Understanding and Targeting Apoptotic Pathways in Ovarian Cancer



Abstract
1. Introduction
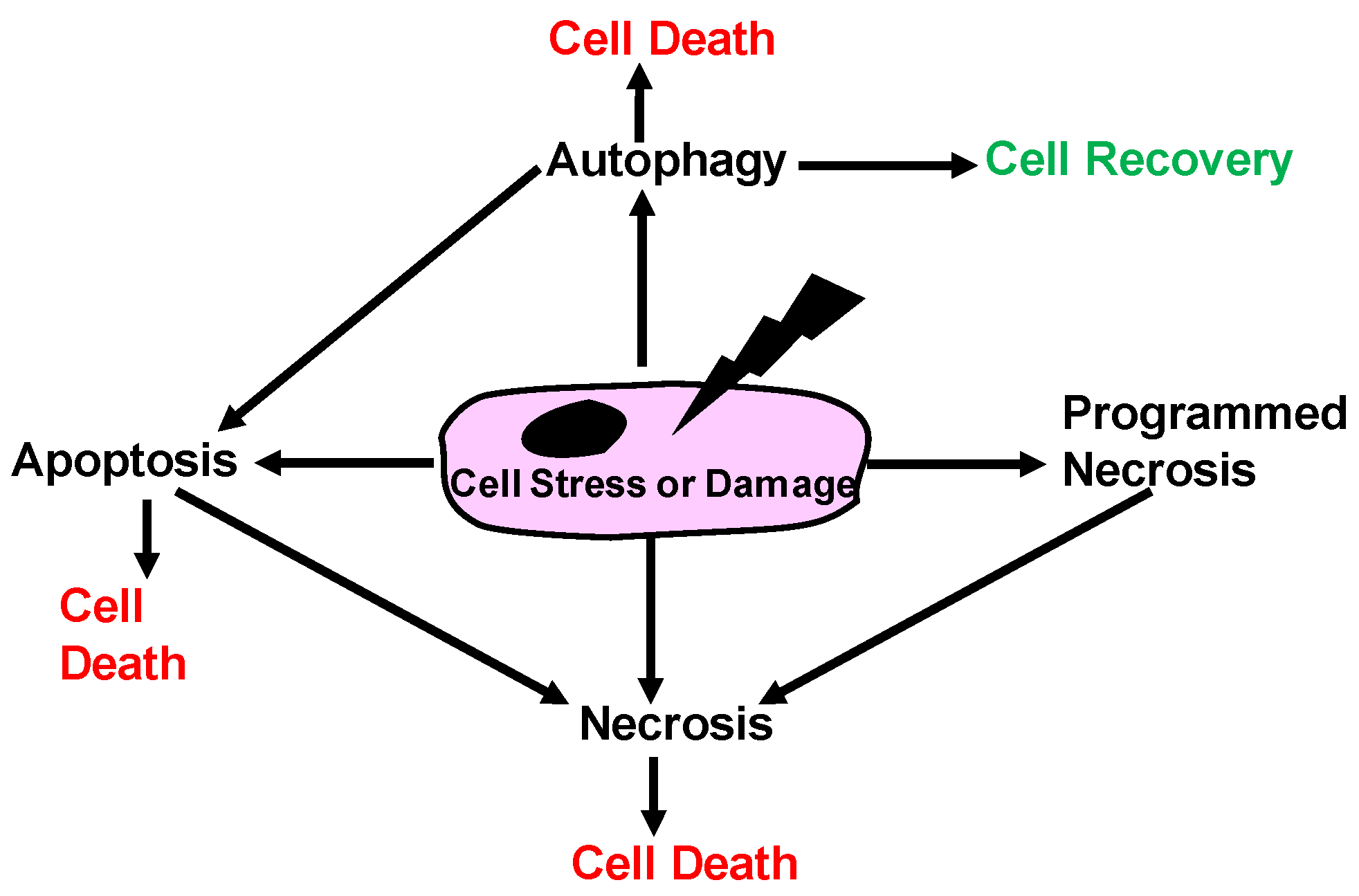
1.1. Historical View of Cell Death
1.2. Apoptosis
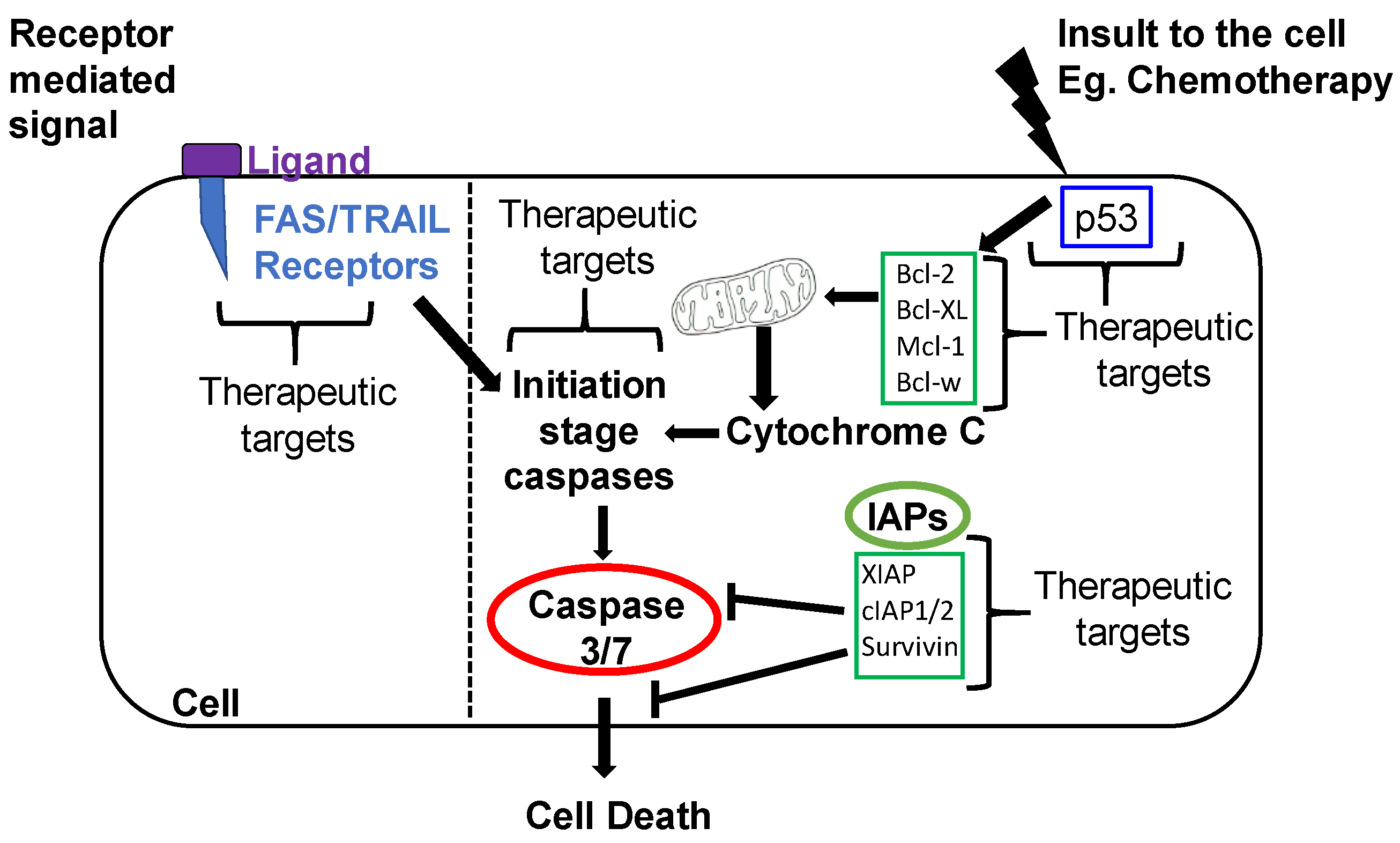
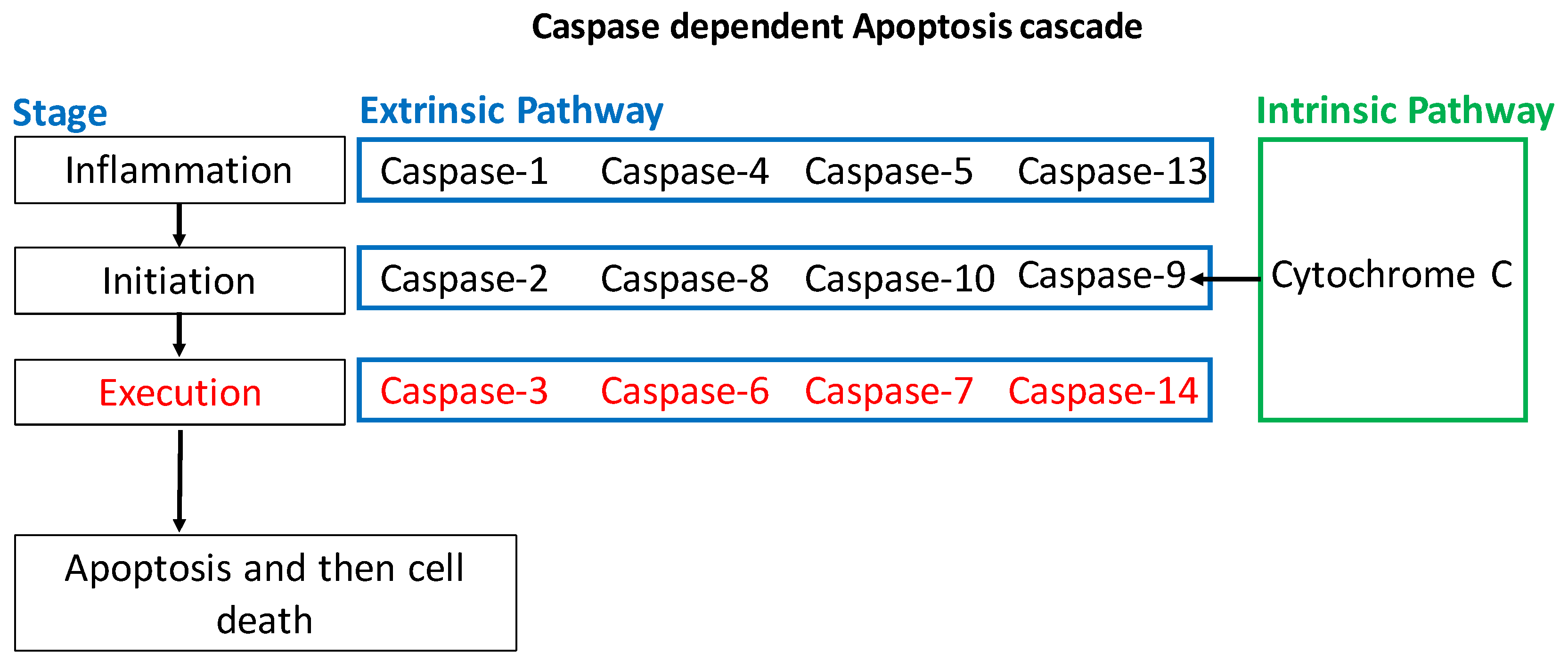
1.2.1. Intrinsic Apoptotic Pathway
1.2.2. Extrinsic Apoptotic Pathway
2. Apoptotic Mechanisms Altered in OvCa
2.1. Activators of Apoptosis in OvCa
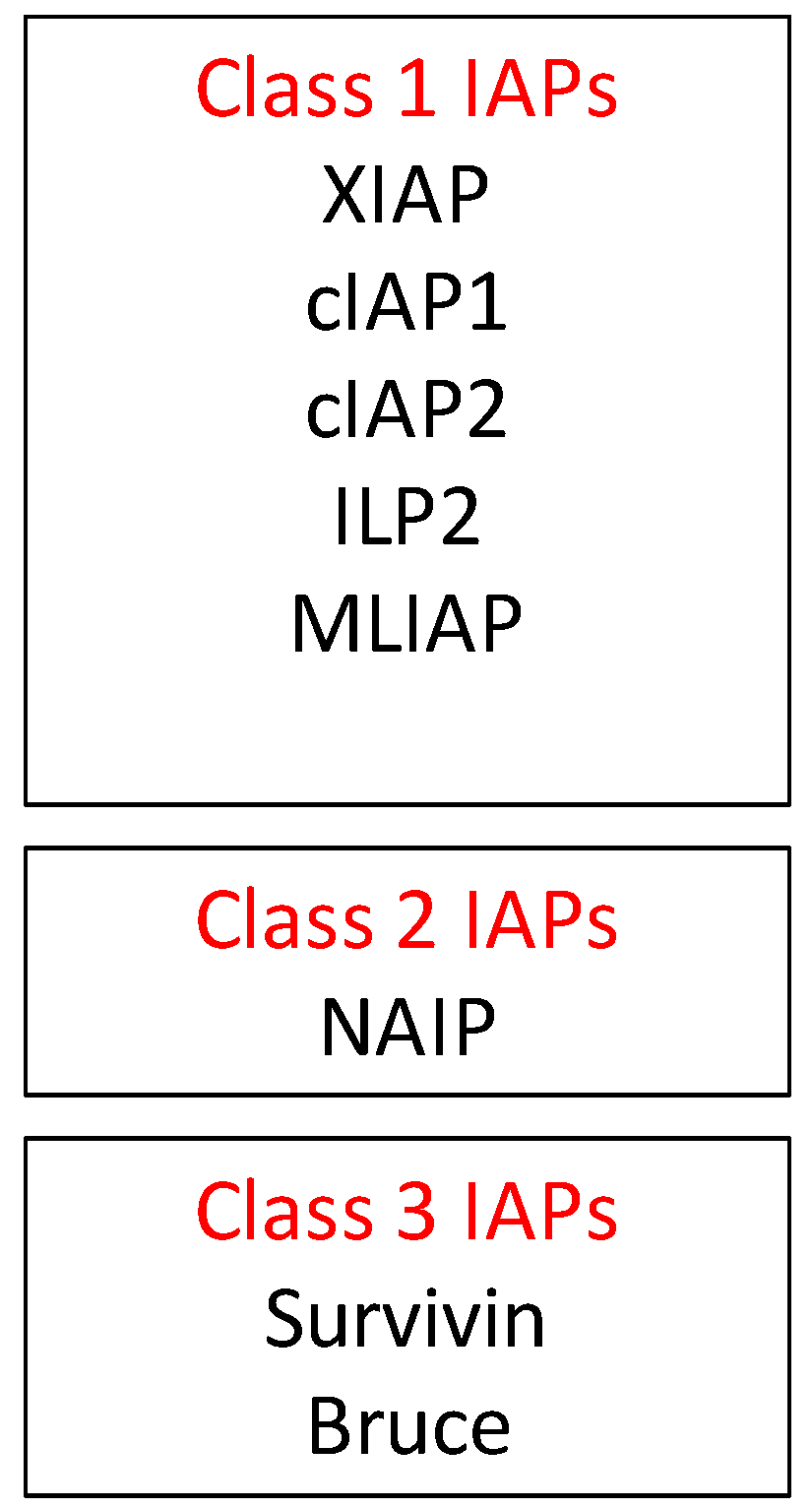
2.2. Inhibitors of Apoptosis Proteins (IAP) in OvCa
2.3. Ubiquitination Mediated Apoptosis in OvCa
3. Regulation of OvCa Apoptosis via Glycosylation and Glycan-Related Proteins
3.1. O-Linked Glycosylation
3.2. N-Linked Glycosylation
4. Apoptosis Regulation by Galectins
5. Epigenetic Modifications and Their Role in OvCa Apoptosis
5.1. miRNAs in OvCa Apoptosis
5.2. DNA Methylation
5.3. Histone Acetylation
6. Clinical Use of Drugs Targeting Apoptosis in Ovarian Cancer
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AMPK | 5′ AMP-activated protein kinase |
| CA125 | Cancer antigen 125, mucin 16 |
| CA-MSCs | Cancer-associated mesenchymal cells |
| CDT2 | Cell division cycle protein 2 |
| CRL | Cullin-RING ubiquitin ligases |
| CRL4 | Cullin-really interesting new gene ubiquitin ligase 4 |
| DcR3 | Decoy Receptor 3 |
| DDB2 | Damage Specific DNA Binding Protein 2 |
| DISC | Death-inducing signal complex |
| DNMT | DNA methyltransferase |
| EF24 | 3,5-bis(2-flurobenzylidene) piperidin-4-one |
| FADD | Fas-associated death domain protein |
| FLIP | IL-1 β-converting enzyme (FLICE)-like inhibitory proteins (FLIP) |
| FasL | Fas ligand |
| FDA | Food and drug administration |
| FLICE | Fas-associated death domain-like interleukin-1β-converting enzyme |
| FLIP | FLICE-like inhibitory protein |
| FITC | Fluorescein isothiocyanate |
| GLUT1 | Glucose transporter 1 |
| hOSE | Human ovarian surface epithelium |
| IAP | Inhibitor of apoptosis |
| KGDH | α-Ketoglutarate dehydrogenase |
| MAPK | Mitogen-activated protein kinase |
| miRNA | Small non-coding RNA |
| MTT | 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide |
| mTOR | Mammalian target of rapamycin |
| NAD | Nicotinamide adenine dinucleotide |
| NAE | NEDD8-activating Enzyme E1 |
| NF-κB | Nuclear factor κB |
| O-GlcNAc | O-linked N-acetylglucosamine |
| OvCa | Ovarian cancer |
| PARP | Poly-ADP ribose polymerase |
| PDK1 | Pyruvate dehydrogenase kinase 1 |
| PI | Propidium iodide |
| PTEN | Phosphatase and tensin homolog |
| ROC1 | Regulator of Cullins-1 |
| ROS | Reactive oxygen species |
| SCF | CRL/Skp Cullin F-box containing complex |
| scFvs | Single chain variable fragments |
| shRNA | Short hairpin RNA |
| SM | Smac-mimetics |
| SNA | Sambucus nigra agglutinin |
| ST6Gal1 | Sialyltransferase β-galactosamide α-2, 6-sialyltransterase 1 |
| TCA | Tricarboxylic acid |
| TLR4 | Toll-Like Receptor 4 |
| TNF | Tumor necrosis factor |
| TRAIL | TNF-related apoptosis-inducing ligand |
| TUNEL | Terminal deoxynucleotidyl transferase dUTP nick end labeling |
| UPS | Ubiquitin-proteasome-system |
| UPS14 | Ubiquitin-specific protease 14 |
| VPRBP | Viral protein R binding protein |
References
- Torre, L.A.; Trabert, B.; DeSantis, C.E.; Miller, K.D.; Samimi, G.; Runowicz, C.D.; Gaudet, M.M.; Jemal, A.; Siegel, R.L. Ovarian cancer statistics, 2018. CA Cancer J. Clin. 2018, 68, 284–296. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef] [PubMed]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef]
- Hayflick, L.; Moorhead, P.S. The serial cultivation of human diploid cell strains. Exp. Cell Res. 1961, 25, 585–621. [Google Scholar] [CrossRef]
- Klionsky, D.J. Autophagy revisited: A conversation with Christian de Duve. Autophagy 2008, 4, 740–743. [Google Scholar] [CrossRef]
- Lockshin, R.A.; Williams, C.M. Programmed Cell Death--I. Cytology of Degeneration in the Intersegmental Muscles of the Pernyi Silkmoth. J. Insect Physiol. 1965, 11, 123–133. [Google Scholar] [CrossRef]
- Kerr, J.F.; Wyllie, A.H.; Currie, A.R. Apoptosis: A basic biological phenomenon with wide-ranging implications in tissue kinetics. Br. J. Cancer 1972, 26, 239–257. [Google Scholar] [CrossRef]
- Dhuriya, Y.K.; Sharma, D. Necroptosis: A regulated inflammatory mode of cell death. J. Neuroinflamm. 2018, 15, 199. [Google Scholar] [CrossRef]
- Alberts, B. Molecular Biology of the Cell, 4th ed.; Garland Science: New York, NY, USA, 2002; 1548p. [Google Scholar]
- Cory, S.; Adams, J.M. The Bcl2 family: Regulators of the cellular life-or-death switch. Nat. Rev. Cancer 2002, 2, 647–656. [Google Scholar] [CrossRef]
- Maddika, S.; Ande, S.R.; Panigrahi, S.; Paranjothy, T.; Weglarczyk, K.; Zuse, A.; Eshraghi, M.; Manda, K.D.; Wiechec, E.; Los, M. Cell survival, cell death and cell cycle pathways are interconnected: Implications for cancer therapy. Drug Resist. Updat. 2007, 10, 13–29. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Vogelstein, B.; Lane, D.; Levine, A.J. Surfing the p53 network. Nature 2000, 408, 307–310. [Google Scholar] [CrossRef] [PubMed]
- Schimmer, A.D. Inhibitor of apoptosis proteins: Translating basic knowledge into clinical practice. Cancer Res. 2004, 64, 7183–7190. [Google Scholar] [CrossRef] [PubMed]
- Salvesen, G.S.; Duckett, C.S. IAP proteins: Blocking the road to death’s door. Nat. Rev. Mol. Cell Biol. 2002, 3, 401–410. [Google Scholar] [CrossRef]
- Yang, Y.; Li, S.; Sun, Y.; Zhang, D.; Zhao, Z.; Liu, L. Reversing platinum resistance in ovarian cancer multicellular spheroids by targeting Bcl-2. Onco Targets Ther. 2019, 12, 897–906. [Google Scholar] [CrossRef]
- Yu, Y.; Xu, L.; Qi, L.; Wang, C.; Xu, N.; Liu, S.; Li, S.; Tian, H.; Liu, W.; Xu, Y.; et al. ABT737 induces mitochondrial pathway apoptosis and mitophagy by regulating DRP1-dependent mitochondrial fission in human ovarian cancer cells. Biomed. Pharmacother. 2017, 96, 22–29. [Google Scholar] [CrossRef]
- Hassan, H.A.; Salem, M.L.; Gouida, M.S.; El-Azab, K.M. Comparative expression of caspases and annexin V in benign and malignant ovarian tumors. J. Cancer Res. Ther. 2018, 14, 1042–1048. [Google Scholar] [CrossRef]
- Yan, X.Y.; Zhong, X.R.; Yu, S.H.; Zhang, L.C.; Liu, Y.N.; Zhang, Y.; Sun, L.K.; Su, J. p62 aggregates mediated Caspase 8 activation is responsible for progression of ovarian cancer. J. Cell Mol. Med. 2019, 23, 4030–4042. [Google Scholar] [CrossRef]
- Kim, M.; Hernandez, L.; Annunziata, C.M. Caspase 8 expression may determine the survival of women with ovarian cancer. Cell Death Dis. 2016, 7, e2045. [Google Scholar] [CrossRef]
