Protein Kinase A Distribution in Meningioma



Abstract
:1. Introduction
2. Results
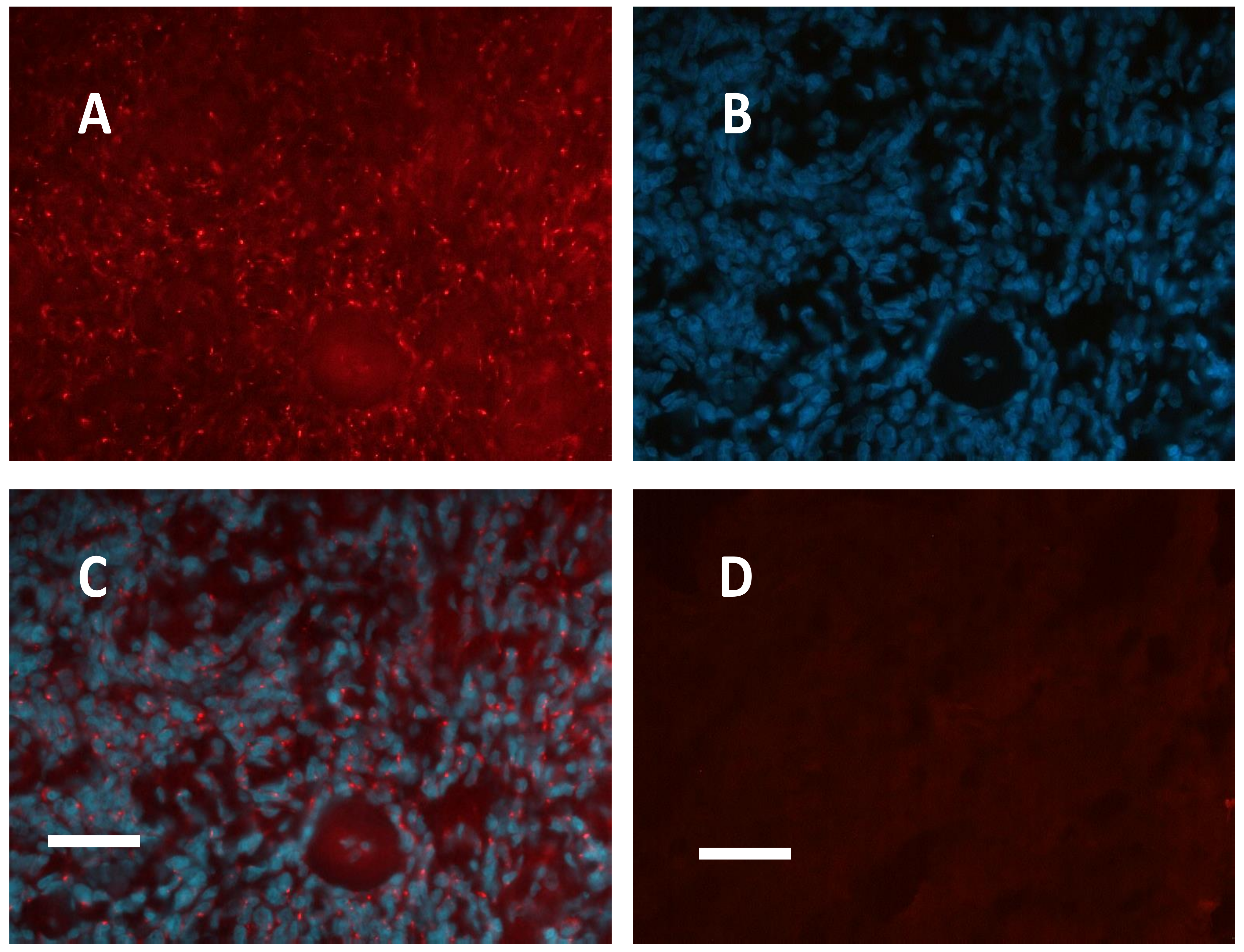
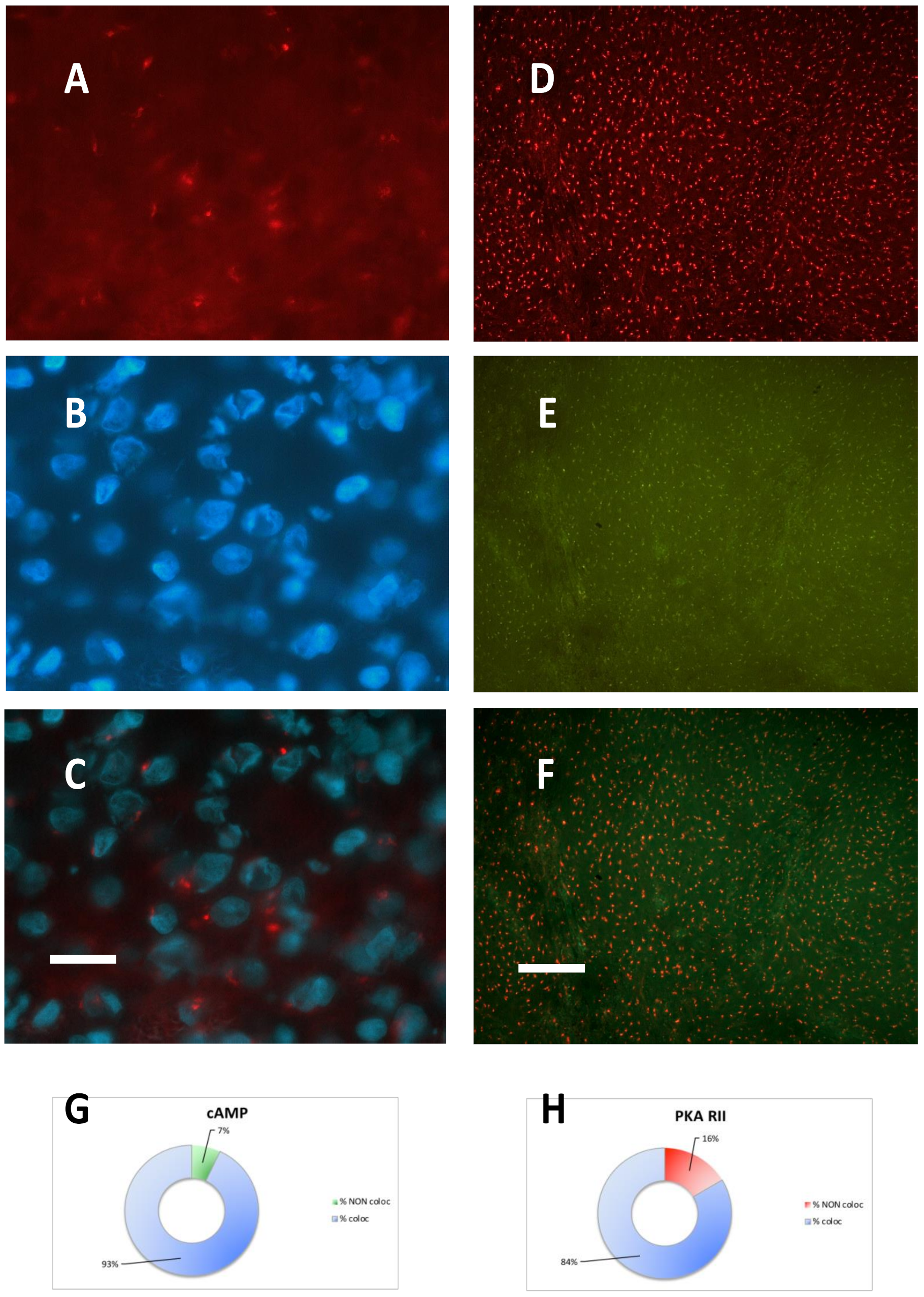
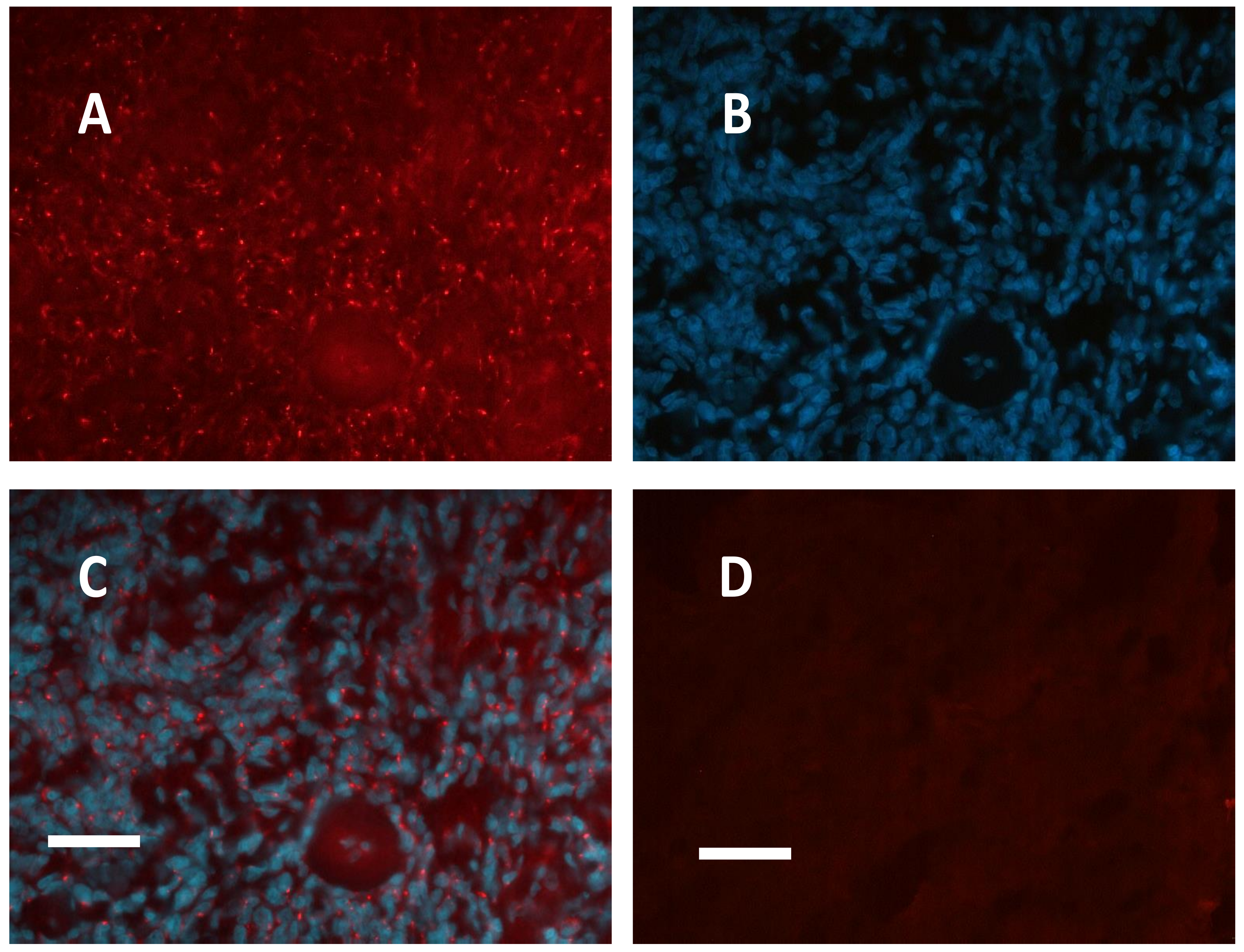
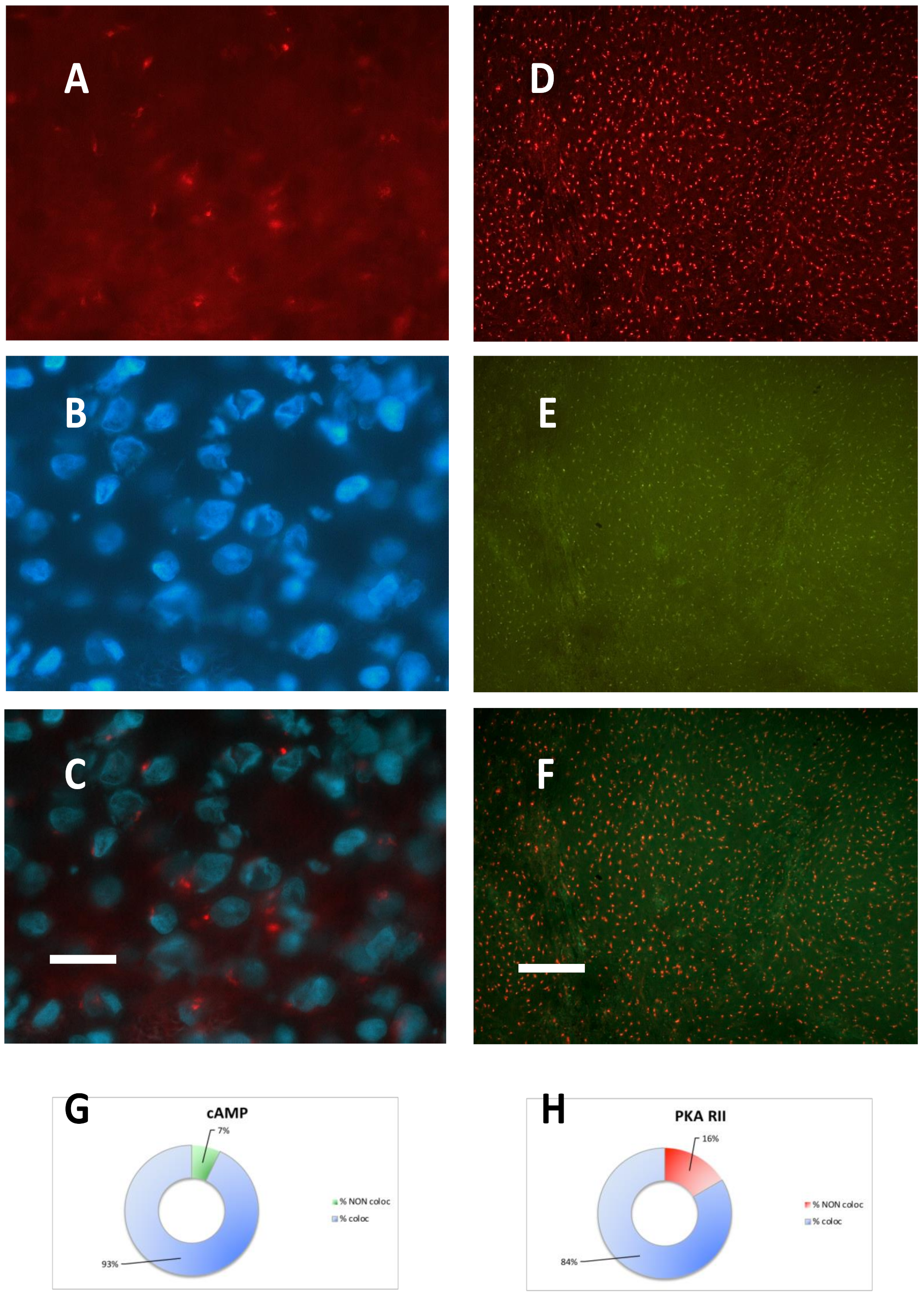
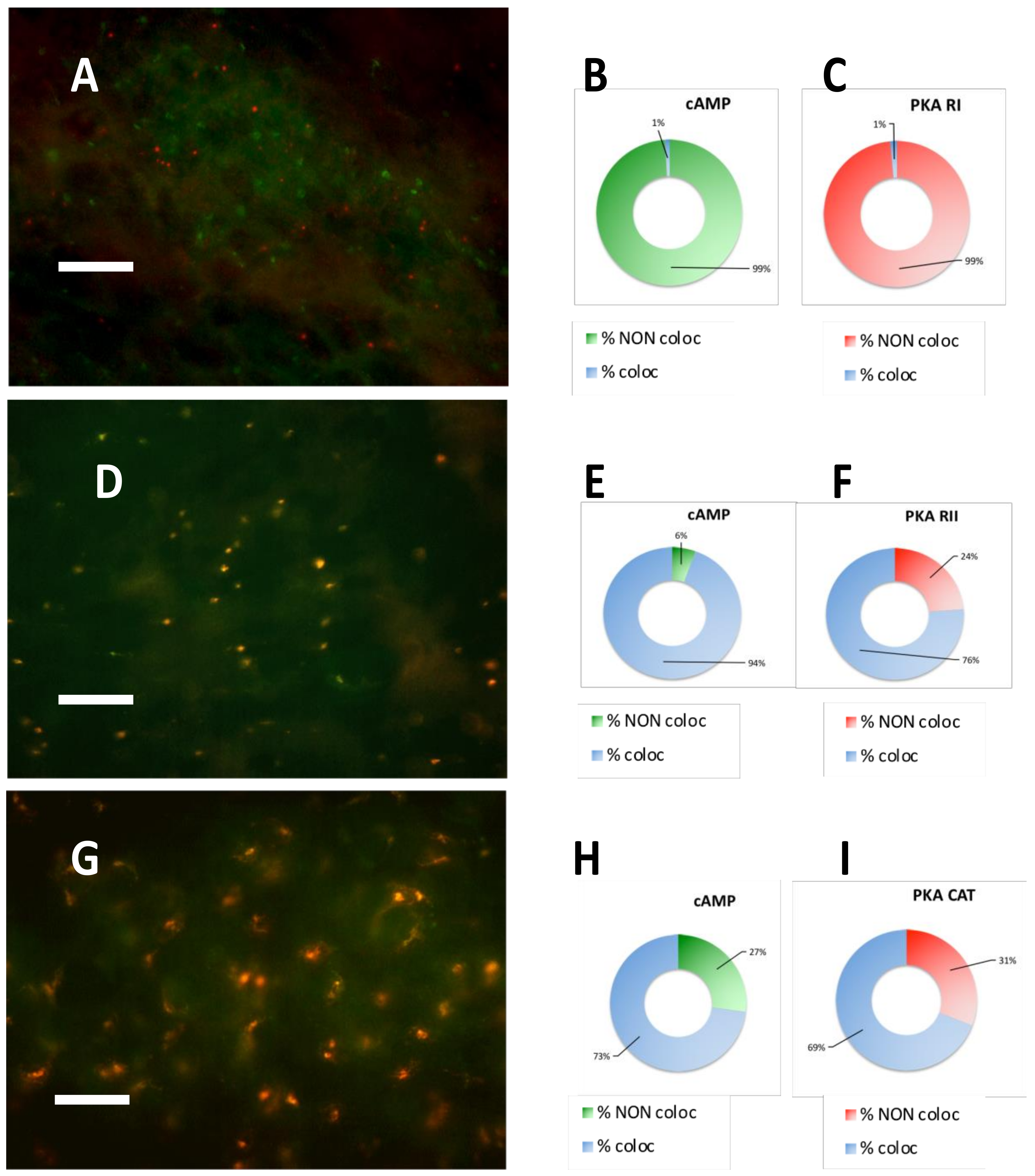
2.1. Meningioma Tissue Binds cAMP
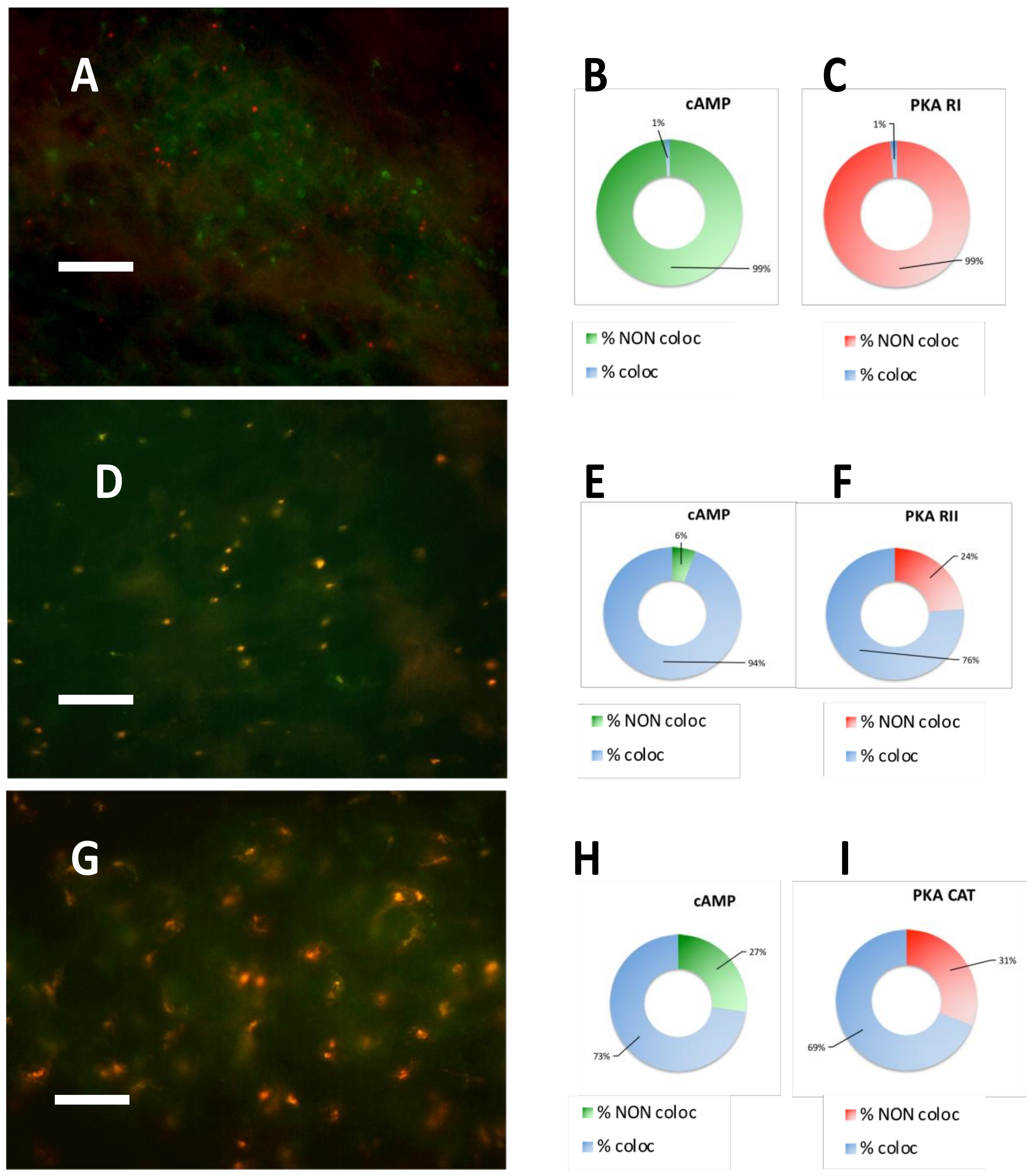
2.2. PKA Regulatory and Catalytic Subunits Immunofluorescence
2.3. PKA RI is Extremely Labile in Meningioma
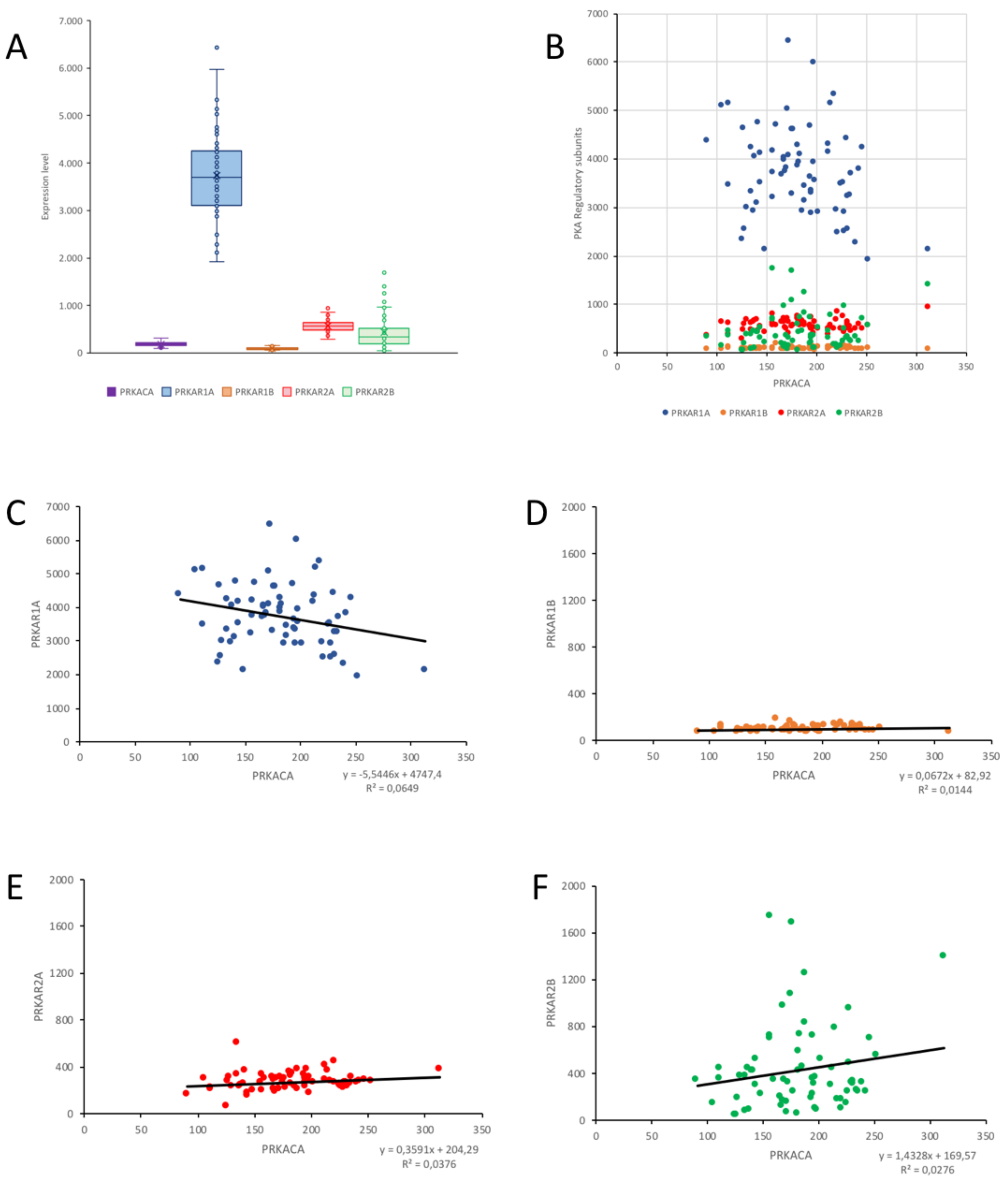
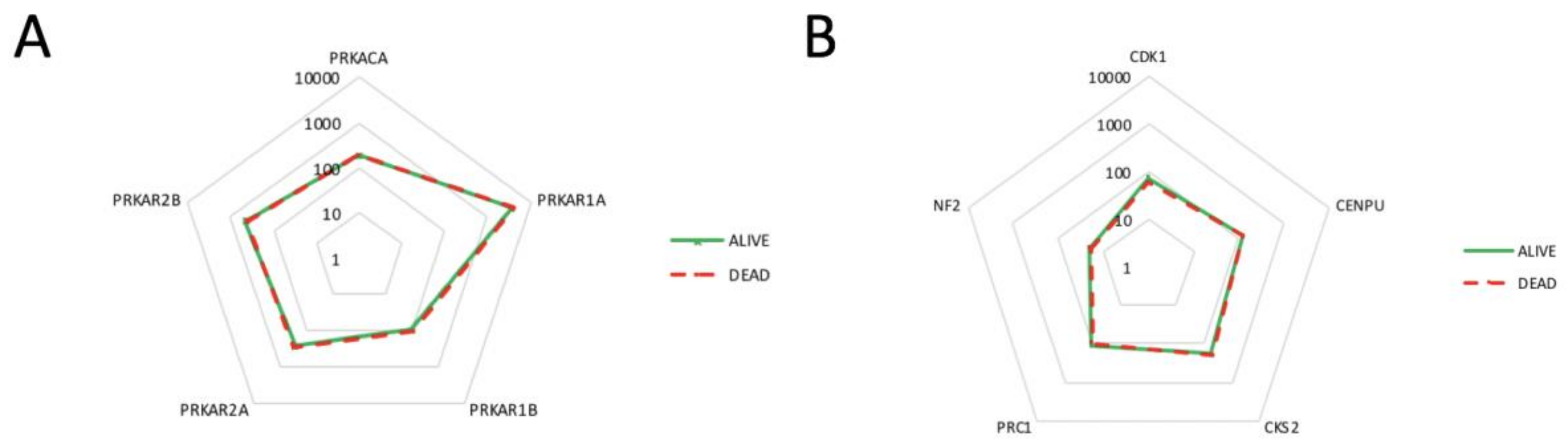
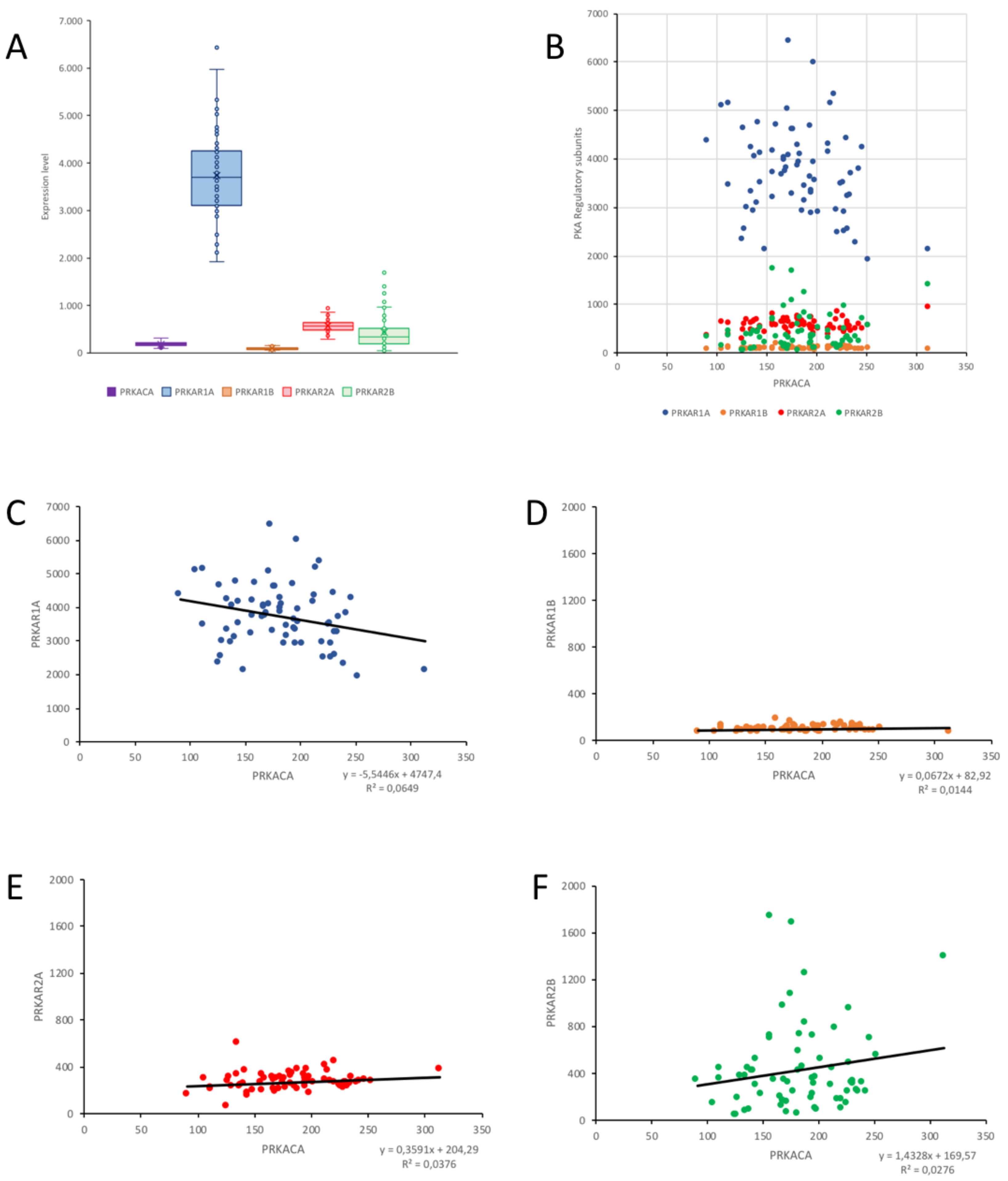
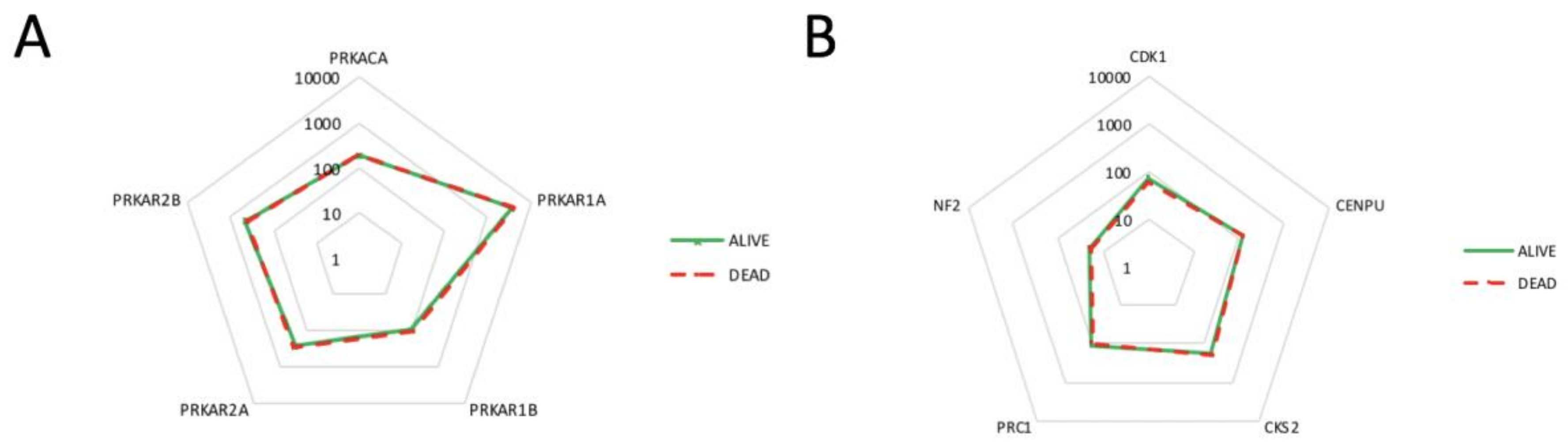
2.4. Correlation of PKA Catalytic Subunit Expression Levels with Different Gene Products
3. Discussion
4. Materials and Methods
4.1. Patients
4.2. Tissue Processing
4.3. Image Analysis
4.4. Expression Data Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| 8-Fluo-cAMP | 8-(2-fluoresceinylthioureidoaminoethylthio)-adenosine 3’,5’ -cyclic monophosphate |
| Alexa488-cAMP | 8-(Alexa488)- adenosine 3’,5’-cyclic monophosphate |
| Alexa555-cAMP | 8-(Alexa555)- adenosine 3’,5’-cyclic monophosphate |
| cAMP | cyclic AMP |
| CNS | Central nervous system |
| PBS | Phosphate buffered saline |
| PKA | Protein kinase A, cAMP-dependent protein kinase |
| R | Regulatory subunits of cAMP dependent protein kinases |
| SAF-cAMP | 8-(5-thioacetamidofluorescein)-adenosine 3’,5’-cyclic monophosphate |
| SAF-cGMP | 8-(5-thioacetamidofluorescein)-guanosin 3’,5’-cyclic monophosphate |
| SAR-cAMP | 8-(5-thioacetamidotetramethylrhodamine)-adenosine 3’,5’-cyclic monophosphate |
References
- Wiemels, J.; Wrensch, M.; Claus, E.B. Epidemiology and etiology of meningioma. J. Neuro Oncol. 2010, 99, 307–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed]
- Pereira, B.J.A.; Oba-Shinjo, M.; Nogueira de Almeida, A.; Marie, S.K.N. Molecular alterations in meningiomas: Literature review. Clin. Neurol. Neurosurg. 2019, 176, 89–96. [Google Scholar] [CrossRef] [PubMed]
