cell lymphoma lacking the t(2;5) translocation. J. Pathol. 2015, 236, 445–456. [Google Scholar] [CrossRef] [PubMed]
- Odejide, O.; Weigert, O.; Lane, A.A.; Toscano, D.; Lunning, M.A.; Kopp, N.; Kim, S.; van Bodegom, D.; Bolla, S.; Schatz, J.H.; et al. A targeted mutational landscape of angioimmunoblastic T-cell lymphoma. Blood 2014, 123, 1293–1296. [Google Scholar] [CrossRef] [PubMed]
- Cayrol, F.; Praditsuktavorn, P.; Fernando, T.M.; Kwiatkowski, N.; Marullo, R.; Calvo-Vidal, M.N.; Phillip, J.; Pera, B.; Yang, S.N.; Takpradit, K.; et al. THZ1 targeting CDK7 suppresses STAT transcriptional activity and sensitizes T-cell lymphomas to BCL2 inhibitors. Nat. Commun. 2017, 8, 14290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moskowitz, A.; Jacobsen, E.; Ruan, J.; Schatz, J.H.; Obadi, O.; Motylinski, K.; Jarjies, C.; Galasso, N.; Hancock, H.; Davey, T.; et al. Durable Responses Observed with JAK Inhibition in T-Cell Lymphomas. Blood 2018, 132, 2922. [Google Scholar] [CrossRef]
- Andersson, E.; Kuusanmaki, H.; Bortoluzzi, S.; Parsons, A.; Rajala, H.; Adrichem, A.V.; Eldfors, S.; Olson, T.; Clemente, M.J.; Laasonen, A.; et al. Activating somatic mutations outside the SH2-domain of STAT3 in LGL-Leukemia. Leukemia 2016, 30, 1204–1208. [Google Scholar] [CrossRef]
- Epling-Burnette, P.K.; Liu, J.H.; Catlett-Falcone, R.; Turkson, J.; Oshiro, M.; Kothapalli, R.; Li, Y.; Wang, J.M.; Yang-Yen, H.F.; Karras, J.; et al. Inhibition of STAT3 signaling leads to apoptosis of leukemic large granular lymphocytes and decreased Mcl-1 expression. J. Clin. Investig. 2001, 107, 351–362. [Google Scholar] [CrossRef]
- Liu, J.; Liang, L.; Li, D.; Nong, L.; Zheng, Y.; Huang, S.; Zhang, B.; Li, T. JAK3/STAT3 oncogenic pathway and PRDM1 expression stratify clinicopathologic features of extranodal NK/T-cell lymphoma, nasal type. Oncol. Rep. 2019, 41, 3219–3232. [Google Scholar] [CrossRef]