- Deveraux, Q.L.; Takahashi, R.; Salvesen, G.S.; Reed, J.C. X-linked IAP is a direct inhibitor of cell-death proteases. Nature 1997, 388, 300–304. [Google Scholar] [CrossRef]
- Li, J.; Feng, Q.; Kim, J.M.; Schneiderman, D.; Liston, P.; Li, M.; Vanderhyden, B.; Faught, W.; Fung, M.F.; Senterman, M.; et al. Human ovarian cancer and cisplatin resistance: Possible role of inhibitor of apoptosis proteins. Endocrinology 2001, 142, 370–380. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Mansouri, A.; Zhang, Q.; Ridgway, L.D.; Tian, L.; Claret, F.X. Cisplatin resistance in an ovarian carcinoma is associated with a defect in programmed cell death control through XIAP regulation. Oncol. Res. 2003, 13, 399–404. [Google Scholar] [CrossRef]
- Sasaki, H.; Sheng, Y.; Kotsuji, F.; Tsang, B.K. Down-regulation of X-linked inhibitor of apoptosis protein induces apoptosis in chemoresistant human ovarian cancer cells. Cancer Res. 2000, 60, 5659–5666. [Google Scholar] [PubMed]
- Korch, C.; Spillman, M.A.; Jackson, T.A.; Jacobsen, B.M.; Murphy, S.K.; Lessey, B.A.; Jordan, V.C.; Bradford, A.P. DNA profiling analysis of endometrial and ovarian cell lines reveals misidentification, redundancy and contamination. Gynecol. Oncol. 2012, 127, 241–248. [Google Scholar] [CrossRef] [PubMed]
- Castells, M.; Milhas, D.; Gandy, C.; Thibault, B.; Rafii, A.; Delord, J.P.; Couderc, B. Microenvironment mesenchymal cells protect ovarian cancer cell lines from apoptosis by inhibiting XIAP inactivation. Cell Death Dis. 2013, 4, e887. [Google Scholar] [CrossRef]
- Shaw, T.J.; Lacasse, E.C.; Durkin, J.P.; Vanderhyden, B.C. Downregulation of XIAP expression in ovarian cancer cells induces cell death in vitro and in vivo. Int. J. Cancer 2008, 122, 1430–1434. [Google Scholar] [CrossRef]
- Zaffaroni, N.; Pennati, M.; Colella, G.; Perego, P.; Supino, R.; Gatti, L.; Pilotti, S.; Zunino, F.; Daidone, M.G. Expression of the anti-apoptotic gene survivin correlates with taxol resistance in human ovarian cancer. Cell Mol. Life Sci. 2002, 59, 1406–1412. [Google Scholar] [CrossRef]
- Chen, L.; Liang, L.; Yan, X.; Liu, N.; Gong, L.; Pan, S.; Lin, F.; Zhang, Q.; Zhao, H.; Zheng, F. Survivin status affects prognosis and chemosensitivity in epithelial ovarian cancer. Int. J. Gynecol. Cancer 2013, 23, 256–263. [Google Scholar] [CrossRef]
- Jiang, L.; Luo, R.Y.; Yang, J.; Cheng, Y.X. Knockdown of survivin contributes to antitumor activity in cisplatin-resistant ovarian cancer cells. Mol. Med. Rep. 2013, 7, 425–430. [Google Scholar] [CrossRef]
- Zheng, F.; Ruan, F.; Xie, X.K.; Liu, S.Y. Apoptosis of drug-resistant human ovarian carcinoma cell line COC1/DDP induced by survivin antisense oligonucleotides. Chin. Med. J. (Engl.) 2006, 119, 1572–1575. [Google Scholar] [CrossRef]
- Abedini, M.R.; Qiu, Q.; Yan, X.; Tsang, B.K. Possible role of FLICE-like inhibitory protein (FLIP) in chemoresistant ovarian cancer cells in vitro. Oncogene 2004, 23, 6997–7004. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bazzaro, M.; Lee, M.K.; Zoso, A.; Stirling, W.L.; Santillan, A.; Shih Ie, M.; Roden, R.B. Ubiquitin-proteasome system stress sensitizes ovarian cancer to proteasome inhibitor-induced apoptosis. Cancer Res. 2006, 66, 3754–3763. [Google Scholar] [CrossRef] [PubMed]
- Miranda, M.; Sorkin, A. Regulation of receptors and transporters by ubiquitination: New insights into surprisingly similar mechanisms. Mol. Interv. 2007, 7, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Mitra, S. Ovarian. Cancer from Pathogenesis to Treatment, 2018th ed.; Devaja, O., Ed.; IntechOpen: London, UK, 2018; pp. 135–154. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, J.; Zhong, J.; Deng, Y.; Xi, Q.; He, S.; Yang, S.; Jiang, L.; Huang, M.; Tang, C.; et al. Ubiquitin-specific protease 14 (USP14) regulates cellular proliferation and apoptosis in epithelial ovarian cancer. Med. Oncol. 2015, 32, 379. [Google Scholar] [CrossRef] [PubMed]
- Pan, W.W.; Zhou, J.J.; Yu, C.; Xu, Y.; Guo, L.J.; Zhang, H.Y.; Zhou, D.; Song, F.Z.; Fan, H.Y. Ubiquitin E3 ligase CRL4(CDT2/DCAF2) as a potential chemotherapeutic target for ovarian surface epithelial cancer. J. Biol. Chem. 2013, 288, 29680–29691. [Google Scholar] [CrossRef] [PubMed]
- MacKay, C.; Carroll, E.; Ibrahim, A.F.M.; Garg, A.; Inman, G.J.; Hay, R.T.; Alpi, A.F. E3 ubiquitin ligase HOIP attenuates apoptotic cell death induced by cisplatin. Cancer Res. 2014, 74, 2246–2257. [Google Scholar] [CrossRef]
- Abedini, M.R.; Muller, E.J.; Brun, J.; Bergeron, R.; Gray, D.A.; Tsang, B.K. Cisplatin induces p53-dependent FLICE-like inhibitory protein ubiquitination in ovarian cancer cells. Cancer Res. 2008, 68, 4511–4517. [Google Scholar] [CrossRef]
- Crowley, L.C.; Marfell, B.J.; Waterhouse, N.J. Analyzing Cell Death by Nuclear Staining with Hoechst 33342. Cold Spring Harb. Protoc. 2016, 2016. [Google Scholar] [CrossRef]
- Selvendiran, K.; Tong, L.; Vishwanath, S.; Bratasz, A.; Trigg, N.J.; Kutala, V.K.; Hideg, K.; Kuppusamy, P. EF24 induces G2/M arrest and apoptosis in cisplatin-resistant human ovarian cancer cells by increasing PTEN expression. J. Biol. Chem. 2007, 282, 28609–28618. [Google Scholar] [CrossRef]
- Yu, H.; Su, J.; Xu, Y.; Kang, J.; Li, H.; Zhang, L.; Yi, H.; Xiang, X.; Liu, F.; Sun, L. p62/SQSTM1 involved in cisplatin resistance in human ovarian cancer cells by clearing ubiquitinated proteins. Eur. J. Cancer 2011, 47, 1585–1594. [Google Scholar] [CrossRef]
- Apweiler, R.; Hermjakob, H.; Sharon, N. On the frequency of protein glycosylation, as deduced from analysis of the SWISS-PROT database. Biochim. Biophys. Acta 1999, 1473, 4–8. [Google Scholar] [CrossRef]
- Lichtenstein, R.G.; Rabinovich, G.A. Glycobiology of cell death: When glycans and lectins govern cell fate. Cell Death Differ. 2013, 20, 976–986. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, I.G.; Pucci, M.; Venturi, G.; Malagolini, N.; Chiricolo, M.; Dall’Olio, F. Glycosylation as a Main Regulator of Growth and Death Factor Receptors Signaling. Int. J. Mol. Sci. 2018, 19, 580. [Google Scholar] [CrossRef]
- Lau, K.S.; Dennis, J.W. N-Glycans in cancer progression. Glycobiology 2008, 18, 750–760. [Google Scholar] [CrossRef] [PubMed]
- Christiansen, M.N.; Chik, J.; Lee, L.; Anugraham, M.; Abrahams, J.L.; Packer, N.H. Cell surface protein glycosylation in cancer. Proteomics 2014, 14, 525–546. [Google Scholar] [CrossRef]
- Hernandez, J.D.; Baum, L.G. Ah, sweet mystery of death! Galectins and control of cell fate. Glycobiology 2002, 12, 127R–136R. [Google Scholar] [CrossRef]
- Felder, M.; Kapur, A.; Gonzalez-Bosquet, J.; Horibata, S.; Heintz, J.; Albrecht, R.; Fass, L.; Kaur, J.; Hu, K.; Shojaei, H.; et al. MUC16 (CA125): Tumor biomarker to cancer therapy, a work in progress. Mol. Cancer 2014, 13, 129. [Google Scholar] [CrossRef]