- Shaikh, N.; Dixit, K.; Raizer, J. Recent advances in managing/understanding meningioma. F1000Research 2018, 7, 490. [Google Scholar] [CrossRef]
- Grönholm, M.; Vossebein, L.; Carlson, C.R.; Kuja-Panula, J.; Teesalu, T.; Alfthan, K.; Vaheri, A.; Rauvala, H.; Herberg, F.W.; Taskén, K.; et al. Merlin links to the cAMP neuronal signaling pathway by anchoring the RIβ subunit of protein kinase A. J. Biol. Chem. 2003, 278, 41167–41172. [Google Scholar] [CrossRef]
- Torres-Quesada, O.; Mayrhofer, J.E.; Stefan, E. The many faces of compartmentalized PKA signalosomes. Cell Signal. 2017, 37, 1–11. [Google Scholar] [CrossRef]
- Roskoski, R., Jr. A historical overview of protein kinases and their targeted small molecule inhibitors. Pharmacol. Res. 2015, 100, 1–23. [Google Scholar] [CrossRef]
- Ilouz, R.; Lev-Ram, V.; Bushong, E.A.; Stiles, T.L.; Friedmann-Morvinski, D.; Douglas, C.; Goldberg, G.; Ellisman, M.H.; Taylor, S.S. Isoform-specific subcellular localization and function of protein kinase A identified by mosaic imaging of mouse brain. eLife 2017, 12, E17681. [Google Scholar] [CrossRef]
- Liu, Q.; Nguyen, E.; Døskeland, S.; Ségal-Bendirdjian, É. Camp-Dependent Protein Kinase A (PKA)-Mediated c-Myc Degradation Is Dependent on the Relative Proportion of PKA-I and PKA-II Isozymes. Mol. Pharmacol. 2015, 88, 469–476. [Google Scholar] [CrossRef]
- Mantovani, G.; Bondioni, S.; Lania, A.G.; Rodolfo, M.; Peverelli, E.; Polentarutti, N.; Veliz Rodriguez, T.; Ferrero, S.; Bosari, S.; Beck-Peccoz, P.; et al. High expression of PKA regulatory subunit 1A protein is related to proliferation of human melanoma cells. Oncogene. 2008, 27, 1834–1843. [Google Scholar] [CrossRef]
- Alfthan, K.; Heiska, L.; Grönholm, M.; Renkema, G.H.; Carpén, O. Cyclic AMP-dependent protein kinase phosphorylates merlin at serine 518 independently of p21-activated kinase and promotes merlin-ezrin heterodimerization. J. Biol. Chem. 2004, 279, 18559–18566. [Google Scholar] [CrossRef] [PubMed]