- Chen, Y.W.; Guo, T.; Shen, L.; Wong, K.Y.; Tao, Q.; Choi, W.W.; Au-Yeung, R.K.; Chan, Y.P.; Wong, M.L.; Tang, J.C.; et al. Receptor-type tyrosine-protein phosphatase k directly targets STAT3 activation for tumor suppression in nasal NK/T-cell lymphoma. Blood 2015, 125, 1589–1600. [Google Scholar] [CrossRef]
- Linlin Song, T.; Nairismägi, M.; Laurensia, Y.; Lim, J.Q.; Tan, J.; Li, Z.M.; Pang, W.L.; Kizhakeyil, A.; Wijaya, G.C.; Huang, D.C.; et al. Oncogenic activation of the STAT3 pathway drives PD-L1 expression in natural killer/T cell lymphoma. Blood 2018, 132, 1146–1158. [Google Scholar] [CrossRef]
- Kwong, Y.L.; Chan, T.S.Y.; Tan, D.; Kim, S.J.; Poon, L.M.; Mow, B.; Khong, P.L.; Loong, F.; Au-Yeung, R.; Iqbal, J.; et al. PD1 blockade with pembrolizumab is highly effective in relapsed or refractory NK/T-cell lymphoma failing l-asparaginase. Blood 2017, 129, 2437–2442. [Google Scholar] [CrossRef] [Green Version]
- Onozawa, E.; Shibayama, H.; Takada, H.; Imadome, K.I.; Aoki, S.; Yoshimori, M.; Shimizu, N.; Fujiwara, S.; Koyama, T.; Miura, O.; et al. STAT3 is constitutively activated in chronic active Epstein-Barr virus infection and can be a therapeutic target. Oncotarget 2018, 9, 31077–31089. [Google Scholar] [CrossRef]
- Woetmann, A.; Nielsen, M.; Christensen, S.T.; Brockdorff, J.; Kaltoft, K.; Engel, A.M.; Skov, S.; Brender, C.; Geisler, C.; Svejgaard, A.; et al. Inhibition of protein phosphatase 2A induces serine/threonine phosphorylation, subcellular redistribution, and functional inhibition of STAT3. Proc. Natl. Acad. Sci. USA 1999, 96, 10620–10625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van der Fits, L.; Out-Luiting, J.J.; van Leeuwen, M.A.; Samsom, J.N.; Willemze, R.; Tensen, C.P.; Vermeer, M.H. Autocrine IL-21 Stimulation Is Involved in the Maintenance of Constitutive STAT3 Activation in Sézary Syndrome. J. Investig. Dermatol. 2012, 132, 440–447. [Google Scholar] [CrossRef]
- Verma, N.K.; Davies, A.; Long, A.; Kelleher, D.; Volkov, Y. STAT3 knockdown by siRNA induces apoptosis in human cutaneous T-cell lymphoma line Hut78 via downregulation of Bcl-xL. Cell Mol. Biol. Lett. 2010, 15, 342–355. [Google Scholar] [CrossRef]