- Olivier, R.I.; Lubsen-Brandsma, M.A.; Verhoef, S.; van Beurden, M. CA125 and transvaginal ultrasound monitoring in high-risk women cannot prevent the diagnosis of advanced ovarian cancer. Gynecol. Oncol. 2006, 100, 20–26. [Google Scholar] [CrossRef]
- Olivier, R.I.; van Beurden, M.; van t Veer, L.J. The role of gene expression profiling in the clinical management of ovarian cancer. Eur. J. Cancer 2006, 42, 2930–2938. [Google Scholar] [CrossRef]
- Reinartz, S.; Failer, S.; Schuell, T.; Wagner, U. CA125 (MUC16) gene silencing suppresses growth properties of ovarian and breast cancer cells. Eur. J. Cancer 2012, 48, 1558–1569. [Google Scholar] [CrossRef]
- Boivin, M.; Lane, D.; Piche, A.; Rancourt, C. CA125 (MUC16) tumor antigen selectively modulates the sensitivity of ovarian cancer cells to genotoxic drug-induced apoptosis. Gynecol. Oncol. 2009, 115, 407–413. [Google Scholar] [CrossRef] [PubMed]
- de Queiroz, R.M.; Madan, R.; Chien, J.; Dias, W.B.; Slawson, C. Changes in O-Linked N-Acetylglucosamine (O-GlcNAc) Homeostasis Activate the p53 Pathway in Ovarian Cancer Cells. J. Biol. Chem. 2016, 291, 18897–18914. [Google Scholar] [CrossRef] [PubMed]
- Connor, J.P.; Felder, M.; Kapur, A.; Onujiogu, N. DcR3 binds to ovarian cancer via heparan sulfate proteoglycans and modulates tumor cells response to platinum with corresponding alteration in the expression of BRCA1. BMC Cancer 2012, 12, 176. [Google Scholar] [CrossRef] [PubMed]
- Wichert, B.; Milde-Langosch, K.; Galatenko, V.; Schmalfeldt, B.; Oliveira-Ferrer, L. Prognostic role of the sialyltransferase ST6GAL1 in ovarian cancer. Glycobiology 2018, 28, 898–903. [Google Scholar] [CrossRef]
- Schultz, M.J.; Swindall, A.F.; Wright, J.W.; Sztul, E.S.; Landen, C.N.; Bellis, S.L. ST6Gal-I sialyltransferase confers cisplatin resistance in ovarian tumor cells. J. Ovarian. Res. 2013, 6, 25. [Google Scholar] [CrossRef]
- Chowdhury, S.R.; Ray, U.; Chatterjee, B.P.; Roy, S.S. Targeted apoptosis in ovarian cancer cells through mitochondrial dysfunction in response to Sambucus nigra agglutinin. Cell Death Dis. 2017, 8, e2762. [Google Scholar] [CrossRef]
- Gwak, H.; Haegeman, G.; Tsang, B.K.; Song, Y.S. Cancer-specific interruption of glucose metabolism by resveratrol is mediated through inhibition of Akt/GLUT1 axis in ovarian cancer cells. Mol. Carcinog 2015, 54, 1529–1540. [Google Scholar] [CrossRef]
- Gwak, H.; Kim, S.; Dhanasekaran, D.N.; Song, Y.S. Resveratrol triggers ER stress-mediated apoptosis by disrupting N-linked glycosylation of proteins in ovarian cancer cells. Cancer Lett. 2016, 371, 347–353. [Google Scholar] [CrossRef]
- Chetry, M.; Thapa, S.; Hu, X.; Song, Y.; Zhang, J.; Zhu, H.; Zhu, X. The Role of Galectins in Tumor Progression, Treatment and Prognosis of Gynecological Cancers. J. Cancer 2018, 9, 4742–4755. [Google Scholar] [CrossRef]
- Zhang, P.; Zhang, P.; Shi, B.; Zhou, M.; Jiang, H.; Zhang, H.; Pan, X.; Gao, H.; Sun, H.; Li, Z. Galectin-1 overexpression promotes progression and chemoresistance to cisplatin in epithelial ovarian cancer. Cell Death Dis. 2014, 5, e991. [Google Scholar] [CrossRef]
- Matarrese, P.; Tinari, A.; Mormone, E.; Bianco, G.A.; Toscano, M.A.; Ascione, B.; Rabinovich, G.A.; Malorni, W. Galectin-1 sensitizes resting human T lymphocytes to Fas (CD95)-mediated cell death via mitochondrial hyperpolarization, budding, and fission. J. Biol. Chem. 2005, 280, 6969–6985. [Google Scholar] [CrossRef] [PubMed]
- Wells, V.; Mallucci, L. Phosphoinositide 3-kinase targeting by the beta galactoside binding protein cytokine negates akt gene expression and leads aggressive breast cancer cells to apoptotic death. Breast Cancer Res. 2009, 11, R2. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, J.T.; Evans, D.P.; Galvan, M.; Pace, K.E.; Leitenberg, D.; Bui, T.N.; Baum, L.G. CD45 modulates galectin-1-induced T cell death: Regulation by expression of core 2 O-glycans. J. Immunol. 2001, 167, 5697–5707. [Google Scholar] [CrossRef] [PubMed]
- Mirandola, L.; Yu, Y.; Cannon, M.J.; Jenkins, M.R.; Rahman, R.L.; Nguyen, D.D.; Grizzi, F.; Cobos, E.; Figueroa, J.A.; Chiriva-Internati, M. Galectin-3 inhibition suppresses drug resistance, motility, invasion and angiogenic potential in ovarian cancer. Gynecol. Oncol. 2014, 135, 573–579. [Google Scholar] [CrossRef]
- Wang, D.; You, D.; Li, L. Galectin-3 regulates chemotherapy sensitivity in epithelial ovarian carcinoma via regulating mitochondrial function. J. Toxicol. Sci. 2019, 44, 47–56. [Google Scholar] [CrossRef]
- Hossein, G.; Keshavarz, M.; Ahmadi, S.; Naderi, N. Synergistic effects of PectaSol-C modified citrus pectin an inhibitor of Galectin-3 and paclitaxel on apoptosis of human SKOV-3 ovarian cancer cells. Asian Pac. J. Cancer Prev. 2013, 14, 7561–7568. [Google Scholar] [CrossRef]
- Lu, H.; Liu, Y.; Wang, D.; Wang, L.; Zhou, H.; Xu, G.; Xie, L.; Wu, M.; Lin, Z.; Yu, Y.; et al. Galectin-3 regulates metastatic capabilities and chemotherapy sensitivity in epithelial ovarian carcinoma via NF-kappaB pathway. Tumour. Biol. 2016, 37, 11469–11477. [Google Scholar] [CrossRef]
- Cai, G.; Ma, X.; Chen, B.; Huang, Y.; Liu, S.; Yang, H.; Zou, W. Galectin-3 induces ovarian cancer cell survival and chemoresistance via TLR4 signaling activation. Tumour. Biol. 2016, 37, 11883–11891. [Google Scholar] [CrossRef]
- Rajput, S.; Volk-Draper, L.D.; Ran, S. TLR4 is a novel determinant of the response to paclitaxel in breast cancer. Mol. Cancer Ther. 2013, 12, 1676–1687. [Google Scholar] [CrossRef]
- Byrd-Leifer, C.A.; Block, E.F.; Takeda, K.; Akira, S.; Ding, A. The role of MyD88 and TLR4 in the LPS-mimetic activity of Taxol. Eur. J. Immunol 2001, 31, 2448–2457. [Google Scholar] [CrossRef]
- Villeneuve, C.; Baricault, L.; Canelle, L.; Barboule, N.; Racca, C.; Monsarrat, B.; Magnaldo, T.; Larminat, F. Mitochondrial proteomic approach reveals galectin-7 as a novel BCL-2 binding protein in human cells. Mol. Biol. Cell 2011, 22, 999–1013. [Google Scholar] [CrossRef] [PubMed]
- Brustmann, H. Epidermal growth factor receptor expression in serous ovarian carcinoma: An immunohistochemical study with galectin-3 and cyclin D1 and outcome. Int. J. Gynecol. Pathol. 2008, 27, 380–389. [Google Scholar] [CrossRef] [PubMed]
