Jak-Stat Signaling Induced by Interleukin-6 Family Cytokines in Hepatocellular Carcinoma



{kind=link}
{kind=link}
Abstract
:1. Introduction
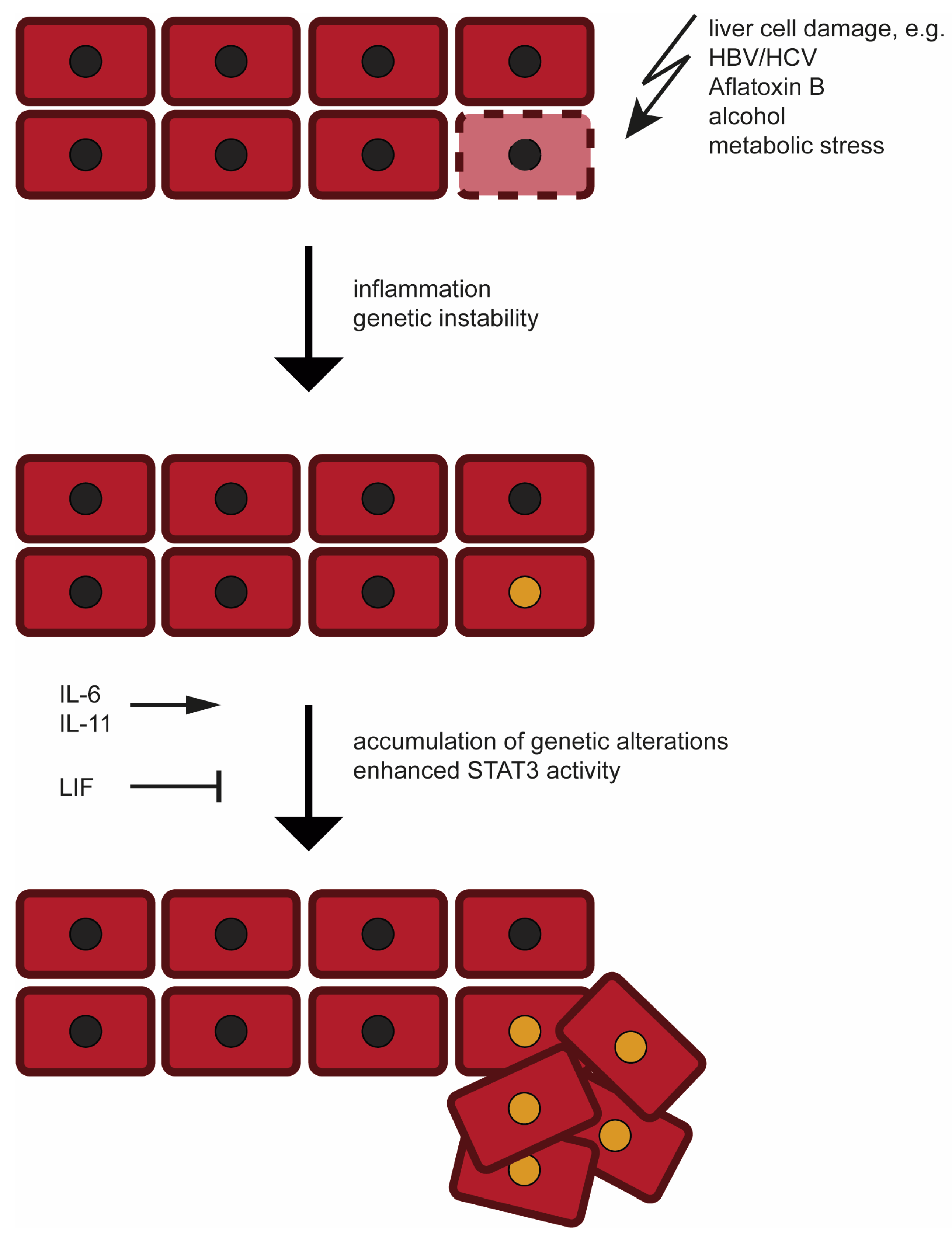
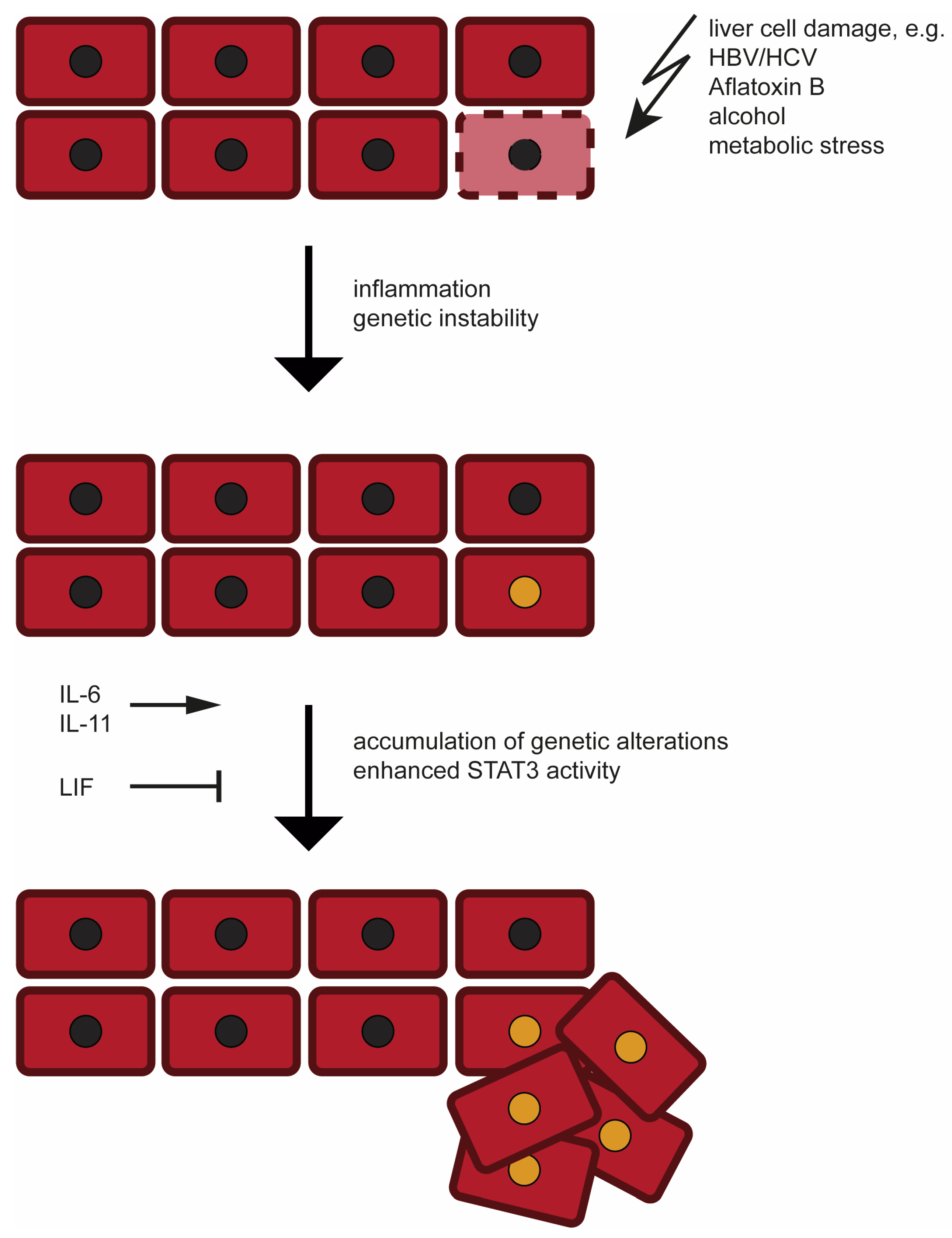
2. Etiology and Development of Hepatocellular Carcinoma (HCC)
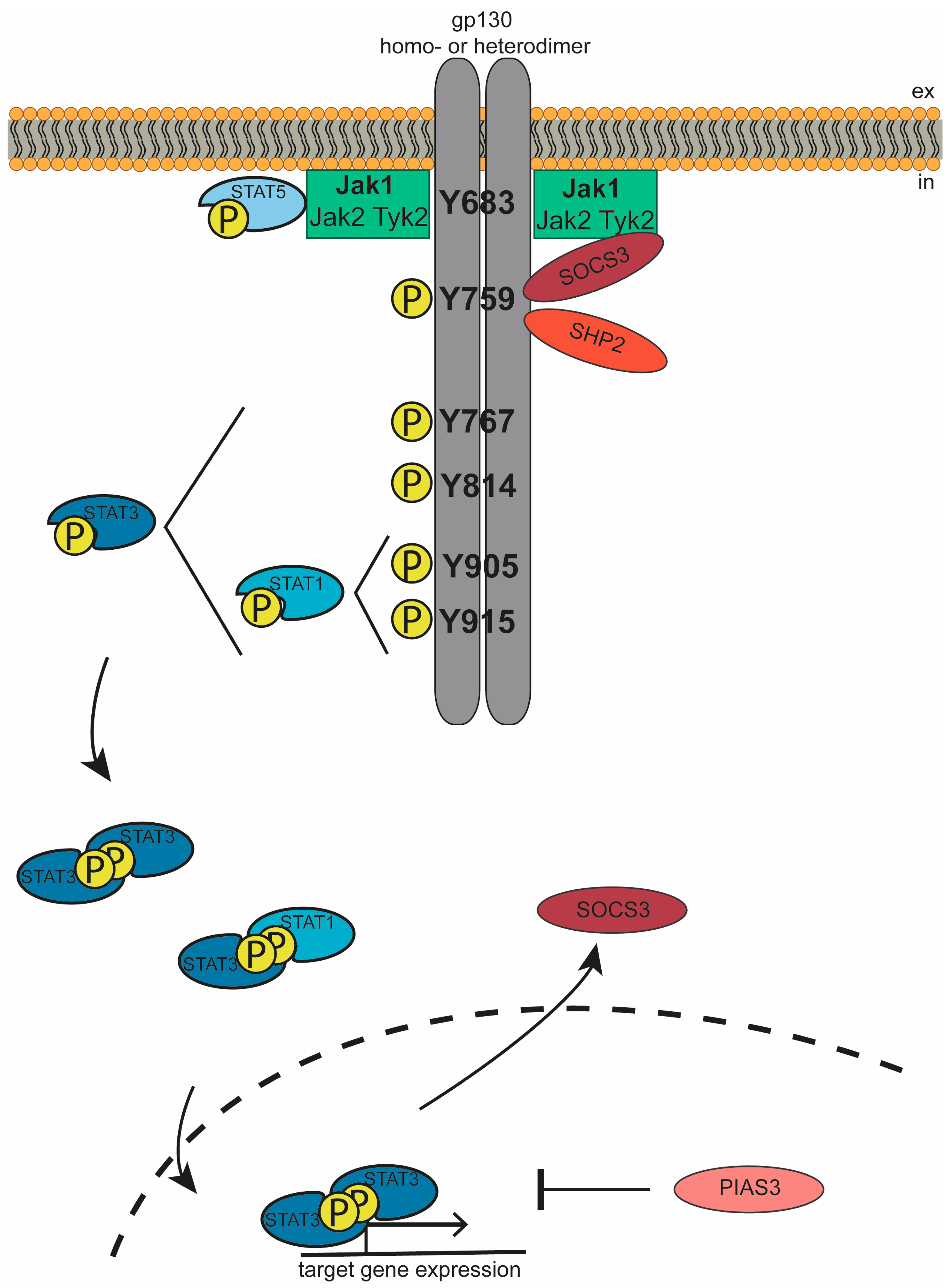
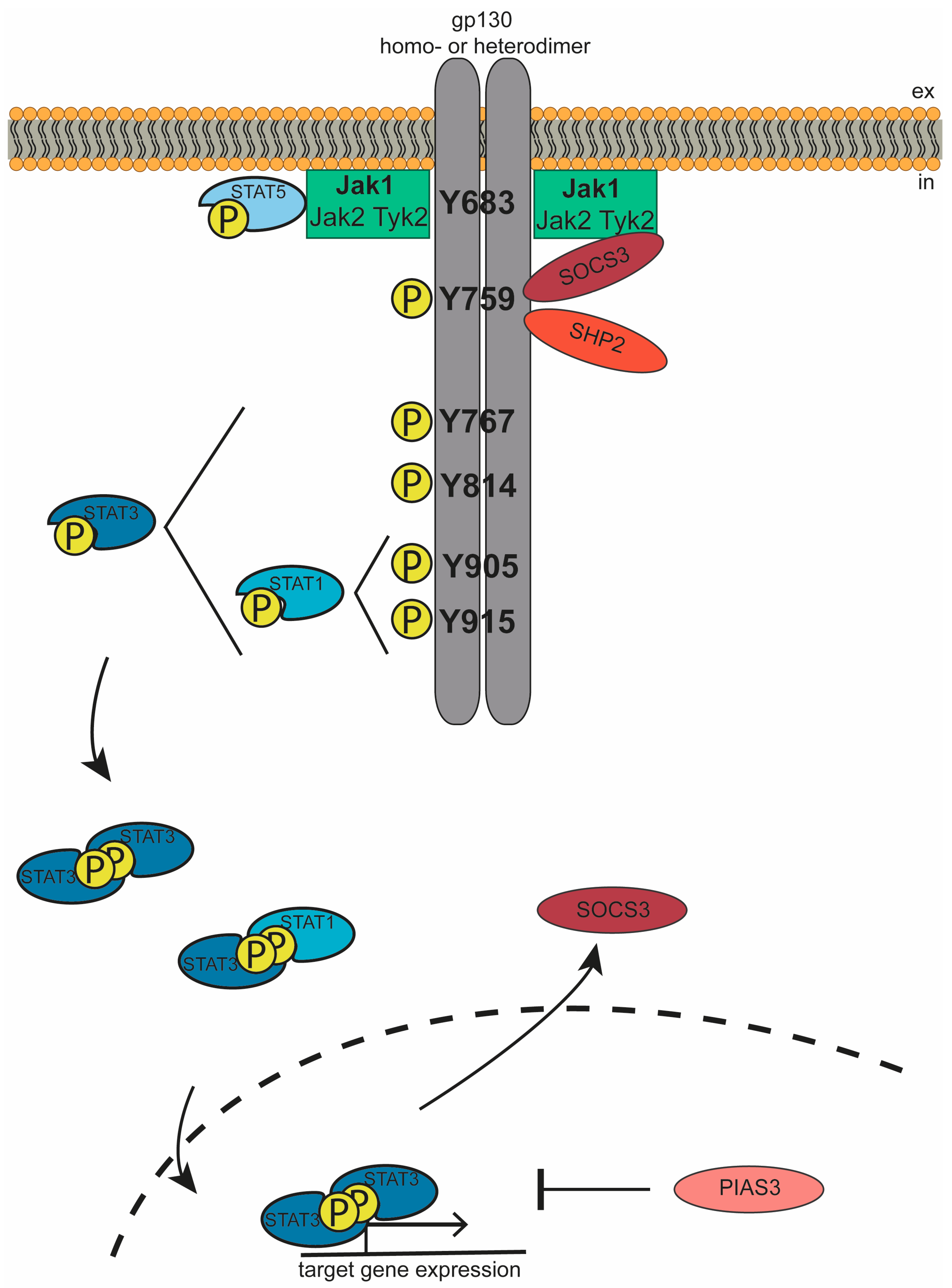
3. The Jak/STAT Signaling Cascade
4. IL-6 Family Cytokines in HCC Development
5. Inborn Mutations in Proteins of the Jak/STAT Cascade in HCC Development
6. Therapeutic Opportunities for HCC Patients
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Villanueva, A. Hepatocellular Carcinoma. N. Engl. J. Med. 2019, 380, 1450–1462. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.; Stepanova, M.; Ong, J.P.; Jacobson, I.M.; Bugianesi, E.; Duseja, A.; Eguchi, Y.; Wong, V.W.; Negro, F.; Yilmaz, Y.; et al. Nonalcoholic Steatohepatitis Is the Fastest Growing Cause of Hepatocellular Carcinoma in Liver Transplant Candidates. Clin. Gastroenterol Hepatol. 2019, 17, 748–755.e743. [Google Scholar] [CrossRef] [PubMed]
- Villarino, A.V.; Kanno, Y.; O’Shea, J.J. Mechanisms and consequences of Jak-STAT signaling in the immune system. Nat. Immunol. 2017, 18, 374–384. [Google Scholar] [CrossRef] [PubMed]
- O’Shea, J.J.; Schwartz, D.M.; Villarino, A.V.; Gadina, M.; McInnes, I.B.; Laurence, A. The JAK-STAT pathway: Impact on human disease and therapeutic intervention. Annu. Rev. Med. 2015, 66, 311–328. [Google Scholar] [CrossRef] [PubMed]
- Lokau, J.; Garbers, C. Activating mutations of the gp130/JAK/STAT pathway in human diseases. Adv. Protein Chem. Struct. Biol. 2019, 116, 283–309. [Google Scholar] [CrossRef] [PubMed]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Global Burden of Disease Liver Cancer Collaboration; Akinyemiju, T.; Abera, S.; Ahmed, M.; Alam, N.; Alemayohu, M.A.; Allen, C.; Al-Raddadi, R.; Alvis-Guzman, N.; Amoako, Y.; et al. The Burden of Primary Liver Cancer and Underlying Etiologies from 1990 to 2015 at the Global, Regional, and National Level: Results from the Global Burden of Disease Study 2015. JAMA Oncol. 2017, 3, 1683–1691. [Google Scholar] [CrossRef]
- Degasperi, E.; Colombo, M. Distinctive features of hepatocellular carcinoma in non-alcoholic fatty liver disease. Lancet Gastroenterol. Hepatol. 2016, 1, 156–164. [Google Scholar] [CrossRef] [Green Version]
- Sanyal, A.J.; Yoon, S.K.; Lencioni, R. The etiology of hepatocellular carcinoma and consequences for treatment. Oncologist 2010, 15 (Suppl. 4), 14–22. [Google Scholar] [CrossRef]
- Raimondi, S.; Bruno, S.; Mondelli, M.U.; Maisonneuve, P. Hepatitis C virus genotype 1b as a risk factor for hepatocellular carcinoma development: A meta-analysis. J. Hepatol. 2009, 50, 1142–1154. [Google Scholar] [CrossRef]
- Kanwal, F.; Kramer, J.R.; Ilyas, J.; Duan, Z.; El-Serag, H.B. HCV genotype 3 is associated with an increased risk of cirrhosis and hepatocellular cancer in a national sample of U.S. Veterans with HCV. Hepatology 2014, 60, 98–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Luan, W.; Villanueva, G.A.; Rahbari, N.N.; Yee, H.T.; Manizate, F.; Hiotis, S.P. Clinical prognostic variables in young patients (under 40 years) with hepatitis B virus-associated hepatocellular carcinoma. J. Dig. Dis. 2012, 13, 214–218. [Google Scholar] [CrossRef] [PubMed]
- Paradis, V.; Zalinski, S.; Chelbi, E.; Guedj, N.; Degos, F.; Vilgrain, V.; Bedossa, P.; Belghiti, J. Hepatocellular carcinomas in patients with metabolic syndrome often develop without significant liver fibrosis: A pathological analysis. Hepatology 2009, 49, 851–859. [Google Scholar] [CrossRef]
- Schlesinger, S.; Aleksandrova, K.; Pischon, T.; Jenab, M.; Fedirko, V.; Trepo, E.; Overvad, K.; Roswall, N.; Tjonneland, A.; Boutron-Ruault, M.C.; et al. Diabetes mellitus, insulin treatment, diabetes duration, and risk of biliary tract cancer and hepatocellular carcinoma in a European cohort. Ann. Oncol. 2013, 24, 2449–2455. [Google Scholar] [CrossRef] [PubMed]
- Calle, E.E.; Rodriguez, C.; Walker-Thurmond, K.; Thun, M.J. Overweight, obesity, and mortality from cancer in a prospectively studied cohort of U.S. adults. N. Engl. J. Med. 2003, 348, 1625–1638. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.Y.; Wu, J.C.; Yu, S.H.; Lin, J.T.; Wu, M.S.; Wu, C.Y. The occurrence of hepatocellular carcinoma in different risk stratifications of clinically noncirrhotic nonalcoholic fatty liver disease. Int. J. Cancer 2017, 141, 1307–1314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, Y.J.; Yang, H.I.; Wu, H.C.; Lee, M.H.; Liu, J.; Wang, L.Y.; Lu, S.N.; Jen, C.L.; You, S.L.; Santella, R.M.; et al. Aflatoxin B1 exposure increases the risk of hepatocellular carcinoma associated with hepatitis C virus infection or alcohol consumption. Eur. J. Cancer 2018, 94, 37–46. [Google Scholar] [CrossRef]
- Seshachalam, V.P.; Sekar, K.; Hui, K.M. Insights into the etiology-associated gene regulatory networks in hepatocellular carcinoma from The Cancer Genome Atlas. J. Gastroenterol. Hepatol. 2018, 33, 2037–2047. [Google Scholar] [CrossRef]