- van der Fits, L.; van Kester, M.S.; Qin, Y.; Out-Luiting, J.J.; Smit, F.; Zoutman, W.H.; Willemze, R.; Tensen, C.P.; Vermeer, M.H. MicroRNA-21 expression in CD4+ T cells is regulated by STAT3 and is pathologically involved in Sézary syndrome. J. Invest. Dermatol. 2011, 131, 762–768. [Google Scholar] [CrossRef] [PubMed]
- Sibbesen, N.A.; Kopp, K.L.; Litvinov, I.V.; Jønson, L.; Willerslev-Olsen, A.; Fredholm, S.; Petersen, D.L.; Nastasi, C.; Krejsgaard, T.; Lindahl, L.M.; et al. Jak3, STAT3, and STAT5 inhibit expression of miR-22, a novel tumor suppressor microRNA, in cutaneous T-Cell lymphoma. Oncotarget 2015, 6, 20555–20569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vieyra-Garcia, P.A.; Wei, T.; Naym, D.G.; Fredholm, S.; Fink-Puches, R.; Cerroni, L.; Odum, N.; O’Malley, J.T.; Gniadecki, R.; Wolf, P. STAT3/5-dependent IL9 overexpression contributes to neoplastic cell survival in mycosis fungoides. Clin. Cancer Res. 2016, 22, 3328–3339. [Google Scholar] [CrossRef]
- Fanok, M.H.; Sun, A.; Fogli, L.K.; Narendran, V.; Eckstein, M.; Kannan, K.; Dolgalev, I.; Lazaris, C.; Heguy, A.; Laird, M.E.; et al. Role of Dysregulated Cytokine Signaling and Bacterial Triggers in the Pathogenesis of Cutaneous T-Cell Lymphoma. J. Invest. Dermatol. 2018, 138, 1116–1125. [Google Scholar] [CrossRef]
- Krejsgaard, T.; Ralfkiaer, U.; Clasen-Linde, E.; Eriksen, K.W.; Kopp, K.L.; Bonefeld, C.M.; Geisler, C.; Dabelsteen, S.; Wasik, M.A.; Ralfkiaer, E.; et al. Malignant cutaneous T-cell lymphoma cells express IL-17 utilizing the Jak3/Stat3 signaling pathway. J. Invest. Dermatol. 2011, 131, 1331–1338. [Google Scholar] [CrossRef]
- Brender, C.; Nielsen, M.; Kaltoft, K.; Mikkelsen, G.; Zhang, Q.; Wasik, M.; Billestrup, N.; Ødum, N. STAT3-mediated constitutive expression of SOCS-3 in cutaneous T-cell lymphoma. Blood 2001, 97, 1056–1062. [Google Scholar] [CrossRef] [Green Version]