- Oishi, T.; Itamochi, H.; Kigawa, J.; Kanamori, Y.; Shimada, M.; Takahashi, M.; Shimogai, R.; Kawaguchi, W.; Sato, S.; Terakawa, N. Galectin-3 may contribute to Cisplatin resistance in clear cell carcinoma of the ovary. Int. J. Gynecol Cancer 2007, 17, 1040–1046. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Jeon, H.K.; Lee, J.K.; Sung, C.O.; Do, I.G.; Choi, C.H.; Kim, T.J.; Kim, B.G.; Bae, D.S.; Lee, J.W. Clinical significance of galectin-7 in epithelial ovarian cancer. Anticancer Res. 2013, 33, 1555–1561. [Google Scholar]
- Jafari, S.M.; Nazri, A.; Shabani, M.; Balajam, N.Z.; Aghaei, M. Galectin-9 induces apoptosis in OVCAR-3 ovarian cancer cell through mitochondrial pathway. Res. Pharm Sci. 2018, 13, 557–565. [Google Scholar] [CrossRef]
- Kinose, Y.; Sawada, K.; Nakamura, K.; Kimura, T. The role of microRNAs in ovarian cancer. Biomed. Res. Int. 2014, 2014, 249393. [Google Scholar] [CrossRef]
- Wang, Y.; Lee, C.G. MicroRNA and cancer--focus on apoptosis. J. Cell Mol. Med. 2009, 13, 12–23. [Google Scholar] [CrossRef]
- Zhang, H.; Zuo, Z.; Lu, X.; Wang, L.; Wang, H.; Zhu, Z. MiR-25 regulates apoptosis by targeting Bim in human ovarian cancer. Oncol. Rep. 2012, 27, 594–598. [Google Scholar] [CrossRef]
- Creighton, C.J.; Fountain, M.D.; Yu, Z.; Nagaraja, A.K.; Zhu, H.; Khan, M.; Olokpa, E.; Zariff, A.; Gunaratne, P.H.; Matzuk, M.M.; et al. Molecular profiling uncovers a p53-associated role for microRNA-31 in inhibiting the proliferation of serous ovarian carcinomas and other cancers. Cancer Res. 2010, 70, 1906–1915. [Google Scholar] [CrossRef]
- Chan, J.K.; Blansit, K.; Kiet, T.; Sherman, A.; Wong, G.; Earle, C.; Bourguignon, L.Y. The inhibition of miR-21 promotes apoptosis and chemosensitivity in ovarian cancer. Gynecol. Oncol. 2014, 132, 739–744. [Google Scholar] [CrossRef]
- Sarkozy, M.; Kahan, Z.; Csont, T. A myriad of roles of miR-25 in health and disease. Oncotarget 2018, 9, 21580–21612. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.; Ma, P.; Wu, D.; Shu, Y.; Gao, W. Functions and mechanisms of microRNA-31 in human cancers. Biomed. Pharmacother. 2018, 108, 1162–1169. [Google Scholar] [CrossRef] [PubMed]
- Mitamura, T.; Watari, H.; Wang, L.; Kanno, H.; Hassan, M.K.; Miyazaki, M.; Katoh, Y.; Kimura, T.; Tanino, M.; Nishihara, H.; et al. Downregulation of miRNA-31 induces taxane resistance in ovarian cancer cells through increase of receptor tyrosine kinase MET. Oncogenesis 2013, 2, e40. [Google Scholar] [CrossRef] [PubMed]
- Lou, Y.; Cui, Z.; Wang, F.; Yang, X.; Qian, J. miR-21 down-regulation promotes apoptosis and inhibits invasion and migration abilities of OVCAR3 cells. Clin. Invest. Med. 2011, 34, E281. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.Y.; Zhang, Y.Y.; Zhu, B.L.; Feng, F.Z.; Yan, H.; Zhang, H.Y.; Zhou, B. miR-21 regulates the proliferation and apoptosis of ovarian cancer cells through PTEN/PI3K/AKT. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 4149–4155. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Chen, W.; Jin, Y.; Xue, R.; Su, J.; Mu, Z.; Li, J.; Jiang, S. miR-142-5p enhances cisplatin-induced apoptosis in ovarian cancer cells by targeting multiple anti-apoptotic genes. Biochem. Pharmacol. 2019, 161, 98–112. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, R.; Nicoloso, M.; Arvizo, R.; Wang, E.; Cortez, A.; Rossi, S.; Calin, G.A.; Mukherjee, P. MiR-15a and MiR-16 control Bmi-1 expression in ovarian cancer. Cancer Res. 2009, 69, 9090–9095. [Google Scholar] [CrossRef]
- Cimmino, A.; Calin, G.A.; Fabbri, M.; Iorio, M.V.; Ferracin, M.; Shimizu, M.; Wojcik, S.E.; Aqeilan, R.I.; Zupo, S.; Dono, M.; et al. miR-15 and miR-16 induce apoptosis by targeting BCL2. Proc. Natl. Acad. Sci. USA 2005, 102, 13944–13949. [Google Scholar] [CrossRef]
- Liu, P.; Qi, X.; Bian, C.; Yang, F.; Lin, X.; Zhou, S.; Xie, C.; Zhao, X.; Yi, T. MicroRNA-18a inhibits ovarian cancer growth via directly targeting TRIAP1 and IPMK. Oncol. Lett. 2017, 13, 4039–4046. [Google Scholar] [CrossRef]
- Jia, Y.; Lin, R.; Jin, H.; Si, L.; Jian, W.; Yu, Q.; Yang, S. MicroRNA-34 suppresses proliferation of human ovarian cancer cells by triggering autophagy and apoptosis and inhibits cell invasion by targeting Notch 1. Biochimie 2019, 160, 193–199. [Google Scholar] [CrossRef]
- Chen, X.; Chen, S.; Xiu, Y.L.; Sun, K.X.; Zong, Z.H.; Zhao, Y. RhoC is a major target of microRNA-93-5P in epithelial ovarian carcinoma tumorigenesis and progression. Mol. Cancer 2015, 14, 31. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Rao, Y.M.; Shi, H.R.; Ji, M.; Chen, C.H. MiR-106a targets Mcl-1 to suppress cisplatin resistance of ovarian cancer A2780 cells. J. Huazhong Univ. Sci. Technol. Med. Sci. 2013, 33, 567–572. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Xu, H.; Shen, H.; Li, H. microRNA-106a modulates cisplatin sensitivity by targeting PDCD4 in human ovarian cancer cells. Oncol. Lett. 2014, 7, 183–188. [Google Scholar] [CrossRef] [PubMed]
- Yuan, L.; Li, S.; Zhou, Q.; Wang, D.; Zou, D.; Shu, J.; Huang, Y. MiR-124 inhibits invasion and induces apoptosis of ovarian cancer cells by targeting programmed cell death 6. Oncol. Lett. 2017, 14, 7311–7317. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zhang, X.; Huang, L.; Zhao, Y.; Tan, W. Downregulation of miR-130a contributes to cisplatin resistance in ovarian cancer cells by targeting X-linked inhibitor of apoptosis (XIAP) directly. Acta Biochim. Biophys. Sin. (Shanghai) 2013, 45, 995–1001. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.; Jiang, Y.; Mu, X.; Xu, L.; Cheng, W.; Wang, X. MiR-135a functions as a tumor suppressor in epithelial ovarian cancer and regulates HOXA10 expression. Cell. Signal. 2014, 26, 1420–1426. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Chen, W.; Zeng, W.; Wan, C.; Duan, S.; Jiang, S. microRNA-137 promotes apoptosis in ovarian cancer cells via the regulation of XIAP. Br. J. Cancer 2017, 116, 66–76. [Google Scholar] [CrossRef]
- Su, J.; Ruan, S.; Dai, S.; Mi, J.; Chen, W.; Jiang, S. NF1 regulates apoptosis in ovarian cancer cells by targeting MCL1 via miR-142-5p. Pharmacogenomics 2019, 20, 155–165. [Google Scholar] [CrossRef]
- Kleemann, M.; Bereuther, J.; Fischer, S.; Marquart, K.; Hanle, S.; Unger, K.; Jendrossek, V.; Riedel, C.U.; Handrick, R.; Otte, K. Investigation on tissue specific effects of pro-apoptotic micro RNAs revealed miR-147b as a potential biomarker in ovarian cancer prognosis. Oncotarget 2017, 8, 18773–18791. [Google Scholar] [CrossRef]
- Sun, L.; Zhai, R.; Zhang, L.; Zhao, S. MicroRNA-149 suppresses the proliferation and increases the sensitivity of ovarian cancer cells to cisplatin by targeting X-linked inhibitor of apoptosis. Oncol. Lett. 2018, 15, 7328–7334. [Google Scholar] [CrossRef]