- Moinzadeh, P.; Breuhahn, K.; Stutzer, H.; Schirmacher, P. Chromosome alterations in human hepatocellular carcinomas correlate with aetiology and histological grade—Results of an explorative CGH meta-analysis. Br. J. Cancer 2005, 92, 935–941. [Google Scholar] [CrossRef]
- The International Consensus Group for Hepatocellular Neoplasia. Pathologic diagnosis of early hepatocellular carcinoma: A report of the international consensus group for hepatocellular neoplasia. Hepatology 2009, 49, 658–664. [Google Scholar] [CrossRef]
- European Association for the Study of the Liver. EASL Clinical Practice Guidelines: Management of hepatocellular carcinoma. J. Hepatol. 2018, 69, 182–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, Y.N. Update on precursor and early lesions of hepatocellular carcinomas. Arch. Pathol. Lab. Med. 2011, 135, 704–715. [Google Scholar] [CrossRef]
- Roncalli, M.; Terracciano, L.; Di Tommaso, L.; David, E.; Colombo, M.; Gruppo Italiano Patologi Apparato Digerente (GIPAD); Società Italiana di Anatomia Patologica e Citopatologia Diagnostica/International Academy of Pathology, Italian division (SIAPEC/IAP). Liver precancerous lesions and hepatocellular carcinoma: The histology report. Dig. Liver Dis. 2011, 43 (Suppl. 4), S361–S372. [Google Scholar] [CrossRef]
- Niu, Z.S.; Niu, X.J.; Wang, W.H.; Zhao, J. Latest developments in precancerous lesions of hepatocellular carcinoma. World J. Gastroenterol. 2016, 22, 3305–3314. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Kondo, F.; Ebara, M.; Sugiura, N.; Okabe, S.; Sunaga, M.; Yoshikawa, M.; Suzuki, E.; Ogasawara, S.; Shinozaki, Y.; et al. Natural history of large regenerative nodules and dysplastic nodules in liver cirrhosis: 28-year follow-up study. Hepatol. Int. 2015, 9, 330–336. [Google Scholar] [CrossRef] [Green Version]
- Tandon, P.; Garcia-Tsao, G. Prognostic indicators in hepatocellular carcinoma: A systematic review of 72 studies. Liver Int. 2009, 29, 502–510. [Google Scholar] [CrossRef]
- Salomao, M.; Remotti, H.; Vaughan, R.; Siegel, A.B.; Lefkowitch, J.H.; Moreira, R.K. The steatohepatitic variant of hepatocellular carcinoma and its association with underlying steatohepatitis. Hum. Pathol. 2012, 43, 737–746. [Google Scholar] [CrossRef]
- Shibahara, J.; Ando, S.; Sakamoto, Y.; Kokudo, N.; Fukayama, M. Hepatocellular carcinoma with steatohepatitic features: A clinicopathological study of Japanese patients. Histopathology 2014, 64, 951–962. [Google Scholar] [CrossRef]
- Jain, D.; Nayak, N.C.; Kumaran, V.; Saigal, S. Steatohepatitic hepatocellular carcinoma, a morphologic indicator of associated metabolic risk factors: A study from India. Arch. Pathol. Lab. Med. 2013, 137, 961–966. [Google Scholar] [CrossRef]
- Calderaro, J.; Couchy, G.; Imbeaud, S.; Amaddeo, G.; Letouze, E.; Blanc, J.F.; Laurent, C.; Hajji, Y.; Azoulay, D.; Bioulac-Sage, P.; et al. Histological subtypes of hepatocellular carcinoma are related to gene mutations and molecular tumour classification. J. Hepatol. 2017, 67, 727–738. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network. Comprehensive and Integrative Genomic Characterization of Hepatocellular Carcinoma. Cell 2017, 169, 1327–1341 e1323. [Google Scholar] [CrossRef] [PubMed]
- Nault, J.C.; Mallet, M.; Pilati, C.; Calderaro, J.; Bioulac-Sage, P.; Laurent, C.; Laurent, A.; Cherqui, D.; Balabaud, C.; Zucman-Rossi, J. High frequency of telomerase reverse-transcriptase promoter somatic mutations in hepatocellular carcinoma and preneoplastic lesions. Nat. Commun. 2013, 4, 2218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nault, J.C.; Calderaro, J.; Di Tommaso, L.; Balabaud, C.; Zafrani, E.S.; Bioulac-Sage, P.; Roncalli, M.; Zucman-Rossi, J. Telomerase reverse transcriptase promoter mutation is an early somatic genetic alteration in the transformation of premalignant nodules in hepatocellular carcinoma on cirrhosis. Hepatology 2014, 60, 1983–1992. [Google Scholar] [CrossRef] [PubMed]
- Torrecilla, S.; Sia, D.; Harrington, A.N.; Zhang, Z.; Cabellos, L.; Cornella, H.; Moeini, A.; Camprecios, G.; Leow, W.Q.; Fiel, M.I.; et al. Trunk mutational events present minimal intra-and inter-tumoral heterogeneity in hepatocellular carcinoma. J. Hepatol. 2017, 67, 1222–1231. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.H.; Oh, B.K.; Yoo, J.E.; Yoon, S.M.; Choi, J.; Kim, K.S.; Park, Y.N. Chromosomal instability, telomere shortening, and inactivation of p21(WAF1/CIP1) in dysplastic nodules of hepatitis B virus-associated multistep hepatocarcinogenesis. Mod. Pathol. 2009, 22, 1121–1131. [Google Scholar] [CrossRef] [PubMed]
- Plentz, R.R.; Park, Y.N.; Lechel, A.; Kim, H.; Nellessen, F.; Langkopf, B.H.; Wilkens, L.; Destro, A.; Fiamengo, B.; Manns, M.P.; et al. Telomere shortening and inactivation of cell cycle checkpoints characterize human hepatocarcinogenesis. Hepatology 2007, 45, 968–976. [Google Scholar] [CrossRef] [PubMed]