- Bellon, M.; Lu, L.; Nicot, C. Constitutive activation of Pim1 kinase is a therapeutic target for adult T-cell leukemia. Blood 2016, 127, 2439–2450. [Google Scholar] [CrossRef] [PubMed]
- Dittmer, J. Mechanisms governing metastatic dormancy in breast cancer. Semin. Cancer Biol. 2017, 44, 72–82. [Google Scholar] [CrossRef] [PubMed]
- Feng, M.G.; Liu, C.; Chen, L.; Feng, W.B.; Liu, M.; Hai, H.; Lu, J.M. MiR-21 attenuates apoptosis-triggered by amyloid-β via modulating PDCD4/PI3K/AKT/GSK-3β pathway in SH-SY5Y cells. Biomed. Pharmacother. 2018, 101, 1003–1007. [Google Scholar] [CrossRef] [PubMed]
- Lawrie, C.H. MicroRNAs and lymphomagenesis: A functional review. Br. J. Haematol. 2013, 160, 571–581. [Google Scholar] [CrossRef] [PubMed]
- Brender, C.; Lovato, P.; Sommer, V.H.; Woetmann, A.; Mathiesen, A.M.; Geisler, C.; Wasik, M.; Ødum, N. Constitutive SOCS-3 expression protects T-cell lymphoma against growth inhibition by IFNalpha. Leukemia 2005, 19, 209–213. [Google Scholar] [CrossRef]
- Debnath, B.; Xu, S.; Neamati, N. Small molecule inhibitors of signal transducer and activator of transcription 3 (Stat3) protein. J. Med. Chem. 2012, 55, 6645–6668. [Google Scholar] [CrossRef]
- Yu, H.; Lee, H.; Herrmann, A.; Buettner, R.; Jove, R. Revisiting STAT3 signalling in cancer: New and unexpected biological functions. Nat. Rev. Cancer 2014, 14, 736–746. [Google Scholar] [CrossRef]
- Zerbini, C.A.; Lomonte, A. Tofacitinib for the treatment of rheumatoid arthritis. Expert Rev. Clin. Immunol. 2012, 8, 319–331. [Google Scholar] [CrossRef]
- Porpaczy, E.; Tripolt, S.; Hoelbl-Kovacic, A.; Gisslinger, B.; Bago-Horvath, Z.; Casanova-Hevia, E.; Clappier, E.; Decker, T.; Fajmann, S.; Fux, D.A.; et al. Aggressive B-cell lymphomas in patients with myelofibrosis receiving JAK1/2 inhibitor therapy. Blood 2018, 132, 694–706. [Google Scholar] [CrossRef]