- Zhan, Y.; Xiang, F.; Wu, R.; Xu, J.; Ni, Z.; Jiang, J.; Kang, X. MiRNA-149 modulates chemosensitivity of ovarian cancer A2780 cells to paclitaxel by targeting MyD88. J. Ovarian. Res. 2015, 8, 48. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Y.; Ma, N.; Wang, D.; Zhang, Y.; Zhou, J.; Wu, G.; Zhao, R.; Huang, H.; Wang, X.; Qiao, Y.; et al. MiR-152 and miR-185 co-contribute to ovarian cancer cells cisplatin sensitivity by targeting DNMT1 directly: A novel epigenetic therapy independent of decitabine. Oncogene 2014, 33, 378–386. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Xu, Q.H.; Dong, Y.H.; Li, G.X.; Yang, L.; Wang, L.W.; Li, H.Y. MiR-181a upregulation is associated with epithelial-to-mesenchymal transition (EMT) and multidrug resistance (MDR) of ovarian cancer cells. Eur Rev. Med. Pharmacol. Sci. 2016, 20, 2004–2010. [Google Scholar] [PubMed]
- Nakano, H.; Yamada, Y.; Miyazawa, T.; Yoshida, T. Gain-of-function microRNA screens identify miR-193a regulating proliferation and apoptosis in epithelial ovarian cancer cells. Int. J. Oncol. 2013, 42, 1875–1882. [Google Scholar] [CrossRef] [PubMed]
- Dai, J.; Wei, R.; Zhang, P.; Kong, B. Overexpression of microRNA-195-5p reduces cisplatin resistance and angiogenesis in ovarian cancer by inhibiting the PSAT1-dependent GSK3beta/beta-catenin signaling pathway. J. Transl. Med. 2019, 17, 190. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Li, Q.; Huang, H.; Li, Y.; Li, L.; Hou, W.; You, Z. Overexpression of miRNA-221 promotes cell proliferation by targeting the apoptotic protease activating factor-1 and indicates a poor prognosis in ovarian cancer. Int. J. Oncol. 2017. [Google Scholar] [CrossRef]
- Liu, X.; Wen, J.; Wang, H.; Wang, Y. Long non-coding RNA LINC00460 promotes epithelial ovarian cancer progression by regulating microRNA-338-3p. Biomed. Pharmacother. 2018, 108, 1022–1028. [Google Scholar] [CrossRef]
- Kleemann, M.; Schneider, H.; Unger, K.; Bereuther, J.; Fischer, S.; Sander, P.; Marion Schneider, E.; Fischer-Posovszky, P.; Riedel, C.U.; Handrick, R.; et al. Induction of apoptosis in ovarian cancer cells by miR-493-3p directly targeting AKT2, STK38L, HMGA2, ETS1 and E2F5. Cell Mol. Life Sci. 2019, 76, 539–559. [Google Scholar] [CrossRef]
- Zhang, J.; Gao, D.; Zhang, H. Upregulation of miR-614 promotes proliferation and inhibits apoptosis in ovarian cancer by suppressing PPP2R2A expression. Mol. Med. Rep. 2018, 17, 6285–6292. [Google Scholar] [CrossRef]
- Eoh, K.J.; Lee, S.H.; Kim, H.J.; Lee, J.Y.; Kim, S.; Kim, S.W.; Kim, Y.T.; Nam, E.J. MicroRNA-630 inhibitor sensitizes chemoresistant ovarian cancer to chemotherapy by enhancing apoptosis. Biochem. Biophys. Res. Commun. 2018, 497, 513–520. [Google Scholar] [CrossRef]
- Zou, Y.T.; Gao, J.Y.; Wang, H.L.; Wang, Y.; Wang, H.; Li, P.L. Downregulation of microRNA-630 inhibits cell proliferation and invasion and enhances chemosensitivity in human ovarian carcinoma. Genet. Mol. Res. 2015, 14, 8766–8777. [Google Scholar] [CrossRef] [PubMed]
- Leng, R.; Zha, L.; Tang, L. MiR-718 represses VEGF and inhibits ovarian cancer cell progression. FEBS Lett. 2014, 588, 2078–2086. [Google Scholar] [CrossRef] [PubMed]
- Kleemann, M.; Schneider, H.; Unger, K.; Sander, P.; Schneider, E.M.; Fischer-Posovszky, P.; Handrick, R.; Otte, K. MiR-744-5p inducing cell death by directly targeting HNRNPC and NFIX in ovarian cancer cells. Sci. Rep. 2018, 8, 9020. [Google Scholar] [CrossRef] [PubMed]
- Pan, C.; Wang, D.; Zhang, Y.; Yu, W. MicroRNA-1284 Inhibits Cell Viability and Induces Apoptosis of Ovarian Cancer Cell Line OVCAR3. Oncol. Res. 2016, 24, 429–435. [Google Scholar] [CrossRef] [PubMed]
- Moufarrij, S.; Dandapani, M.; Arthofer, E.; Gomez, S.; Srivastava, A.; Lopez-Acevedo, M.; Villagra, A.; Chiappinelli, K.B. Epigenetic therapy for ovarian cancer: Promise and progress. Clin. Epigenetics 2019, 11, 7. [Google Scholar] [CrossRef]
- Maradeo, M.E.; Cairns, P. Translational application of epigenetic alterations: Ovarian cancer as a model. FEBS Lett. 2011, 585, 2112–2120. [Google Scholar] [CrossRef]
- Nervi, C.; De Marinis, E.; Codacci-Pisanelli, G. Epigenetic treatment of solid tumours: A review of clinical trials. Clin. Epigenetics 2015, 7, 127. [Google Scholar] [CrossRef]
- Ahluwalia, A.; Yan, P.; Hurteau, J.A.; Bigsby, R.M.; Jung, S.H.; Huang, T.H.; Nephew, K.P. DNA methylation and ovarian cancer. I. Analysis of CpG island hypermethylation in human ovarian cancer using differential methylation hybridization. Gynecol. Oncol. 2001, 82, 261–268. [Google Scholar] [CrossRef]
- Ehrlich, M.; Woods, C.B.; Yu, M.C.; Dubeau, L.; Yang, F.; Campan, M.; Weisenberger, D.J.; Long, T.; Youn, B.; Fiala, E.S.; et al. Quantitative analysis of associations between DNA hypermethylation, hypomethylation, and DNMT RNA levels in ovarian tumors. Oncogene 2006, 25, 2636–2645. [Google Scholar] [CrossRef]
- Tam, K.F.; Liu, V.W.; Liu, S.S.; Tsang, P.C.; Cheung, A.N.; Yip, A.M.; Ngan, H.Y. Methylation profile in benign, borderline and malignant ovarian tumors. J. Cancer Res. Clin. Oncol. 2007, 133, 331–341. [Google Scholar] [CrossRef]
- Balch, C.; Fang, F.; Matei, D.E.; Huang, T.H.; Nephew, K.P. Minireview: Epigenetic changes in ovarian cancer. Endocrinology 2009, 150, 4003–4011. [Google Scholar] [CrossRef] [PubMed]
- Tomar, T.; Alkema, N.G.; Schreuder, L.; Meersma, G.J.; de Meyer, T.; van Criekinge, W.; Klip, H.G.; Fiegl, H.; van Nieuwenhuysen, E.; Vergote, I.; et al. Methylome analysis of extreme chemoresponsive patients identifies novel markers of platinum sensitivity in high-grade serous ovarian cancer. BMC Med. 2017, 15, 116. [Google Scholar] [CrossRef] [PubMed]
- Hua, K.T.; Wang, M.Y.; Chen, M.W.; Wei, L.H.; Chen, C.K.; Ko, C.H.; Jeng, Y.M.; Sung, P.L.; Jan, Y.H.; Hsiao, M.; et al. The H3K9 methyltransferase G9a is a marker of aggressive ovarian cancer that promotes peritoneal metastasis. Mol. Cancer 2014, 13, 189. [Google Scholar] [CrossRef] [PubMed]
- Smith, H.J.; Straughn, J.M.; Buchsbaum, D.J.; Arend, R.C. Epigenetic therapy for the treatment of epithelial ovarian cancer: A clinical review. Gynecol. Oncol. Rep. 2017, 20, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Hu, W.; Shen, D.Y.; Kavanagh, J.J.; Fu, S. Azacitidine enhances sensitivity of platinum-resistant ovarian cancer cells to carboplatin through induction of apoptosis. Am. J. Obstet. Gynecol. 2009, 200, 177.e1–177.e9. [Google Scholar] [CrossRef]
- Konovalov, S.; Garcia-Bassets, I. Analysis of the levels of lysine-specific demethylase 1 (LSD1) mRNA in human ovarian tumors and the effects of chemical LSD1 inhibitors in ovarian cancer cell lines. J. Ovarian. Res. 2013, 6, 75. [Google Scholar] [CrossRef]