- Mucignat-Caretta, C.; Caretta, A. Localization of Triton-insoluble cAMP-dependent kinase type RIβ in rat and mouse brain. J. Neurocytol. 2001, 30, 885–894. [Google Scholar] [CrossRef] [PubMed]
- Mucignat-Caretta, C.; Caretta, A. Clustered distribution of cAMP-dependent protein kinase regulatory isoform RIα during the development of the rat brain. J. Comp. Neurol. 2002, 451, 324–333. [Google Scholar] [CrossRef] [PubMed]
- Mucignat-Caretta, C.; Caretta, A. Regional variations in the localization of insoluble kinase A regulatory isoforms during rodent brain development. J. Chem. Neuroanat. 2004, 27, 201–212. [Google Scholar] [CrossRef]
- Mucignat-Caretta, C.; Cavaggioni, A.; Redaelli, M.; Malatesta, M.; Zancanaro, C.; Caretta, A. Selective Distribution of Protein Kinase A Regulatory Subunit RIIα in Rodent Gliomas. Neuro Oncol. 2008, 10, 958–967. [Google Scholar] [CrossRef]
- Mucignat-Caretta, C.; Denaro, L.; Redaelli, M.; D’Avella, D.; Caretta, A. Protein kinase A regulatory subunit distribution in medulloblastoma. BMC Cancer 2010, 10, 141. [Google Scholar] [CrossRef]
- Mucignat-Caretta, C.; Denaro, L.; D’Avella, D.; Caretta, A. Protein Kinase A Distribution Differentiates Human Glioblastoma from Brain Tissue. Cancers 2018, 10, 2. [Google Scholar] [CrossRef]
- Caretta, A.; Mucignat-Caretta, C. Protein kinase a in Cancer. Cancers 2011, 3, 913–926. [Google Scholar] [CrossRef]
- Zorzan, M.; Giordan, E.; Redaelli, M.; Caretta, A.; Mucignat-Caretta, C. Molecular targets in glioblastoma. Future Oncol. 2015, 11, 1407–1420. [Google Scholar] [CrossRef]
- Mucignat-Caretta, C. Aggregates of cAMP-dependent kinase RIα characterize a type of cholinergic neurons in the rat brain. Brain Res. Mol. Brain Res. 2000, 80, 233–236. [Google Scholar] [CrossRef]
- Mucignat-Caretta, C.; Caretta, A. Distribution of insoluble cAMP-dependent kinase type RI and RII in the lizard and turtle central nervous system. Brain Res. 2007, 1154, 84–94. [Google Scholar] [CrossRef] [PubMed]