- Turkson, J.; Ryan, D.; Kim, J.S.; Zhang, Y.; Chen, Z.; Haura, E.; Laudano, A.; Sebti, S.; Hamilton, A.D.; Jove, R. Phosphotyrosyl peptides block Stat3-mediated DNA binding activity, gene regulation, and cell transformation. J. Biol. Chem. 2001, 276, 45443–45455. [Google Scholar] [CrossRef]
- Timofeeva, O.A.; Gaponenko, V.; Lockett, S.J.; Tarasov, S.G.; Jiang, S.; Michejda, C.J.; Perantoni, A.O.; Tarasova, N.I. Rationally designed inhibitors identify STAT3 N-domain as a promising anticancer drug target. ACS Chem. Biol. 2007, 2, 799–809. [Google Scholar] [CrossRef] [PubMed]
- Fontaine, F.; Overman, J.; François, M. Pharmacological manipulation of transcription factor protein-protein interactions: Opportunities and obstacles. Cell Regen. 2015, 4, 2. [Google Scholar] [CrossRef] [PubMed]
- Bendell, J.C.; Hong, D.S.; Burris, H.A., 3rd; Naing, A.; Jones, S.F.; Falchook, G.; Bricmont, P.; Elekes, A.; Rock, E.P.; Kurzrock, R. Phase 1, open-label, dose-escalation, and pharmacokinetic study of STAT3 inhibitor OPB-31121 in subjects with advanced solid tumors. Cancer Chemother. Pharmacol. 2014, 74, 125–130. [Google Scholar] [CrossRef] [PubMed]
- Wong, A.L.A.; Hirpara, J.L.; Pervaiz, S.; Eu, J.Q.; Sethi, G.; Goh, B.C. Do STAT3 inhibitors have potential in the future for cancer therapy? Expert Opin. Investig. Drugs 2017, 26, 883–887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, A.L.; Soo, R.A.; Tan, D.S.; Lee, S.C.; Lim, J.S.; Marban, P.C.; Kong, L.R.; Lee, Y.J.; Wang, L.Z.; Thuya, W.L.; et al. Phase I and biomarker study of OPB-51602, a novel signal transducer and activator of transcription (STAT) 3 inhibitor, in patients with refractory solid malignancies. Ann. Oncol. 2015, 26, 998–1005. [Google Scholar] [CrossRef] [PubMed]
- Miyoshi, K.; Takaishi, M.; Nakajima, K.; Ikeda, M.; Kanda, T.; Tarutani, M.; Iiyama, T.; Asao, N.; DiGiovanni, J.; Sano, S. Stat3 as a therapeutic target for the treatment of psoriasis: A clinical feasibility study with STA-21, a Stat3 inhibitor. J. Invest. Dermatol. 2011, 131, 108–117. [Google Scholar] [CrossRef]
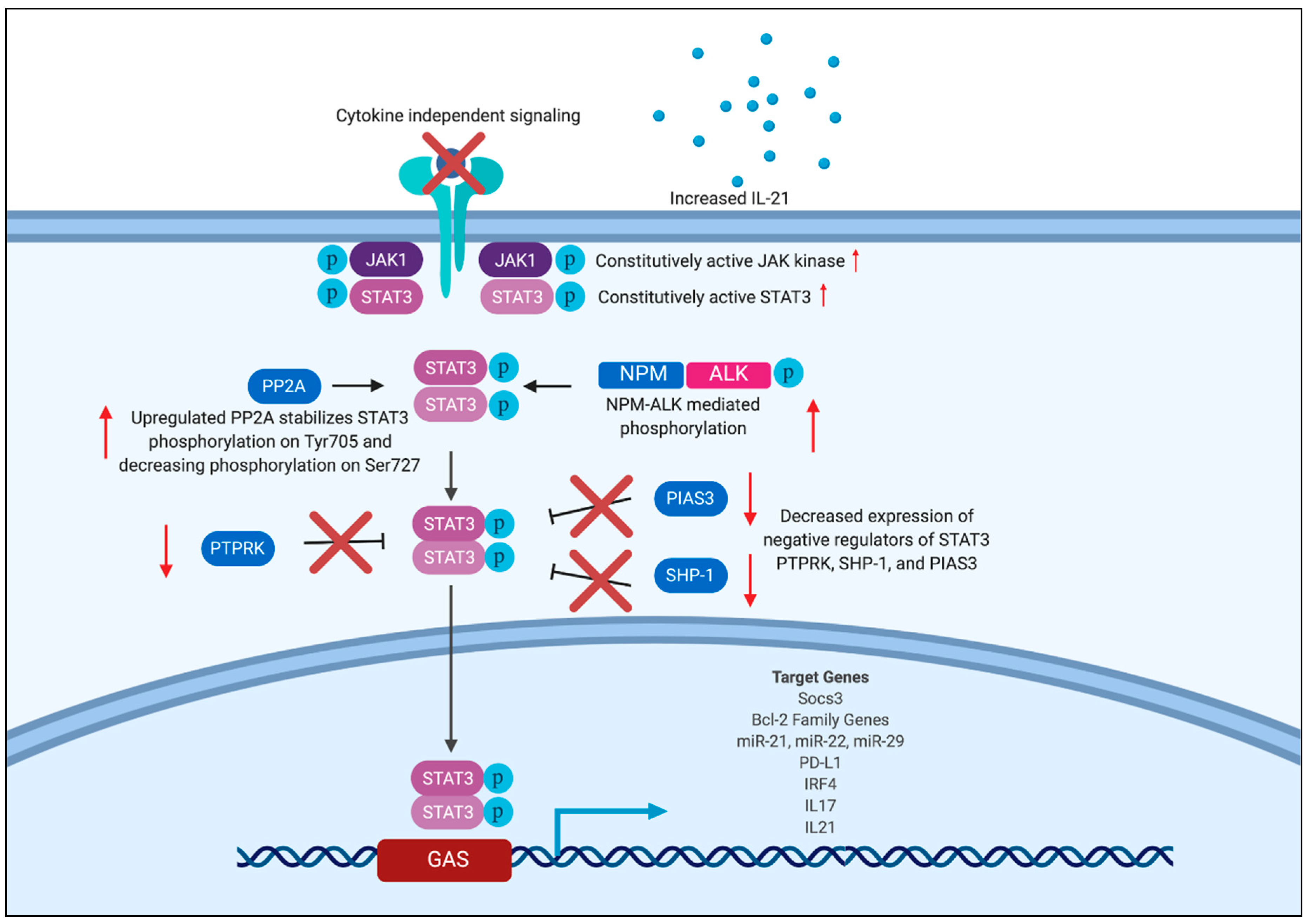