- Feng, S.; Jin, Y.; Cui, M.; Zheng, J. Lysine-Specific Demethylase 1 (LSD1) Inhibitor S2101 Induces Autophagy via the AKT/mTOR Pathway in SKOV3 Ovarian Cancer Cells. Med. Sci. Monit. 2016, 22, 4742–4748. [Google Scholar] [CrossRef]
- Tomek, S.; Horak, P.; Pribill, I.; Haller, G.; Rossler, M.; Zielinski, C.C.; Pils, D.; Krainer, M. Resistance to TRAIL-induced apoptosis in ovarian cancer cell lines is overcome by co-treatment with cytotoxic drugs. Gynecol. Oncol. 2004, 94, 107–114. [Google Scholar] [CrossRef]
- Horak, P.; Pils, D.; Haller, G.; Pribill, I.; Roessler, M.; Tomek, S.; Horvat, R.; Zeillinger, R.; Zielinski, C.; Krainer, M. Contribution of epigenetic silencing of tumor necrosis factor-related apoptosis inducing ligand receptor 1 (DR4) to TRAIL resistance and ovarian cancer. Mol. Cancer Res. 2005, 3, 335–343. [Google Scholar] [CrossRef]
- Guo, J.; Cai, J.; Yu, L.; Tang, H.; Chen, C.; Wang, Z. EZH2 regulates expression of p57 and contributes to progression of ovarian cancer in vitro and in vivo. Cancer Sci. 2011, 102, 530–539. [Google Scholar] [CrossRef]
- Rao, Z.Y.; Cai, M.Y.; Yang, G.F.; He, L.R.; Mai, S.J.; Hua, W.F.; Liao, Y.J.; Deng, H.X.; Chen, Y.C.; Guan, X.Y.; et al. EZH2 supports ovarian carcinoma cell invasion and/or metastasis via regulation of TGF-beta1 and is a predictor of outcome in ovarian carcinoma patients. Carcinogenesis 2010, 31, 1576–1583. [Google Scholar] [CrossRef] [PubMed]
- Alldredge, J.K.; Eskander, R.N. EZH2 inhibition in ARID1A mutated clear cell and endometrioid ovarian and endometrioid endometrial cancers. Gynecol. Oncol. Res. Pract. 2017, 4, 17. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zhang, R. Role of EZH2 in Epithelial Ovarian Cancer: From Biological Insights to Therapeutic Target. Front. Oncol. 2013, 3, 47. [Google Scholar] [CrossRef]
- Li, H.; Cai, Q.; Godwin, A.K.; Zhang, R. Enhancer of zeste homolog 2 promotes the proliferation and invasion of epithelial ovarian cancer cells. Mol. Cancer Res. 2010, 8, 1610–1618. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.; Yang, X.; Zhuang, L.; Jiang, X.; Chen, W.; Lee, P.L.; Karuturi, R.K.; Tan, P.B.; Liu, E.T.; Yu, Q. Pharmacologic disruption of Polycomb-repressive complex 2-mediated gene repression selectively induces apoptosis in cancer cells. Genes Dev. 2007, 21, 1050–1063. [Google Scholar] [CrossRef]
- Shen, L.; Cui, J.; Pang, Y.X.; Ma, Y.H.; Liu, P.S. 3-Deazaneplanocin A is a promising therapeutic agent for ovarian cancer cells. Asian Pac. J. Cancer Prev. 2013, 14, 2915–2918. [Google Scholar] [CrossRef]
- Matei, D.; Fang, F.; Shen, C.; Schilder, J.; Arnold, A.; Zeng, Y.; Berry, W.A.; Huang, T.; Nephew, K.P. Epigenetic resensitization to platinum in ovarian cancer. Cancer Res. 2012, 72, 2197–2205. [Google Scholar] [CrossRef]
- Kim, H.J.; Bae, S.C. Histone deacetylase inhibitors: Molecular mechanisms of action and clinical trials as anti-cancer drugs. Am. J. Transl. Res. 2011, 3, 166–179. [Google Scholar]
- Bitler, B.G.; Wu, S.; Park, P.H.; Hai, Y.; Aird, K.M.; Wang, Y.; Zhai, Y.; Kossenkov, A.V.; Vara-Ailor, A.; Rauscher, F.J., 3rd; et al. ARID1A-mutated ovarian cancers depend on HDAC6 activity. Nat. Cell Biol. 2017, 19, 962–973. [Google Scholar] [CrossRef]
- Caslini, C.; Capo-chichi, C.D.; Roland, I.H.; Nicolas, E.; Yeung, A.T.; Xu, X.X. Histone modifications silence the GATA transcription factor genes in ovarian cancer. Oncogene 2006, 25, 5446–5461. [Google Scholar] [CrossRef]
- Rosato, R.R.; Almenara, J.A.; Grant, S. The histone deacetylase inhibitor MS-275 promotes differentiation or apoptosis in human leukemia cells through a process regulated by generation of reactive oxygen species and induction of p21CIP1/WAF1 1. Cancer Res. 2003, 63, 3637–3645. [Google Scholar] [PubMed]
- Karaca, B.; Atmaca, H.; Bozkurt, E.; Kisim, A.; Uzunoglu, S.; Karabulut, B.; Sezgin, C.; Sanli, U.A.; Uslu, R. Combination of AT-101/cisplatin overcomes chemoresistance by inducing apoptosis and modulating epigenetics in human ovarian cancer cells. Mol. Biol. Rep. 2013, 40, 3925–3933. [Google Scholar] [CrossRef] [PubMed]
- Xiang, X.Y.; Kang, J.S.; Yang, X.C.; Su, J.; Wu, Y.; Yan, X.Y.; Xue, Y.N.; Xu, Y.; Liu, Y.H.; Yu, C.Y.; et al. SIRT3 participates in glucose metabolism interruption and apoptosis induced by BH3 mimetic S1 in ovarian cancer cells. Int. J. Oncol. 2016, 49, 773–784. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A.; Issa, J.P.; Baylin, S. Targeting the cancer epigenome for therapy. Nat. Rev. Genet. 2016, 17, 630–641. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Su, J.; Xu, H.; Yu, S.; Liu, Y.; Zhang, Y.; Sun, L.; Yue, Y.; Zhou, X. Dicumarol inhibits PDK1 and targets multiple malignant behaviors of ovarian cancer cells. PLoS ONE 2017, 12, e0179672. [Google Scholar] [CrossRef] [PubMed]
- Rogalska, A.; Marczak, A. Epothilone B induces human ovarian cancer OV-90 cell apoptosis via external pathway. Environ. Toxicol. Pharmacol. 2015, 39, 700–712. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Liu, V.W.; Chan, D.W.; Yao, K.M.; Ngan, H.Y. LY294002 and metformin cooperatively enhance the inhibition of growth and the induction of apoptosis of ovarian cancer cells. Int. J. Gynecol. Cancer 2012, 22, 15–22. [Google Scholar] [CrossRef]
- Fulda, S. Promises and Challenges of Smac Mimetics as Cancer Therapeutics. Clin. Cancer Res. 2015, 21, 5030–5036. [Google Scholar] [CrossRef]
- Thibault, B.; Genre, L.; Le Naour, A.; Broca, C.; Mery, E.; Vuagniaux, G.; Delord, J.P.; Wiedemann, N.; Couderc, B. DEBIO 1143, an IAP inhibitor, reverses carboplatin resistance in ovarian cancer cells and triggers apoptotic or necroptotic cell death. Sci. Rep. 2018, 8, 17862. [Google Scholar] [CrossRef]
- Hurwitz, H.I.; Smith, D.C.; Pitot, H.C.; Brill, J.M.; Chugh, R.; Rouits, E.; Rubin, J.; Strickler, J.; Vuagniaux, G.; Sorensen, J.M.; et al. Safety, pharmacokinetics, and pharmacodynamic properties of oral DEBIO1143 (AT-406) in patients with advanced cancer: Results of a first-in-man study. Cancer Chemother. Pharmacol. 2015, 75, 851–859. [Google Scholar] [CrossRef]
- Runckel, K.; Barth, M.J.; Mavis, C.; Gu, J.J.; Hernandez-Ilizaliturri, F.J. The SMAC mimetic LCL-161 displays antitumor activity in preclinical models of rituximab-resistant B-cell lymphoma. Blood Adv. 2018, 2, 3516–3525. [Google Scholar] [CrossRef] [PubMed]
- Noonan, A.M.; Bunch, K.P.; Chen, J.Q.; Herrmann, M.A.; Lee, J.M.; Kohn, E.C.; O’Sullivan, C.C.; Jordan, E.; Houston, N.; Takebe, N.; et al. Pharmacodynamic markers and clinical results from the phase 2 study of the SMAC mimetic birinapant in women with relapsed platinum-resistant or -refractory epithelial ovarian cancer. Cancer 2016, 122, 588–597. [Google Scholar] [CrossRef] [PubMed]