- Mucignat-Caretta, C.; Caretta, A. Aggregates of cAMP-dependent kinase isoforms characterize different areas in the developing central nervous system of the chicken, Gallus gallus. Dev. Neurosci. 2011, 33, 144–158. [Google Scholar] [CrossRef] [PubMed]
- Mucignat-Caretta, C.; Bondi, M.; Caretta, A. Animal models of depression: Olfactory lesions affect amygdala, subventricular zone, and aggression. Neurobiol. Dis. 2004, 16, 386–395. [Google Scholar] [CrossRef] [PubMed]
- Mucignat-Caretta, C.; Caretta, A. Drug-induced Parkinson’s disease modulates protein kinase A and Olfactory Marker Protein in the mouse olfactory bulb. Behav. Brain Funct. 2017, 13, 1. [Google Scholar] [CrossRef] [PubMed]
- Mucignat-Caretta, C.; Caretta, A. Binding of two fluorescent cAMP analogues to type I and II regulatory subunits of cAMP-dependent protein kinases. Biochim. Biophys. Acta 1997, 1357, 81–90. [Google Scholar] [CrossRef]
- Mucignat-Caretta, C.; Conti, V.; Caretta, A. cAMP-dependent protein kinase type RI is found in clusters in the rat detergent-insoluble neuronal fraction. Neurosci. Lett. 1999, 273, 1–4. [Google Scholar] [CrossRef]
- Lee, Y.; Liu, J.; Patel, S.; Cloughesy, T.; Lai, A.; Farooqi, H.; Seligson, D.; Dong, J.; Liau, L.; Becker, D.; et al. Genomic landscape of meningiomas. Brain Pathol. 2010, 20, 751–762. [Google Scholar] [CrossRef]
- Sahm, F.; Schrimpf, D.; Stichel, D.; Jones, D.T.W.; Hielscher, T.; Schefzyk, S.; Okonechnikov, K.; Koelsche, C.; Reuss, D.E.; Capper, D.; et al. DNA methylation-based classification and grading system for meningioma: A multicentre, retrospective analysis. Lancet Oncol. 2017, 18, 682–694. [Google Scholar] [CrossRef]
- Hilton, D.A.; Shivane, A.; Kirk, L.; Bassiri, K.; Enki, D.G.; Hanemann, C.O. Activation of multiple growth factor signalling pathways is frequent in meningiomas. Neuropathology 2016, 36, 250–261. [Google Scholar] [CrossRef]
- Peyre, M.; Salaud, C.; Clermont-Taranchon, E.; Niwa-Kawakita, M.; Goutagny, S.; Mawrin, C.; Giovannini, M.; Kalamarides, M. PDGF activation in PGDS-positive arachnoid cells induces meningioma formation in mice promoting tumor progression in combination with Nf2 and Cdkn2ab loss. Oncotarget. 2015, 6, 32713–32722. [Google Scholar] [CrossRef]