- Marchio, A.; Terris, B.; Meddeb, M.; Pineau, P.; Duverger, A.; Tiollais, P.; Bernheim, A.; Dejean, A. Chromosomal abnormalities in liver cell dysplasia detected by comparative genomic hybridisation. Mol. Pathol. 2001, 54, 270–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marquardt, J.U.; Seo, D.; Andersen, J.B.; Gillen, M.C.; Kim, M.S.; Conner, E.A.; Galle, P.R.; Factor, V.M.; Park, Y.N.; Thorgeirsson, S.S. Sequential transcriptome analysis of human liver cancer indicates late stage acquisition of malignant traits. J. Hepatol. 2014, 60, 346–353. [Google Scholar] [CrossRef] [PubMed]
- Garbers, C.; Hermanns, H.; Schaper, F.; Müller-Newen, G.; Grötzinger, J.; Rose-John, S.; Scheller, J. Plasticity and cross-talk of Interleukin 6-type cytokines. Cytokine Growth Factor Rev. 2012, 23, 85–97. [Google Scholar] [CrossRef]
- Aparicio-Siegmund, S.; Garbers, C. The biology of interleukin-27 reveals unique pro-and anti-inflammatory functions in immunity. Cytokine Growth Factor Rev. 2015, 26, 579–586. [Google Scholar] [CrossRef]
- Garbers, C.; Scheller, J. Interleukin-6 and interleukin-11: Same same but different. Biol. Chem. 2013, 394, 1145–1161. [Google Scholar] [CrossRef] [PubMed]
- Hermanns, H.M. Oncostatin M and interleukin-31: Cytokines, receptors, signal transduction and physiology. Cytokine Growth Factor Rev. 2015, 26, 545–558. [Google Scholar] [CrossRef] [PubMed]
- Eulenfeld, R.; Dittrich, A.; Khouri, C.; Muller, P.J.; Mutze, B.; Wolf, A.; Schaper, F. Interleukin-6 signalling: More than Jaks and STATs. Eur. J. Cell Biol. 2012, 91, 486–495. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, K.; Wu, L.-W.; Grivennikov, S.I.; de Jong, P.R.; Lian, I.; Yu, F.-X.; Wang, K.; Ho, S.B.; Boland, B.S.; Chang, J.T.; et al. A gp130-Src-YAP module links inflammation to epithelial regeneration. Nature 2015, 519, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Stahl, N.; Boulton, T.G.; Farruggella, T.; Ip, N.Y.; Davis, S.; Witthuhn, B.A.; Quelle, F.W.; Silvennoinen, O.; Barbieri, G.; Pellegrini, S.; et al. Association and activation of Jak-Tyk kinases by CNTF-LIF-OSM-IL-6 beta receptor components. Science 1994, 263, 92–95. [Google Scholar] [CrossRef] [PubMed]
- Murakami, M.; Narazaki, M.; Hibi, M.; Yawata, H.; Yasukawa, K.; Hamaguchi, M.; Taga, T.; Kishimoto, T. Critical cytoplasmic region of the interleukin 6 signal transducer gp130 is conserved in the cytokine receptor family. Proc. Natl. Acad. Sci. USA 1991, 88, 11349–11353. [Google Scholar] [CrossRef] [PubMed]
- Morris, R.; Kershaw, N.J.; Babon, J.J. The molecular details of cytokine signaling via the JAK/STAT pathway. Protein Sci. 2018, 27, 1984–2009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greiser, J.S.; Stross, C.; Heinrich, P.C.; Behrmann, I.; Hermanns, H.M. Orientational constraints of the gp130 intracellular juxtamembrane domain for signaling. J. Biol. Chem. 2002, 277, 26959–26965. [Google Scholar] [CrossRef]
- Guschin, D.; Rogers, N.; Briscoe, J.; Witthuhn, B.; Watling, D.; Horn, F.; Pellegrini, S.; Yasukawa, K.; Heinrich, P.; Stark, G. A major role for the protein tyrosine kinase JAK1 in the JAK/STAT signal transduction pathway in response to interleukin-6. EMBO J. 1995, 14, 1421–1429. [Google Scholar] [CrossRef]
- Rodig, S.J.; Meraz, M.A.; White, M.J.; Lampe, P.A.; Riley, J.K.; Arthur, C.D.; King, K.L.; Sheehan, K.C.F.; Yin, L.; Pennica, D.; et al. Disruption of the Jak1 Gene Demonstrates Obligatory and Nonredundant Roles of the Jaks in Cytokine-Induced Biologic Responses. Cell 1998, 93, 373–383. [Google Scholar] [CrossRef] [Green Version]
- Schaper, F.; Gendo, C.; Eck, M.; Schmitz, J.; Grimm, C.; Anhuf, D.; Kerr, I.; Heinrich, P. Activation of the protein tyrosine phosphatase SHP2 via the interleukin-6 signal transducing receptor protein gp130 requires tyrosine kinase Jak1 and limits acute-phase protein expression. Biochem. J. 1998, 335, 557–565. [Google Scholar] [CrossRef] [PubMed]
- Karaghiosoff, M.; Neubauer, H.; Lassnig, C.; Kovarik, P.; Schindler, H.; Pircher, H.; McCoy, B.; Bogdan, C.; Decker, T.; Brem, G.; et al. Partial impairment of cytokine responses in Tyk2-deficient mice. Immunity 2000, 13, 549–560. [Google Scholar] [CrossRef]
- Matadeen, R.; Hon, W.-C.; Heath, J.K.; Jones, Y.E.; Fuller, S. The dynamics of signal triggering in a gp130-receptor complex. Structure 2007, 15, 441–448. [Google Scholar] [CrossRef] [PubMed]
- Stahl, N.; Farruggella, T.J.; Boulton, T.G.; Zhong, Z.; Darnell, J.J.; GD, Y. Choice of STATs and other substrates specified by modular tyrosine-based motifs in cytokine receptors. Science 1995, 267, 1349–1353. [Google Scholar] [CrossRef]
- Gerhartz, C.; Heesel, B.; Sasse, J.; Hemmann, U.; Landgraf, C.; Schneider-Mergener, J.; Horn, F.; Heinrich, P.C.; Graeve, L. Differential activation of acute phase response factor/STAT3 and STAT1 via the cytoplasmic domain of the interleukin 6 signal transducer gp130. I. Definition of a novel phosphotyrosine motif mediating STAT1 activation. J. Biol. Chem. 1996, 271, 12991–12998. [Google Scholar] [CrossRef]