- Oltersdorf, T.; Elmore, S.W.; Shoemaker, A.R.; Armstrong, R.C.; Augeri, D.J.; Belli, B.A.; Bruncko, M.; Deckwerth, T.L.; Dinges, J.; Hajduk, P.J.; et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature 2005, 435, 677–681. [Google Scholar] [CrossRef]
- Nicholson, D.W. From bench to clinic with apoptosis-based therapeutic agents. Nature 2000, 407, 810–816. [Google Scholar] [CrossRef] [PubMed]
- Reles, A.; Wen, W.H.; Schmider, A.; Gee, C.; Runnebaum, I.B.; Kilian, U.; Jones, L.A.; El-Naggar, A.; Minguillon, C.; Schonborn, I.; et al. Correlation of p53 mutations with resistance to platinum-based chemotherapy and shortened survival in ovarian cancer. Clin. Cancer Res. 2001, 7, 2984–2997. [Google Scholar] [PubMed]
- Robles, A.I.; Harris, C.C. Clinical outcomes and correlates of TP53 mutations and cancer. Cold Spring Harb Perspect. Biol. 2010, 2, a001016. [Google Scholar] [CrossRef]
- Lambert, J.M.; Gorzov, P.; Veprintsev, D.B.; Soderqvist, M.; Segerback, D.; Bergman, J.; Fersht, A.R.; Hainaut, P.; Wiman, K.G.; Bykov, V.J. PRIMA-1 reactivates mutant p53 by covalent binding to the core domain. Cancer Cell. 2009, 15, 376–388. [Google Scholar] [CrossRef]
- Bykov, V.J.; Issaeva, N.; Shilov, A.; Hultcrantz, M.; Pugacheva, E.; Chumakov, P.; Bergman, J.; Wiman, K.G.; Selivanova, G. Restoration of the tumor suppressor function to mutant p53 by a low-molecular-weight compound. Nat. Med. 2002, 8, 282–288. [Google Scholar] [CrossRef]
- Lambert, J.M.; Moshfegh, A.; Hainaut, P.; Wiman, K.G.; Bykov, V.J. Mutant p53 reactivation by PRIMA-1MET induces multiple signaling pathways converging on apoptosis. Oncogene 2010, 29, 1329–1338. [Google Scholar] [CrossRef]
- Mohell, N.; Alfredsson, J.; Fransson, A.; Uustalu, M.; Bystrom, S.; Gullbo, J.; Hallberg, A.; Bykov, V.J.; Bjorklund, U.; Wiman, K.G. APR-246 overcomes resistance to cisplatin and doxorubicin in ovarian cancer cells. Cell Death Dis. 2015, 6, e1794. [Google Scholar] [CrossRef]
- Astorgues-Xerri, L.; Riveiro, M.E.; Tijeras-Raballand, A.; Serova, M.; Rabinovich, G.A.; Bieche, I.; Vidaud, M.; de Gramont, A.; Martinet, M.; Cvitkovic, E.; et al. OTX008, a selective small-molecule inhibitor of galectin-1, downregulates cancer cell proliferation, invasion and tumour angiogenesis. Eur. J. Cancer 2014, 50, 2463–2477. [Google Scholar] [CrossRef] [PubMed]
- Zachar, Z.; Marecek, J.; Maturo, C.; Gupta, S.; Stuart, S.D.; Howell, K.; Schauble, A.; Lem, J.; Piramzadian, A.; Karnik, S.; et al. Non-redox-active lipoate derivates disrupt cancer cell mitochondrial metabolism and are potent anticancer agents in vivo. J. Mol. Med. (Berl.) 2011, 89, 1137–1148. [Google Scholar] [CrossRef] [PubMed]
- Han, C.Y.; Patten, D.A.; Richardson, R.B.; Harper, M.E.; Tsang, B.K. Tumor metabolism regulating chemosensitivity in ovarian cancer. Genes Cancer 2018, 9, 155–175. [Google Scholar] [CrossRef] [PubMed]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- Hay, N. Reprogramming glucose metabolism in cancer: Can it be exploited for cancer therapy? Nat. Rev. Cancer 2016, 16, 635–649. [Google Scholar] [CrossRef] [PubMed]
- Feron, O. Pyruvate into lactate and back: From the Warburg effect to symbiotic energy fuel exchange in cancer cells. Radiother. Oncol. 2009, 92, 329–333. [Google Scholar] [CrossRef]
- Semenza, G.L. Tumor metabolism: Cancer cells give and take lactate. J. Clin. Investig. 2008, 118, 3835–3837. [Google Scholar] [CrossRef]
- Onozuka, H.; Tsuchihara, K.; Esumi, H. Hypoglycemic/hypoxic condition in vitro mimicking the tumor microenvironment markedly reduced the efficacy of anticancer drugs. Cancer Sci. 2011, 102, 975–982. [Google Scholar] [CrossRef]
- Stuart, S.D.; Schauble, A.; Gupta, S.; Kennedy, A.D.; Keppler, B.R.; Bingham, P.M.; Zachar, Z. A strategically designed small molecule attacks alpha-ketoglutarate dehydrogenase in tumor cells through a redox process. Cancer Metab. 2014, 2, 4. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| microRNA (miRNA) | Function | Methods Used | Citation |
|---|---|---|---|
| miRNA-15a | Bmi-1 is elevated in OvCa cells and tissue samples There is an inverse correlation between miRNA15a and Bmi-1 over expressing miRNA-15a down regulates Bmi-1 protein in OvCa reducing cell proliferation | qPCR, western blot analysis, reporter assay | Bhattacharya et al., 2009 [89] |
| miRNA-16 | miRNA-16 down regulates Bmi-1 (Battacharya, 2009) and Bcl-2 (Cimmino, 2005) protein expression | Western blot analysis, TUNEL | Bhattacharya et al., 2009 [89], Cimmino et al., 2005 [90] |
| miRNA-18a | Overexpression of miRNA-18a in OvCa cell lines decreases levels of TRIAP1, IPMK, and cleaved Caspase 3 and increases apoptosis in vivo | Western blot analysis, TUNEL | Liu et al., 2017 [91] |
| miRNA-21 | Inhibition of miRNA 21 resulted in reduced pAKT and upregulated PTEN (Liu, 2019) and c-IAP2 (Chan, 2014) which interferes, with caspase activation in human OvCa cell lines. | Reporter assay, western blot analysis, qPCR, Annexin V | Chan et al., 2014 [82], Lou et al., 2011 [86], Liu et al., 2019 [87] |
| miRNA-25 | Knockdown of miRNA-25 in OvCa cell lines decreases Bim and Bcl2 levels, increases, Bax, cleaved Caspase-3 levels and apoptosis | Annexin V, western blot analysis | Zhang et al., 2012 [80], Sarkozy et al., 2018 [83] |
| miRNA-31 | In p53 deficient OvCa cell lines, miRNA-31 can induce p53-mediated apoptosis indirectly by targeting E2F2, MIR31, and CDKN2A genes. | qPCR, molecular profiling | Creighton et al., 2010 [81] |
| miRNA-34 | OvCa cell lines transfected with miRNA-34 mimics increase levels of Bax, while decreasing levels of Bcl-2 leading to increased apoptosis | Annexin V, western blot analysis | Jia et al., 2019 [92] |
| miRNA-93-5P | Overexpression of miRNA-93-5P in OvCa cell lines decreases levels of Bcl/xl, cleaved PARP, and increases levels of p53 and apoptosis. | Annexin V, western blot analysis | Chen et al., 2015 [93] |
| miRNA-106a | Inhibition of miR-106a enhanced the sensitivity of the OvCa cells to chemotherapy and increased apoptosis. Increasing miRNA-106a decreased PDCD4 levels. PDCD4 silencing led to a decrease in cleaved pro-Caspasse-3 and -9. | Flow Cytometry, western blot analysis | Rao et al 2013 [94], Li et al, 2014 [95] |
| miRNA-124 | Overexpression levels of miRNA-124 increases apoptosis and decreased levels of PDCD6 in OvCa cell lines | Annexin V, qPCR | Yuan et al., 2017 [96] |
| miRNA-130a | miRNA-130a downregulates XIAP in human A2780 cells. | qPCR, western blot analysis, flow cytometric analysis, reporter assay, Annexin V | Zhang et al., 2013 [97] |