- Mawrin, C.; Sasse, T.; Kirches, E.; Kropf, S.; Schneider, T.; Grimm, C.; Pambor, C.; Vorwerk, C.K.; Firsching, R.; Lendeckel, U.; et al. Different activation of mitogen-activated protein kinase and Akt signaling is associated with aggressive phenotype of human meningiomas. Clin. Cancer Res. 2005, 11, 4074–4082. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wang, W.; Xu, S.; Wang, S.; Tu, Y.; Xiong, Y.; Mei, J.; Wang, C. The role of MAPK signaling pathway in the Her-2-positive meningiomas. Oncol. Rep. 2016, 36, 685–695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, M.D.; Reeder, J.E.; O’Connell, M. p38MAPK activation and DUSP10 expression in meningiomas. J. Clin. Neurosci. 2016, 30, 110–114. [Google Scholar] [CrossRef] [PubMed]
- Surace, E.I.; Lusis, E.; Haipek, C.A.; Gutmann, D.H. Functional significance of S6K overexpression in meningioma progression. Ann. Neurol. 2004, 56, 295–298. [Google Scholar] [CrossRef]
- Clark, V.E.; Erson-Omay, E.Z.; Serin, A.; Yin, J.; Cotney, J.; Ozduman, K.; Avşar, T.; Li, J.; Murray, P.B.; Henegariu, O.; et al. Genomic analysis of non-NF2 meningiomas reveals mutations in TRAF7, KLF4, AKT1, and SMO. Science 2013, 339, 1077–1080. [Google Scholar] [CrossRef]
- Gupta, S.; Bi, W.L.; Dunn, I.F. Medical management of meningioma in the era of precision medicine. Neurosurg. Focus 2018, 44, E3. [Google Scholar] [CrossRef]
- Karsy, M.; Guan, J.; Cohen, A.; Colman, H.; Jensen, R.L. Medical Management of Meningiomas: Current Status, Failed Treatments, and Promising Horizons. Neurosurg. Clin. N. Am. 2016, 27, 249–260. [Google Scholar] [CrossRef]
- Warrington, N.M.; Sun, T.; Rubin, J.B. Targeting brain tumor cAMP: The case for sex-specific therapeutics. Front. Pharmacol. 2015, 6, 153. [Google Scholar] [CrossRef]
- Pećina-Šlaus, N.; Kafka, A.; Lechpammer, M. Molecular Genetics of Intracranial Meningiomas with Emphasis on Canonical Wnt Signalling. Cancers 2016, 8, E67. [Google Scholar] [CrossRef]