- Heinrich, P.C.; Behrmann, I.; Haan, S.; Hermanns, H.M.; Muller-Newen, G.; Schaper, F. Principles of interleukin (IL)-6-type cytokine signalling and its regulation. Biochem. J. 2003, 374, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Fujitani, Y.; Hibi, M.; Fukada, T.; Takahashi-Tezuka, M.; Yoshida, H.; Yamaguchi, T.; Sugiyama, K.; Yamanaka, Y.; Nakajima, K.; Hirano, T. An alternative pathway for STAT activation that is mediated by the direct interaction between JAK and STAT. Oncogene 1997, 14, 751–761. [Google Scholar] [CrossRef] [Green Version]
- Kaptein, A.; Paillard, V.; Saunders, M. Dominant negative stat3 mutant inhibits interleukin-6-induced Jak-STAT signal transduction. J. Biol. Chem. 1996, 271, 5961–5964. [Google Scholar] [CrossRef]
- Gouilleux, F.; Wakao, H.; Mundt, M.; Groner, B. Prolactin induces phosphorylation of Tyr694 of Stat5 (MGF), a prerequisite for DNA binding and induction of transcription. EMBO J. 1994, 13, 4361–4369. [Google Scholar] [CrossRef]
- Delgoffe, G.M.; Vignali, D.A. STAT heterodimers in immunity: A mixed message or a unique signal? JAKSTAT 2013, 2, e23060. [Google Scholar] [CrossRef]
- Shuai, K.; Stark, G.R.; Kerr, I.M.; Darnell, J.E., Jr. A single phosphotyrosine residue of Stat91 required for gene activation by interferon-gamma. Science 1993, 261, 1744–1746. [Google Scholar] [CrossRef] [PubMed]
- Babon, J.J.; Varghese, L.N.; Nicola, N.A. Inhibition of IL-6 family cytokines by SOCS3. Semin. Immunol. 2014, 26, 13–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshimura, A.; Naka, T.; Kubo, M. SOCS proteins, cytokine signalling and immune regulation. Nat. Rev. Immunol. 2007, 7, 454–465. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, J.; Weissenbach, M.; Haan, S.; Heinrich, P.C.; Schaper, F. SOCS3 exerts its inhibitory function on interleukin-6 signal transduction through the SHP2 recruitment site of gp130. J. Biol. Chem. 2000, 275, 12848–12856. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, S.E.; De Souza, D.; Fabri, L.J.; Corbin, J.; Willson, T.A.; Zhang, J.G.; Silva, A.; Asimakis, M.; Farley, A.; Nash, A.D.; et al. Suppressor of cytokine signaling-3 preferentially binds to the SHP-2-binding site on the shared cytokine receptor subunit gp130. Proc. Natl. Acad. Sci. USA 2000, 97, 6493–6498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sasaki, A.; Yasukawa, H.; Suzuki, A.; Kamizono, S.; Syoda, T.; Kinjyo, I.; Sasaki, M.; Johnston, J.A.; Yoshimura, A. Cytokine-inducible SH2 protein-3 (CIS3/SOCS3) inhibits Janus tyrosine kinase by binding through the N-terminal kinase inhibitory region as well as SH2 domain. Genes Cells 1999, 4, 339–351. [Google Scholar] [CrossRef]
- Yasukawa, H.; Misawa, H.; Sakamoto, H.; Masuhara, M.; Sasaki, A.; Wakioka, T.; Ohtsuka, S.; Imaizumi, T.; Matsuda, T.; Ihle, J.N.; et al. The JAK-binding protein JAB inhibits Janus tyrosine kinase activity through binding in the activation loop. EMBO J. 1999, 18, 1309–1320. [Google Scholar] [CrossRef]
- Kershaw, N.J.; Murphy, J.M.; Liau, N.P.; Varghese, L.N.; Laktyushin, A.; Whitlock, E.L.; Lucet, I.S.; Nicola, N.A.; Babon, J.J. SOCS3 binds specific receptor-JAK complexes to control cytokine signaling by direct kinase inhibition. Nat. Struct Mol. Biol. 2013, 20, 469–476. [Google Scholar] [CrossRef]
- Kershaw, N.J.; Laktyushin, A.; Nicola, N.A.; Babon, J.J. Reconstruction of an active SOCS3-based E3 ubiquitin ligase complex in vitro: Identification of the active components and JAK2 and gp130 as substrates. Growth Factors 2014, 32, 1–10. [Google Scholar] [CrossRef]
- Kamura, T.; Sato, S.; Haque, D.; Liu, L.; Kaelin, W.G., Jr.; Conaway, R.C.; Conaway, J.W. The Elongin BC complex interacts with the conserved SOCS-box motif present in members of the SOCS, ras, WD-40 repeat, and ankyrin repeat families. Genes Dev. 1998, 12, 3872–3881. [Google Scholar] [CrossRef] [Green Version]
- Tebbutt, N.; Giraud, A.; Inglese, M.; Jenkins, B.; Waring, P.; Clay, F.; Malki, S.; Alderman, B.; Grail, D.; Hollande, F.; et al. Reciprocal regulation of gastrointestinal homeostasis by SHP2 and STAT-mediated trefoil gene activation in gp130 mutant mice. Nat. Med. 2002, 8, 1089–1097. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, U.; Schmitz, J.; Weissenbach, M.; Sobota, R.M.; Hortner, M.; Friederichs, K.; Behrmann, I.; Tsiaris, W.; Sasaki, A.; Schneider-Mergener, J.; et al. SHP2 and SOCS3 contribute to Tyr-759-dependent attenuation of interleukin-6 signaling through gp130. J. Biol. Chem. 2003, 278, 661–671. [Google Scholar] [CrossRef] [PubMed]
- Dittrich, A.; Quaiser, T.; Khouri, C.; Gortz, D.; Monnigmann, M.; Schaper, F. Model-driven experimental analysis of the function of SHP-2 in IL-6-induced Jak/STAT signaling. Mol. Biosyst. 2012, 8, 2119–2134. [Google Scholar] [CrossRef] [PubMed]