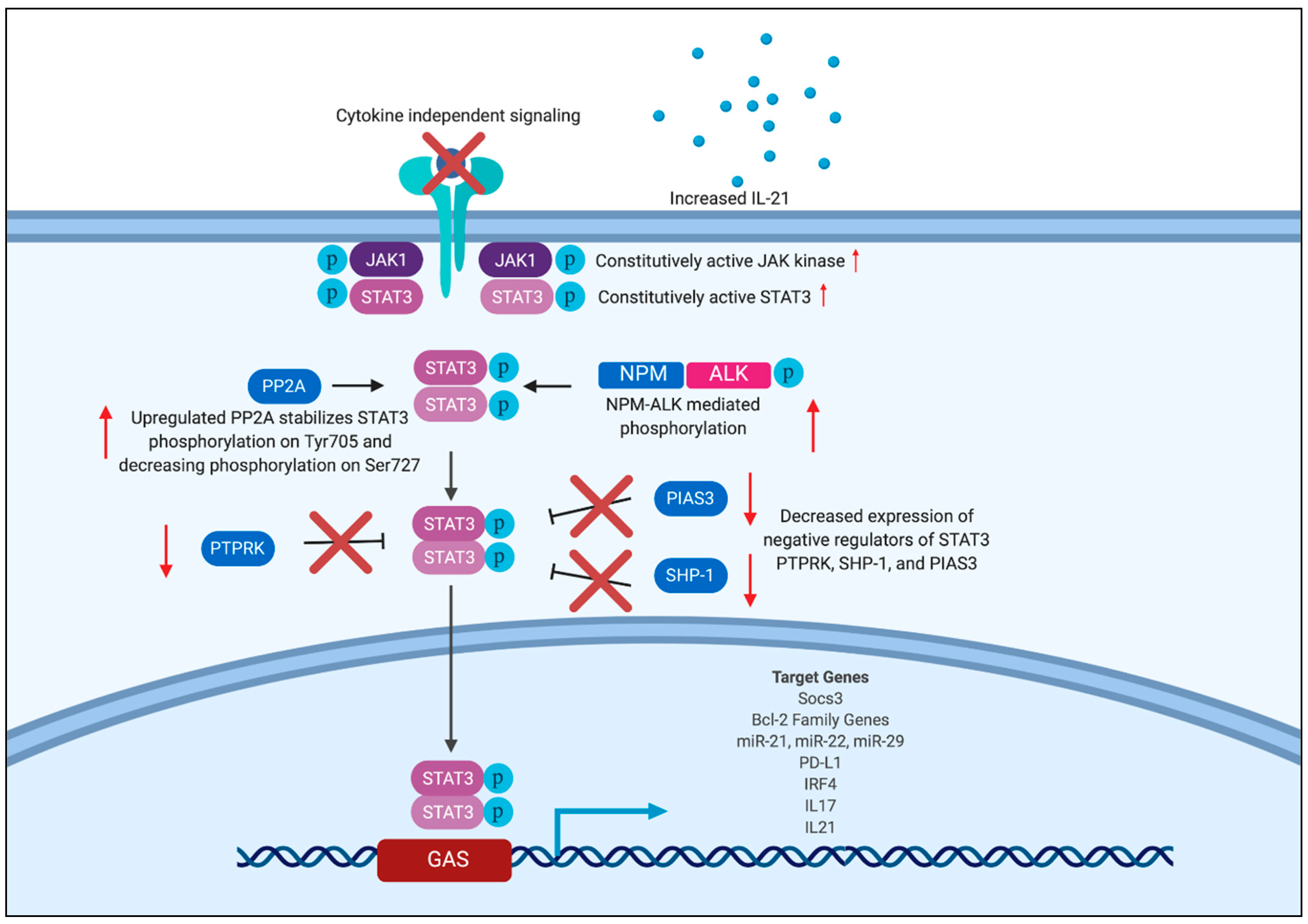{kind=link}
| Mature T and NK Cell Neoplasms | Relevant Literature |
|---|---|
| Anaplastic large-cell lymphoma, ALK+ | [22,24,25,26,46,47,48,49,50,51,52,53,54,55,56,57,58,59] |
| Anaplastic large-cell lymphoma, ALK− | [22,25,46,52,56,59,60,61] |
| Breast Implant Associated anaplastic large-cell lymphoma | [27,28,60] |
| Angioimmunoblastic T cell lymphoma | [22,46,62] |
| Peripheral T cell lymphoma, NOS | [22,46,63,64] |
| T cell large granular Lymphocytic Leukemia | [27,30,31,32,46,65,66] |
| NK/T cell lymphoma, nasal type and NK/T cell lymphoma | [27,34,36,46,67,68,69,70,71] |
| Hepatosplenic T cell lymphoma | [27,33,35] |
| Primary cutaneous γδ T cell lymphomas | [34] |
| Peripheral γδ T cell lymphomas | [34] |
| Enteropathy Associated T cell lymphoma | [37] |
| Cutaneous T cell lymphoma (Mycosis fungoides and Sézary syndrome) | [41,42,43,44,45,46,51,64,72,73,74,75,76,77,78,79,80] |
| Adult T cell leukemia/lymphoma | [29,81] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Seffens, A.; Herrera, A.; Tegla, C.; Buus, T.B.; Hymes, K.B.; Ødum, N.; Geskin, L.J.; Koralov, S.B. STAT3 Dysregulation in Mature T and NK Cell Lymphomas. Cancers 2019, 11, 1711. https://doi.org/10.3390/cancers11111711
Seffens A, Herrera A, Tegla C, Buus TB, Hymes KB, Ødum N, Geskin LJ, Koralov SB. STAT3 Dysregulation in Mature T and NK Cell Lymphomas. Cancers. 2019; 11(11):1711. https://doi.org/10.3390/cancers11111711
Chicago/Turabian StyleSeffens, Angelina, Alberto Herrera, Cosmin Tegla, Terkild B. Buus, Kenneth B. Hymes, Niels Ødum, Larisa J. Geskin, and Sergei B. Koralov. 2019. "STAT3 Dysregulation in Mature T and NK Cell Lymphomas" Cancers 11, no. 11: 1711. https://doi.org/10.3390/cancers11111711
APA StyleSeffens, A., Herrera, A., Tegla, C., Buus, T. B., Hymes, K. B., Ødum, N., Geskin, L. J., & Koralov, S. B. (2019). STAT3 Dysregulation in Mature T and NK Cell Lymphomas. Cancers, 11(11), 1711. https://doi.org/10.3390/cancers11111711