- Vasudevan, H.N.; Braunstein, S.E.; Phillips, J.J.; Pekmezci, M.; Tomlin, B.A.; Wu, A.; Reis, G.F.; Magill, S.T.; Zhang, J.; Feng, F.Y.; et al. Comprehensive Molecular Profiling Identifies FOXM1 as a Key Transcription Factor for Meningioma Proliferation. Cell Rep. 2018, 22, 3672–3683. [Google Scholar] [CrossRef] [Green Version]
- Katoh, M.; Katoh, M. Molecular genetics and targeted therapy of WNT-related human diseases. Int. J. Mol. Med. 2017, 40, 587–606. [Google Scholar] [CrossRef] [PubMed]
- Goldhoff, P.; Warrington, N.M.; Limbrick, D.D., Jr.; Hope, A.; Woerner, B.M.; Jackson, E. Targeted inhibition of cyclic AMP phosphodiesterase-4 promotes brain tumor regression. Clin. Cancer Res. 2008, 14, 7717–7725. [Google Scholar] [CrossRef] [PubMed]
- Schulten, H.J.; Hussein, D.; Al-Adwani, F.; Karim, S.; Al-Maghrabi, J.; Al-Sharif, M.; Jamal, A.; Al-Ghamdi, F.; Baeesa, S.S.; Bangash, M.; et al. Microarray Expression Data Identify DCC as a Candidate Gene for Early Meningioma Progression. PLoS ONE 2016, 11, e0153681. [Google Scholar] [CrossRef] [PubMed]
- Warrington, N.M.; Gianino, S.M.; Jackson, E.; Goldhoff, P.; Garbow, J.R.; Piwnica-Worms, D. Cyclic AMP supppression is sufficient to induce gliomagenesis in a mouse model of Neurofibromatosis-1. Cancer Res. 2010, 70, 5717–5727. [Google Scholar] [CrossRef]
- Klutzny, S.; Anurin, A.; Nicke, B.; Regan, J.L.; Lange, M.; Schulze, L.; Parczyk, K.; Steigemann, P. PDE5 inhibition eliminates cancer stem cells via induction of PKA signaling. Cell Death Dis. 2018, 9, 192. [Google Scholar] [CrossRef]
- Parada, C.A.; Osbun, J.; Kaur, S.; Yakkioui, Y.; Shi, M.; Pan, C.; Busald, T.; Karasozen, Y.; Gonzalez-Cuyar, L.F.; Rostomily, R.; et al. Kinome and phosphoproteome of high-grade meningiomas reveal AKAP12 as a central regulator of aggressiveness and its possible role in progression. Sci. Rep. 2018, 8, 2098. [Google Scholar] [CrossRef]