- Chung, C.D.; Liao, J.; Liu, B.; Rao, X.; Jay, P.; Berta, P.; Shuai, K. Specific inhibition of Stat3 signal transduction by PIAS3. Science 1997, 278, 1803–1805. [Google Scholar] [CrossRef]
- Shuai, K.; Liu, B. Regulation of gene-activation pathways by PIAS proteins in the immune system. Nat. Rev. Immunol. 2005, 5, 593–605. [Google Scholar] [CrossRef]
- Sun, Y.; Tokushige, K.; Isono, E.; Yamauchi, K.; Obata, H. Elevated serum interleukin-6 levels in patients with acute hepatitis. J. Clin. Immunol. 1992, 12, 197–200. [Google Scholar] [CrossRef]
- Deviere, J.; Content, J.; Denys, C.; Vandenbussche, P.; Schandene, L.; Wybran, J.; Dupont, E. High interleukin-6 serum levels and increased production by leucocytes in alcoholic liver cirrhosis. Correlation with IgA serum levels and lymphokines production. Clin. Exp. Immunol. 1989, 77, 221–225. [Google Scholar]
- Kakumu, S.; Shinagawa, T.; Ishikawa, T.; Yoshioka, K.; Wakita, T.; Ida, N. Interleukin 6 production by peripheral blood mononuclear cells in patients with chronic hepatitis B virus infection and primary biliary cirrhosis. Gastroenterol. Jpn. 1993, 28, 18–24. [Google Scholar] [CrossRef]
- Shakiba, E.; Ramezani, M.; Sadeghi, M. Evaluation of serum interleukin-6 levels in hepatocellular carcinoma patients: A systematic review and meta-analysis. Clin. Exp. Hepatol. 2018, 4, 182–190. [Google Scholar] [CrossRef]
- Shao, Y.Y.; Lin, H.; Li, Y.S.; Lee, Y.H.; Chen, H.M.; Cheng, A.L.; Hsu, C.H. High plasma interleukin-6 levels associated with poor prognosis of patients with advanced hepatocellular carcinoma. Jpn. J. Clin. Oncol. 2017, 47, 949–953. [Google Scholar] [CrossRef]
- Naugler, W.E.; Sakurai, T.; Kim, S.; Maeda, S.; Kim, K.; Elsharkawy, A.M.; Karin, M. Gender disparity in liver cancer due to sex differences in MyD88-dependent IL-6 production. Science 2007, 317, 121–124. [Google Scholar] [CrossRef] [PubMed]
- Wan, S.; Zhao, E.; Kryczek, I.; Vatan, L.; Sadovskaya, A.; Ludema, G.; Simeone, D.M.; Zou, W.; Welling, T.H. Tumor-associated macrophages produce interleukin 6 and signal via STAT3 to promote expansion of human hepatocellular carcinoma stem cells. Gastroenterology 2014, 147, 1393–1404. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.Y.; Zhou, Y.L.; Qian, M.J.; Fang, Y.Z.; Ye, S.; Xin, W.X.; Yang, X.C.; Wu, H.H. Interleukin-6 induced by YAP in hepatocellular carcinoma cells recruits tumor-associated macrophages. J. Pharmacol. Sci. 2018, 138, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Giannitrapani, L.; Cervello, M.; Soresi, M.; Notarbartolo, M.; La Rosa, M.; Virruso, L.; D’Alessandro, N.; Montalto, G. Circulating IL-6 and sIL-6R in patients with hepatocellular carcinoma. Ann. N. Y. Acad. Sci. 2002, 963, 46–52. [Google Scholar] [CrossRef]
- Bergmann, J.; Müller, M.; Baumann, N.; Reichert, M.; Heneweer, C.; Bolik, J.; Lücke, K.; Gruber, S.; Carambia, A.; Boretius, S.; et al. IL-6 trans-signaling is essential for the development of hepatocellular carcinoma in mice. Hepatology 2017, 65, 89–103. [Google Scholar] [CrossRef]
- Xiang, Z.L.; Zeng, Z.C.; Fan, J.; Tang, Z.Y.; Zeng, H.Y. Expression of connective tissue growth factor and interleukin-11 in intratumoral tissue is associated with poor survival after curative resection of hepatocellular carcinoma. Mol. Biol. Rep. 2012, 39, 6001–6006. [Google Scholar] [CrossRef]
- Xiang, Z.L.; Zeng, Z.C.; Tang, Z.Y.; Fan, J.; He, J.; Zeng, H.Y.; Zhu, X.D. Potential prognostic biomarkers for bone metastasis from hepatocellular carcinoma. Oncologist 2011, 16, 1028–1039. [Google Scholar] [CrossRef]
- Xiang, Z.L.; Zeng, Z.C.; Fan, J.; Wu, W.Z.; He, J.; Zeng, H.Y.; Tang, Z.Y. A clinicopathological model to predict bone metastasis in hepatocellular carcinoma. J. Cancer Res. Clin. Oncol. 2011, 137, 1791–1797. [Google Scholar] [CrossRef]
- Yuan, J.-H.; Yang, F.; Wang, F.; Ma, J.-Z.; Guo, Y.-J.; Tao, Q.-F.; Liu, F.; Pan, W.; Wang, T.-T.; Zhou, C.-C.; et al. A Long Noncoding RNA Activated by TGF-β Promotes the Invasion-Metastasis Cascade in Hepatocellular Carcinoma. Cancer Cell 2014, 25, 666–681. [Google Scholar] [CrossRef]
- Zhang, L.; Niu, H.; Ma, J.; Yuan, B.Y.; Chen, Y.H.; Zhuang, Y.; Chen, G.W.; Zeng, Z.C.; Xiang, Z.L. The molecular mechanism of LncRNA34a-mediated regulation of bone metastasis in hepatocellular carcinoma. Mol. Cancer 2019, 18, 120. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Yang, Y.; Han, J.; Jiang, W.H.; Chen, C.; Wang, M.C.; Gao, R.; Li, S.; Tian, T.; Wang, J.; et al. TMED3 promotes hepatocellular carcinoma progression via IL-11/STAT3 signaling. Sci. Rep. 2016, 6, 37070. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Zheng, X.; Fu, B.; Nian, Z.; Qian, Y.; Sun, R.; Tian, Z.; Wei, H. Hepatectomy promotes recurrence of liver cancer by enhancing IL-11-STAT3 signaling. EBioMedicine 2019, 46, 119–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.F.; He, M.L.; Fu, W.M.; Wang, H.; Chen, L.Z.; Zhu, X.; Chen, Y.; Xie, D.; Lai, P.; Chen, G.; et al. Primate-specific microRNA-637 inhibits tumorigenesis in hepatocellular carcinoma by disrupting signal transducer and activator of transcription 3 signaling. Hepatology 2011, 54, 2137–2148. [Google Scholar] [CrossRef] [PubMed]