| miRNA-135a | Transfection with an miRNA-135a mimic increases Caspase-3 activity and p53 levels and decreases Bcl-2 levels in OvCa cell lines. | Western blot, Caspase-3 activity assay, Annexin V | Tang et al., 2014 [98] |
| miRNA-137 | miR-137 knockout by CRISPR/Cas9 increases XIAP levels and inhibits apoptosis in OvCa cell lines | TUNEL, DAPI, western blot analysis | Li et al., 2017 [99] |
| miR-142-5p | miRNA-142-5p inhibits XIAP in human OvCa cell lines. miR-142-5p targeted anti-apoptotic genes (Birc3, Bcl2, Bcl2L2 and Mcl1) | Dual luciferase assay, western blot analysis, flow cytometric analysis | Li et al., 2019 [88], Su et al. 2019 [100] |
| miR-147b | Elevated miRNA-147b results in increased Bak1 and Bax levels and reduces levels of Bcl-2 and Bcl-xl in SKOV3 cells. | Molecular profiling, western blot analysis, Caspase-3 and 7 activity, mitochondrial potential | Kleemann et al., 2017 [101] |
| miRNA-149 | Downregulation of miRNA-149 decreases apoptosis in OvCa cells pre-treated with paclitaxel. Downregulation of miRNA-149 decreases Bax mRNA and protein and increases Bcl-2 mRNA and protein expression inhibits XIAP expression in human OvCa cells. Overexpression of miRNA-149 decreases XIAP mRNA and protein expression and increases apoptosis in OvCa cell lines. | Annexin V, qPCR, western blot analysis | Sun et al., 2018 [102], Zhan et al., 2015 [103] |
| miRNA-152 | Overexpression of miRNA-152 decreased DNMT1 and increased apoptosis and in OvCa cell lines. | Annexin V, western blot analysis, qPCR | Xiang et al., 2014 [104] |
| miRNA-181a | Overexpression of miRNA-181a decreased apoptosis in OvCa cells | Annexin V | Li et al., 2016 [105] |
| miRNA-193a and miR-193b | Overexpression of miRNA-193a or miRNA-193b increases activity of Caspase-3 and -7 in OvCa cells. miRNA-193a also decreases levels of anti-apoptotic factor MCL1 in OvCa cells | Caspase-3 and -7 activity, western blot analysis | Nakano et al., 2013 [106] |
| miRNA-195-5p | Overexpression of miRNA-195-5p increases apoptosis in OvCa cells and in a in vivo model. | Annexin V, TUNEL | Dai et al., 2019 [107] |
| miRNA-221 | Inhibiting miRNA-221 increases APAF1 and apoptosis in OvCa cells | Annexin V, western blot analysis, Hoechst 33342 staining | Li et al., 2017 [108] |
| miRNA-338-3p | miRNA-338-3p induces apoptosis by binding to long non-coding RNA LINC00460. | Luciferase reporter assay, western blot analysis, flow cytometric assay | Liu et al., 2018 [109] |
| miRNA-493-3p | Overexpression of miRNA-493-3p in OvCa cell lines increased Bak levels, release of cytochrome C, cleavage of Caspase-3 and PARP and decreased Bcl-XL levels | Western blot analysis, free cytochrome C staining, Annexin V | Kleeman et al., 2019 [110] |
| miRNA-614 | Overexpression of miRNA-614 decreases level of Bad and increases apoptosis in OvCa cells. | Annexin V, western blot analysis | Zhang et al., 2018 [111] |
| miRNA-630 | Silencing of miRNA-630 in OvCa cell lines increases cleaved Caspase-3, PTEN levels and apoptosis. | Annexin V, western blot analysis | Eoh et al., 2018 [112], Zou et al., 2015 [113] |
| miRNA-718 | Overexpression of miRNA-718 in OvCa cell lines decreases VEGF levels and increases apoptosis. This is reversed when VEGF is restored | Annexin V, western blot analysis | Leng et al., 2014 [114] |
| miRNA-744-5p | Increased expression of miRNA-744-5p increased levels of cleaved Caspase-3, and PARP and decreased levels of Bcl2 in OvCa cell lines. | AnnexinV, western blot analysis, Caspase-3 and 7 activity, mitochondrial membrane potential | Kleemann et al., 2018 [115] |
| miRNA-1284 | Inhibiting miRNA-1284 results in increased Bcl-2 levels and decreased Bax, and cleaved Caspase-3 levels in OvCa cell line. An miRNA-1284 mimic increases apoptosis | Annexin V, western blot analysis | Pan et al., 2016 [116] |
| Drug Treatment | Suspected Target Related to Apoptosis | Last Reported Phase | Cancer Type | NCT |
|---|---|---|---|---|
| Epothilone B | TRAIL and Caspase-8 | Phase II | Recurrent ovarian | NCT00035100 |
| Epothilone B versus Doxorubicin | TRAIL and Caspase-8 | Phase III | Ovarian, Primary Fallopian, or Peritoneal Cancer | NCT00262990 |
| Epothilone B + Omeprazole + Midalzolam | TRAIL and Caspase-8 | Phase I | Advanced malignancies | NCT00420615 |
| Metformin + Paclitaxel + Carboplatin | mTOR pathway circumventing p53-induced | Phase II | Advanced stage ovarian carcinoma | NCT02437812 |
| DEBIO 1143+ Carboplatin + Paclitaxel | cIAP1 | Phase II | Epithelial ovarian cancer | NCT01930292, NCT03270176 |
| DEBIO 1143+ Avelumab | cIAP1 | Phase I | Advanced solid malignancies | NCT03270176 |
| LCL161 | XIAP, cIAP1 and cIAP2 | Phase II | Solid tumors | NCT02649673 |
| Birinapant | IAPs, cIAP1 and cIAP2 and activate Caspase-3 | Phase I/II | Solid tumors | NCT01940172 |
| ABT 737/ABT 263 (navitoclax) | Bcl-2/Bcl-XL | Ex vivo study | Ovarian tumors | NCT01440504 |
| Venclexta (ABT-199) | Bcl-2 inhibitor | Approved | Chronic lymphocytic leukemia | - |
| PRIMA-1MET (APR-246) | p53 | Phase II | High-grade serous ovarian cancer, high grade serious ovarian cancer (Platinum-Resistant) | NCT02098343, NCT03268382 (biomarker ID) |
| Kevetrin | p53 | Phase II | Ovarian cancers | NCT03042702 |
| DPX-Survivac | Survivin peptide attached to an adjuvant | Phase II | Ovarian cancers | NCT03836352, NCT03029403 |
| OTX008 | Galectin-1 | Phase I | Solid tumors | NCT01724320 |
| CPI-613 | Alternative metabolic pathways | Phase III | Solid tumors | NCT03504423 (pancreatic) |
| CPI-613 | Alternative metabolic pathways | Phase II | Lymphoma/Leukemia | NCT03793140 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al-Alem, L.F.; Baker, A.T.; Pandya, U.M.; Eisenhauer, E.L.; Rueda, B.R. Understanding and Targeting Apoptotic Pathways in Ovarian Cancer. Cancers 2019, 11, 1631. https://doi.org/10.3390/cancers11111631
Al-Alem LF, Baker AT, Pandya UM, Eisenhauer EL, Rueda BR. Understanding and Targeting Apoptotic Pathways in Ovarian Cancer. Cancers. 2019; 11(11):1631. https://doi.org/10.3390/cancers11111631
Chicago/Turabian StyleAl-Alem, Linah F., Andrew T. Baker, Unnati M. Pandya, Eric L. Eisenhauer, and Bo R. Rueda. 2019. "Understanding and Targeting Apoptotic Pathways in Ovarian Cancer" Cancers 11, no. 11: 1631. https://doi.org/10.3390/cancers11111631
APA StyleAl-Alem, L. F., Baker, A. T., Pandya, U. M., Eisenhauer, E. L., & Rueda, B. R. (2019). Understanding and Targeting Apoptotic Pathways in Ovarian Cancer. Cancers, 11(11), 1631. https://doi.org/10.3390/cancers11111631