- Dunn, J.; Ferluga, S.; Sharma, V.; Futschik, M.; Hilton, D.A.; Adams, C.L.; Lasonder, E.; Hanemann, C.O. Proteomic analysis discovers the differential expression of novel proteins and phosphoproteins in meningioma including NEK9, HK2 and SET and deregulation of RNA metabolism. EBioMedicine. 2019, 40, 77–91. [Google Scholar] [CrossRef]
- Hüttner, A.; Lei, T.; Fahlbusch, R.; Schrell, W.; Adams, E.F. Relationship between cAMP induced inhibition of human meningioma cell proliferation and autocrine secretion of interleukin-6. Life Sci. 1996, 58, 1323–1329. [Google Scholar] [CrossRef]
- Koper, J.W.; Markstein, R.; Kohler, C.; Kwekkeboom, D.J.; Avezaat, C.J.; Lamberts, S.W.; Reubi, J.C. Somatostatin inhibits the activity of adenylate cyclase in cultured human meningioma cells and stimulates their growth. J. Clin. Endocrinol. Metab. 1992, 74, 543–547. [Google Scholar]
- Büschges, R.; Ichimura, K.; Weber, R.G.; Reifenberger, G.; Collins, V.P. Allelic gain and amplification on the long arm of chromosome 17 in anaplastic meningiomas. Brain Pathol. 2002, 12, 145–153. [Google Scholar] [CrossRef]
- Clark, V.E.; Harmanc, A.S.; Bai, H.; Youngblood, M.W.; Lee, T.I.; Baranoski, J.F.; Ercan-Sencicek, A.G.; Abraham, B.J.; Weintraub, A.S.; Hnisz, D.; et al. Recurrent somatic mutations in POLR2A define a distinct subset of meningiomas. Nat. Genet. 2016, 48, 1253. [Google Scholar] [CrossRef] [PubMed]
- Caretta, A.; Saibil, H. Visualization of cyclic nucleotide binding sites in the vertebrate retina by fluorescence microscopy. J. Cell. Biol. 1989, 108, 1517–1522. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Orozco, C.; Boyer, J.; Leglise, M.; Goodale, J.; Batalov, S.; Hodge, C.L.; Haase, J.; Janes, J.; Huss, J.W. BioGPS: An extensible and customizable portal for querying and organizing gene annotation resources. Genome Biol. 2009, 10, R130. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; MacLeod, I.; Su, A.I. BioGPS and MyGene.info: Organizing online, gene-entric information. Nucl. Acids Res. 2013, 41, D561–D565. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Jin, X.; Tsueng, G.; Afrasiabi, C.; Su, A.I. BioGPS: Building your own mash-up of gene annotations and expression profiles. Nucl. Acids Res. 2016, 44, D313–D316. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CASE N° | cAMP equilibrium binding | PKA RI IF | PKA RII IF | PKA CATALYTIC IF |
|---|---|---|---|---|
| 1 | + | + Mostly on vessels | +++ | +++ |
| 2 | +/− Only in some zones | + | +++ Only in some zones | +++ Only in some zones |
| 3 | +++ | + | +++ | +++ |
| 4 | ++ | + | +++ | +++ |
| 5 | ++ | ++ | ++ | ++ |
| 6 | + | + | ++ | ++ |
| 7 | + | NA | NA | ++ |
| 8 | ++ | + | +++ | +++ |
| 9 | - | + | ++ Only in some zones | ++ Only in some zones |
| 10 | + | + Only in some zones | +++ | +++ |
| 11 | +/− | - | +++ | +++ |
| 12 | +++ | + | +++ | +++ |
| 13 | +++ | - | +++ | +++ |
| PRKACA | PRKAR1A | PRKAR1B | PRKAR2A | PRKAR2B | |
|---|---|---|---|---|---|
| NF2 | 0.2994 ** | 0.0679 | −0.0406 | 0.0965 | 0.1524 |
| CDC2 | −0.2973 ** | 0.0518 | −0.0246 | 0.1128 | −0.3763 *** |
| MLF1IP | −0.2908 ** | 0.0363 | −0.0844 | 0.2692 * | −0.3098 ** |
| CKS2 | −0.3188 ** | −0.1402 | −0.1074 | 0.0945 | −0.3754 *** |
| PRC1 | −0.3622 *** | −0.0497 | −0.0640 | 0.1630 | −0.3563 *** |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Caretta, A.; Denaro, L.; D’Avella, D.; Mucignat-Caretta, C. Protein Kinase A Distribution in Meningioma. Cancers 2019, 11, 1686. https://doi.org/10.3390/cancers11111686
Caretta A, Denaro L, D’Avella D, Mucignat-Caretta C. Protein Kinase A Distribution in Meningioma. Cancers. 2019; 11(11):1686. https://doi.org/10.3390/cancers11111686
Chicago/Turabian StyleCaretta, Antonio, Luca Denaro, Domenico D’Avella, and Carla Mucignat-Caretta. 2019. "Protein Kinase A Distribution in Meningioma" Cancers 11, no. 11: 1686. https://doi.org/10.3390/cancers11111686
APA StyleCaretta, A., Denaro, L., D’Avella, D., & Mucignat-Caretta, C. (2019). Protein Kinase A Distribution in Meningioma. Cancers, 11(11), 1686. https://doi.org/10.3390/cancers11111686