- Luo, Q.; Zhang, Y.; Wang, N.; Jin, G.; Jin, H.; Gu, D.; Tao, X.; Huo, X.; Ge, T.; Cong, W.; et al. Leukemia inhibitory factor receptor is a novel immunomarker in distinction of well-differentiated HCC from dysplastic nodules. Oncotarget 2015, 6, 6989–6999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okamura, Y.; Nomoto, S.; Kanda, M.; Li, Q.; Nishikawa, Y.; Sugimoto, H.; Kanazumi, N.; Takeda, S.; Nakao, A. Leukemia inhibitory factor receptor (LIFR) is detected as a novel suppressor gene of hepatocellular carcinoma using double-combination array. Cancer Lett. 2010, 289, 170–177. [Google Scholar] [CrossRef]
- Luo, Q.; Wang, C.; Jin, G.; Gu, D.; Wang, N.; Song, J.; Jin, H.; Hu, F.; Zhang, Y.; Ge, T.; et al. LIFR functions as a metastasis suppressor in hepatocellular carcinoma by negatively regulating phosphoinositide 3-kinase/AKT pathway. Carcinogenesis 2015, 36, 1201–1212. [Google Scholar] [CrossRef]
- Liang, H.; Block, T.M.; Wang, M.; Nefsky, B.; Long, R.; Hafner, J.; Mehta, A.S.; Marrero, J.; Gish, R.; Norton, P.A. Interleukin-6 and oncostatin M are elevated in liver disease in conjunction with candidate hepatocellular carcinoma biomarker GP73. Cancer Biomark 2012, 11, 161–171. [Google Scholar] [CrossRef] [Green Version]
- Rolvering, C.; Zimmer, A.D.; Kozar, I.; Hermanns, H.M.; Letellier, E.; Vallar, L.; Nazarov, P.V.; Nicot, N.; Ginolhac, A.; Haan, S.; et al. Crosstalk between different family members: IL27 recapitulates IFNgamma responses in HCC cells, but is inhibited by IL6-type cytokines. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 516–526. [Google Scholar] [CrossRef]
- Song, Q.; Chen, X.; Hu, W.; Mei, G.; Yang, X.; Wu, H. Downregulation of Epstein-Barr virus-induced gene 3 is associated with poor prognosis of hepatocellular carcinoma after curative resection. Oncol. Lett. 2018, 15, 7751–7759. [Google Scholar] [CrossRef]
- Li, B.; Su, H.; Cao, J.; Zhang, L. CXCL13 rather than IL-31 is a potential indicator in patients with hepatocellular carcinoma. Cytokine 2017, 89, 91–97. [Google Scholar] [CrossRef]
- Schulze, K.; Nault, J.C.; Villanueva, A. Genetic profiling of hepatocellular carcinoma using next-generation sequencing. J. Hepatol. 2016, 65, 1031–1042. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.X.; Zhu, Q.G.; Zhang, S.M.; Guan, L.; Li, T.; Zhang, L.; Wang, S.Y.; Ren, W.L.; Chen, X.M.; Zhao, J.; et al. Precision medicine for hepatocellular carcinoma: Driver mutations and targeted therapy. Oncotarget 2017, 8, 55715–55730. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S. The mutational landscape of hepatocellular carcinoma. Clin. Mol. Hepatol. 2015, 21, 220–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kan, Z.; Zheng, H.; Liu, X.; Li, S.; Barber, T.D.; Gong, Z.; Gao, H.; Hao, K.; Willard, M.D.; Xu, J.; et al. Whole-genome sequencing identifies recurrent mutations in hepatocellular carcinoma. Genome Res. 2013, 23, 1422–1433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rebouissou, S.; Amessou, M.; Couchy, G.; Poussin, K.; Imbeaud, S.; Pilati, C.; Izard, T.; Balabaud, C.; Bioulac-Sage, P.; Zucman-Rossi, J. Frequent in-frame somatic deletions activate gp130 in inflammatory hepatocellular tumours. Nature 2009, 457, 200–204. [Google Scholar] [CrossRef]
- Poussin, K.; Pilati, C.; Couchy, G.; Calderaro, J.; Bioulac-Sage, P.; Bacq, Y.; Paradis, V.; Leteurtre, E.; Sturm, N.; Ramos, J.; et al. Biochemical and functional analyses of gp130 mutants unveil JAK1 as a novel therapeutic target in human inflammatory hepatocellular adenoma. Oncoimmunology 2013, 2. [Google Scholar] [CrossRef]
- Hatting, M.; Spannbauer, M.; Peng, J.; Al Masaoudi, M.; Sellge, G.; Nevzorova, Y.A.; Gassler, N.; Liedtke, C.; Cubero, F.J.; Trautwein, C. Lack of gp130 expression in hepatocytes attenuates tumor progression in the DEN model. Cell Death Dis 2015, 6, e1667. [Google Scholar] [CrossRef]
- Galicia, J.; Tai, H.; Komatsu, Y.; Shimada, Y.; Akazawa, K.; Yoshie, H. Polymorphisms in the IL-6 receptor (IL-6R) gene: Strong evidence that serum levels of soluble IL-6R are genetically influenced. Genes Immun. 2004, 5, 513–516. [Google Scholar] [CrossRef]
- Rafiq, S.; Frayling, T.; Murray, A.; Hurst, A.; Stevens, K.; Weedon, M.; Henley, W.; Ferrucci, L.; Bandinelli, S.; Corsi, A.M.; et al. A common variant of the interleukin 6 receptor (IL-6r) gene increases IL-6r and IL-6 levels, without other inflammatory effects. Genes Immun. 2007, 8, 552–559. [Google Scholar] [CrossRef]
- Garbers, C.; Monhasery, N.; Aparicio-Siegmund, S.; Lokau, J.; Baran, P.; Nowell, M.A.; Jones, S.A.; Rose-John, S.; Scheller, J. The Interleukin-6 Receptor Asp358Ala Single Nucleotide Polymorphism rs2228145 Confers Increased Proteolytic Conversion Rates by ADAM Proteases. Biochim. Biophys. Acta 2014, 1842, 1485–1494. [Google Scholar] [CrossRef]
- Ferreira, R.; Freitag, D.; Cutler, A.; Howson, J.M.; Rainbow, D.; Smyth, D.; Kaptoge, S.; Clarke, P.; Boreham, C.; Coulson, R.; et al. Functional IL6R 358Ala Allele Impairs Classical IL-6 Receptor Signaling and Influences Risk of Diverse Inflammatory Diseases. PLoS Genet. 2013, 9, e1003444. [Google Scholar] [CrossRef] [PubMed]
- Agthe, M.; Brugge, J.; Garbers, Y.; Wandel, M.; Kespohl, B.; Arnold, P.; Flynn, C.M.; Lokau, J.; Aparicio-Siegmund, S.; Bretscher, C.; et al. Mutations in Craniosynostosis Patients Cause Defective Interleukin-11 Receptor Maturation and Drive Craniosynostosis-like Disease in Mice. Cell Rep. 2018, 25, 10–18 e15. [Google Scholar] [CrossRef] [PubMed]
- Keupp, K.; Li, Y.; Vargel, I.; Hoischen, A.; Richardson, R.; Neveling, K.; Alanay, Y.; Uz, E.; Elcioğlu, N.; Rachwalski, M.; et al. Mutations in the interleukin receptor IL11RA cause autosomal recessive Crouzon-like craniosynostosis. Mol. Genet. Genomic Med. 2013, 1, 223–237. [Google Scholar] [CrossRef] [PubMed]
- Nieminen, P.; Morgan, N.; Fenwick, A.; Parmanen, S.; Veistinen, L.; Mikkola, M.; van der Spek, P.; Giraud, A.; Judd, L.; Arte, S.; et al. Inactivation of IL11 signaling causes craniosynostosis, delayed tooth eruption, and supernumerary teeth. Am. J. Hum. Genet. 2011, 89, 67–81. [Google Scholar] [CrossRef] [PubMed]
- Xie, H.J.; Bae, H.J.; Noh, J.H.; Eun, J.W.; Kim, J.K.; Jung, K.H.; Ryu, J.C.; Ahn, Y.M.; Kim, S.Y.; Lee, S.H.; et al. Mutational analysis of JAK1 gene in human hepatocellular carcinoma. Neoplasma 2009, 56, 136–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, S.; Luo, C.; Gu, Q.; Xu, Q.; Wang, G.; Sun, H.; Qian, Z.; Tan, Y.; Qin, Y.; Shen, Y.; et al. Activating JAK1 mutation may predict the sensitivity of JAK-STAT inhibition in hepatocellular carcinoma. Oncotarget 2016, 7, 5461–5469. [Google Scholar] [CrossRef] [PubMed]
- Ungureanu, D.; Wu, J.; Pekkala, T.; Niranjan, Y.; Young, C.; Jensen, O.N.; Xu, C.F.; Neubert, T.A.; Skoda, R.C.; Hubbard, S.R.; et al. The pseudokinase domain of JAK2 is a dual-specificity protein kinase that negatively regulates cytokine signaling. Nat. Struct. Mol. Biol. 2011, 18, 971–976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, G.; Yu, G.Y.; Temkin, V.; Ogata, H.; Kuntzen, C.; Sakurai, T.; Sieghart, W.; Peck-Radosavljevic, M.; Leffert, H.L.; Karin, M. Hepatocyte IKKbeta/NF-kappaB inhibits tumor promotion and progression by preventing oxidative stress-driven STAT3 activation. Cancer Cell 2010, 17, 286–297. [Google Scholar] [CrossRef]
- Calvisi, D.F.; Ladu, S.; Gorden, A.; Farina, M.; Conner, E.A.; Lee, J.S.; Factor, V.M.; Thorgeirsson, S.S. Ubiquitous activation of Ras and Jak/Stat pathways in human HCC. Gastroenterology 2006, 130, 1117–1128. [Google Scholar] [CrossRef]
- Pilati, C.; Amessou, M.; Bihl, M.; Balabaud, C.; Nhieu, J.; Paradis, V.; Nault, J.; Izard, T.; Bioulac-Sage, P.; Couchy, G.; et al. Somatic mutations activating STAT3 in human inflammatory hepatocellular adenomas. J. Exp. Med. 2011, 208, 1359–1366. [Google Scholar] [CrossRef]
- Pilati, C.; Zucman-Rossi, J. Mutations leading to constitutive active gp130/JAK1/STAT3 pathway. Cytokine Growth Factor Rev. 2015, 26, 499–506. [Google Scholar] [CrossRef]
- Zhang, J.; Li, Z.; Liu, L.; Wang, Q.; Li, S.; Chen, D.; Hu, Z.; Yu, T.; Ding, J.; Li, J.; et al. Long noncoding RNA TSLNC8 is a tumor suppressor that inactivates the interleukin-6/STAT3 signaling pathway. Hepatology 2018, 67, 171–187. [Google Scholar] [CrossRef]
- Chen, G.; Wang, H.; Xie, S.; Ma, J.; Wang, G. STAT1 negatively regulates hepatocellular carcinoma cell proliferation. Oncol. Rep. 2013, 29, 2303–2310. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Wang, H.; Wang, J.; Huang, S.; Zhang, W. STAT1 inhibits human hepatocellular carcinoma cell growth through induction of p53 and Fbxw7. Cancer Cell Int. 2015, 15, 111. [Google Scholar] [CrossRef] [Green Version]
- Ma, B.; Chen, K.; Liu, P.; Li, M.; Liu, J.; Sideras, K.; Sprengers, D.; Biermann, K.; Wang, W.; Ijzermans, J.N.M.; et al. Dichotomal functions of phosphorylated and unphosphorylated STAT1 in hepatocellular carcinoma. J. Mol. Med. (Berl.) 2019, 97, 77–88. [Google Scholar] [CrossRef]
- Hoan, N.X.; Van Tong, H.; Giang, D.P.; Cuong, B.K.; Toan, N.L.; Wedemeyer, H.; Bock, C.T.; Kremsner, P.G.; Song, L.H.; Velavan, T.P. SOCS3 genetic variants and promoter hypermethylation in patients with chronic hepatitis B. Oncotarget 2017, 8, 17127–17139. [Google Scholar] [CrossRef]
- Zhang, X.; You, Q.; Zhang, X.; Chen, X. SOCS3 Methylation Predicts a Poor Prognosis in HBV Infection-Related Hepatocellular Carcinoma. Int J. Mol. Sci. 2015, 16, 22662–22675. [Google Scholar] [CrossRef] [Green Version]
- Jiang, B.G.; Wang, N.; Huang, J.; Yang, Y.; Sun, L.L.; Pan, Z.Y.; Zhou, W.P. Tumor SOCS3 methylation status predicts the treatment response to TACE and prognosis in HCC patients. Oncotarget 2017, 8, 28621–28627. [Google Scholar] [CrossRef]
- Riehle, K.J.; Campbell, J.S.; McMahan, R.S.; Johnson, M.M.; Beyer, R.P.; Bammler, T.K.; Fausto, N. Regulation of liver regeneration and hepatocarcinogenesis by suppressor of cytokine signaling 3. J. Exp. Med. 2008, 205, 91–103. [Google Scholar] [CrossRef]
- Daher, S.; Massarwa, M.; Benson, A.A.; Khoury, T. Current and Future Treatment of Hepatocellular Carcinoma: An Updated Comprehensive Review. J. Clin. Transl Hepatol. 2018, 6, 69–78. [Google Scholar] [CrossRef]
- Tian, G.; Yang, S.; Yuan, J.; Threapleton, D.; Zhao, Q.; Chen, F.; Cao, H.; Jiang, T.; Li, L. Comparative efficacy of treatment strategies for hepatocellular carcinoma: Systematic review and network meta-analysis. BMJ Open 2018, 8, e021269. [Google Scholar] [CrossRef]
- Marrero, J.A.; Kulik, L.M.; Sirlin, C.B.; Zhu, A.X.; Finn, R.S.; Abecassis, M.M.; Roberts, L.R.; Heimbach, J.K. Diagnosis, Staging, and Management of Hepatocellular Carcinoma: 2018 Practice Guidance by the American Association for the Study of Liver Diseases. Hepatology 2018, 68, 723–750. [Google Scholar] [CrossRef] [Green Version]
- Lencioni, R.; de Baere, T.; Soulen, M.C.; Rilling, W.S.; Geschwind, J.F. Lipiodol transarterial chemoembolization for hepatocellular carcinoma: A systematic review of efficacy and safety data. Hepatology 2016, 64, 106–116. [Google Scholar] [CrossRef] [Green Version]
- Loosen, S.H.; Schulze-Hagen, M.; Leyh, C.; Benz, F.; Vucur, M.; Kuhl, C.; Trautwein, C.; Tacke, F.; Bruners, P.; Roderburg, C.; et al. IL-6 and IL-8 Serum Levels Predict Tumor Response and Overall Survival after TACE for Primary and Secondary Hepatic Malignancies. Int. J. Mol. Sci. 2018, 19, 1766. [Google Scholar] [CrossRef]
- Garbers, C.; Heink, S.; Korn, T.; Rose-John, S. Interleukin-6: Designing specific therapeutics for a complex cytokine. Nat. Rev. Drug Discov. 2018, 17, 395–412. [Google Scholar] [CrossRef]
- Garbers, C.; Aparicio-Siegmund, S.; Rose-John, S. The IL-6/gp130/STAT3 signaling axis: Recent advances towards specific inhibition. Curr. Opin. Immunol. 2015, 34, 75–82. [Google Scholar] [CrossRef]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.F.; de Oliveira, A.C.; Santoro, A.; Raoul, J.L.; Forner, A.; et al. Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef] [PubMed]
- Kudo, M.; Finn, R.S.; Qin, S.; Han, K.H.; Ikeda, K.; Piscaglia, F.; Baron, A.; Park, J.W.; Han, G.; Jassem, J.; et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: A randomised phase 3 non-inferiority trial. Lancet 2018, 391, 1163–1173. [Google Scholar] [CrossRef]
- Schwartz, D.M.; Kanno, Y.; Villarino, A.; Ward, M.; Gadina, M.; O’Shea, J.J. JAK inhibition as a therapeutic strategy for immune and inflammatory diseases. Nat. Rev. Drug Discov. 2017, 16, 843–862. [Google Scholar] [CrossRef]
- Wilson, G.S.; Tian, A.; Hebbard, L.; Duan, W.; George, J.; Li, X.; Qiao, L. Tumoricidal effects of the JAK inhibitor Ruxolitinib (INC424) on hepatocellular carcinoma in vitro. Cancer Lett. 2013, 341, 224–230. [Google Scholar] [CrossRef]
- Ma, H.; Yan, D.; Wang, Y.; Shi, W.; Liu, T.; Zhao, C.; Huo, S.; Duan, J.; Tao, J.; Zhai, M.; et al. Bazedoxifene exhibits growth suppressive activity by targeting interleukin-6/glycoprotein 130/signal transducer and activator of transcription 3 signaling in hepatocellular carcinoma. Cancer Sci. 2019, 110, 950–961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saad, M.I.; Rose-John, S.; Jenkins, B.J. ADAM17: An Emerging Therapeutic Target for Lung Cancer. Cancers 2019, 11, 1218. [Google Scholar] [CrossRef] [PubMed]
- Saad, M.I.; Alhayyani, S.; McLeod, L.; Yu, L.; Alanazi, M.; Deswaerte, V.; Tang, K.; Jarde, T.; Smith, J.A.; Prodanovic, Z.; et al. ADAM17 selectively activates the IL-6 trans-signaling/ERK MAPK axis in KRAS-addicted lung cancer. EMBO Mol. Med. 2019, 11. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Li, Y.; Tsung, A.; Huang, H.; Du, Q.; Yang, M.; Deng, M.; Xiong, S.; Wang, X.; Zhang, L.; et al. iNOS promotes CD24(+)CD133(+) liver cancer stem cell phenotype through a TACE/ADAM17-dependent Notch signaling pathway. Proc. Natl. Acad. Sci. USA 2018, 115, E10127–E10136. [Google Scholar] [CrossRef]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lokau, J.; Schoeder, V.; Haybaeck, J.; Garbers, C. Jak-Stat Signaling Induced by Interleukin-6 Family Cytokines in Hepatocellular Carcinoma. Cancers 2019, 11, 1704. https://doi.org/10.3390/cancers11111704
Lokau J, Schoeder V, Haybaeck J, Garbers C. Jak-Stat Signaling Induced by Interleukin-6 Family Cytokines in Hepatocellular Carcinoma. Cancers. 2019; 11(11):1704. https://doi.org/10.3390/cancers11111704
Chicago/Turabian StyleLokau, Juliane, Victor Schoeder, Johannes Haybaeck, and Christoph Garbers. 2019. "Jak-Stat Signaling Induced by Interleukin-6 Family Cytokines in Hepatocellular Carcinoma" Cancers 11, no. 11: 1704. https://doi.org/10.3390/cancers11111704
APA StyleLokau, J., Schoeder, V., Haybaeck, J., & Garbers, C. (2019). Jak-Stat Signaling Induced by Interleukin-6 Family Cytokines in Hepatocellular Carcinoma. Cancers, 11(11), 1704. https://doi.org/10.3390/cancers11111704