Glutamine Metabolism in Brain Tumors

Abstract
:1. Introduction
2. Glutamate/Glutamine Metabolism in the Central Nervous System
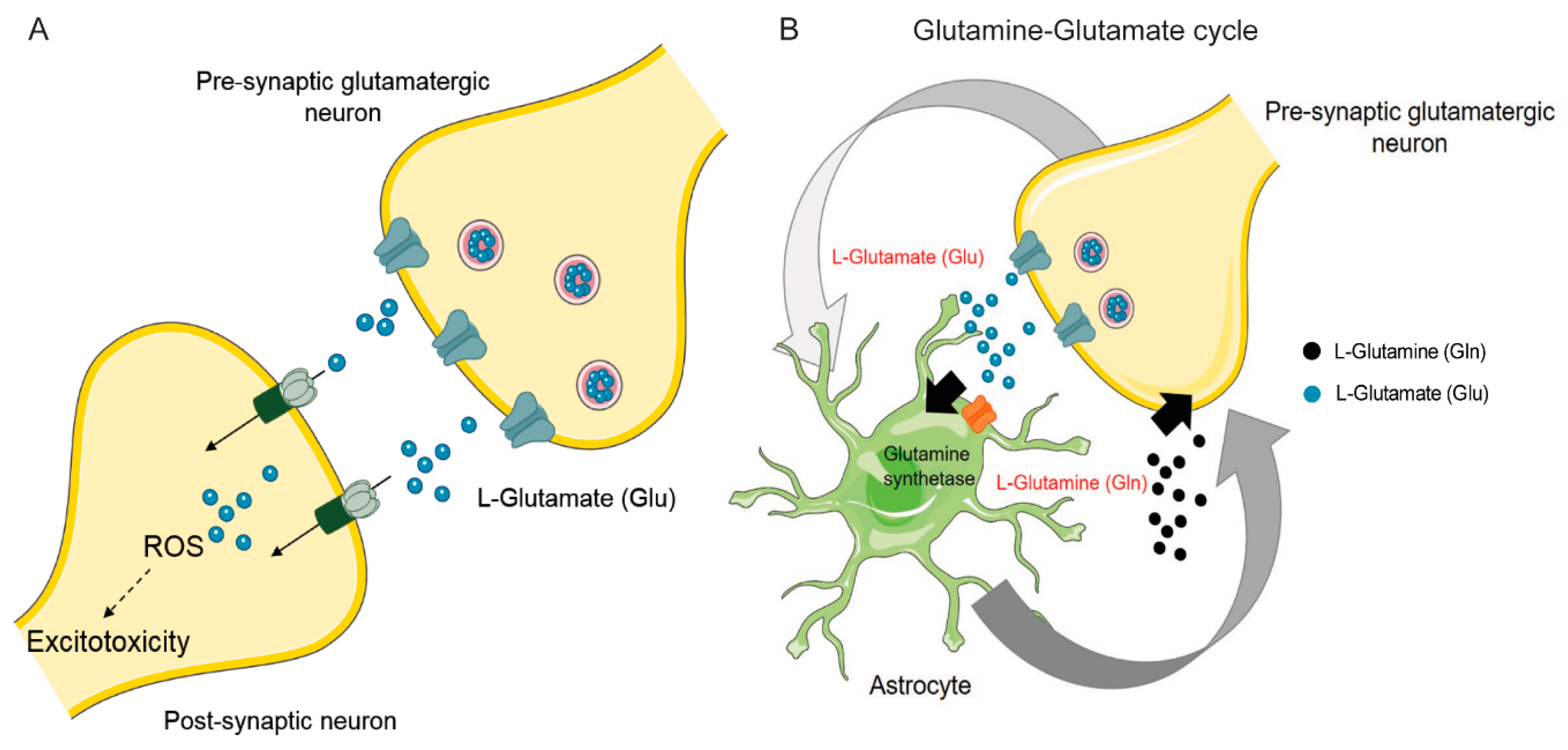
2.1. Synaptic Glutamate/Glutamine Cycle is Critical for Brain Function
2.2. Neurons Express Glutamatergic Receptors That Mediate Synaptic Signaling
2.3. Astrocytes Take Up Glutamate through Excitatory Amino Acid Transporters (EAATs)
2.4. System Xc- and Sodium-Coupled Neutral Amino Acid Transporters (SNATs) can Regulate the Glutamine Glutamate Cycle
2.5. Brain Tumor Cells can Promote Their Growth Via Glutamine/Glutamate Transporters
Transcriptional Control of Glutamine Metabolism
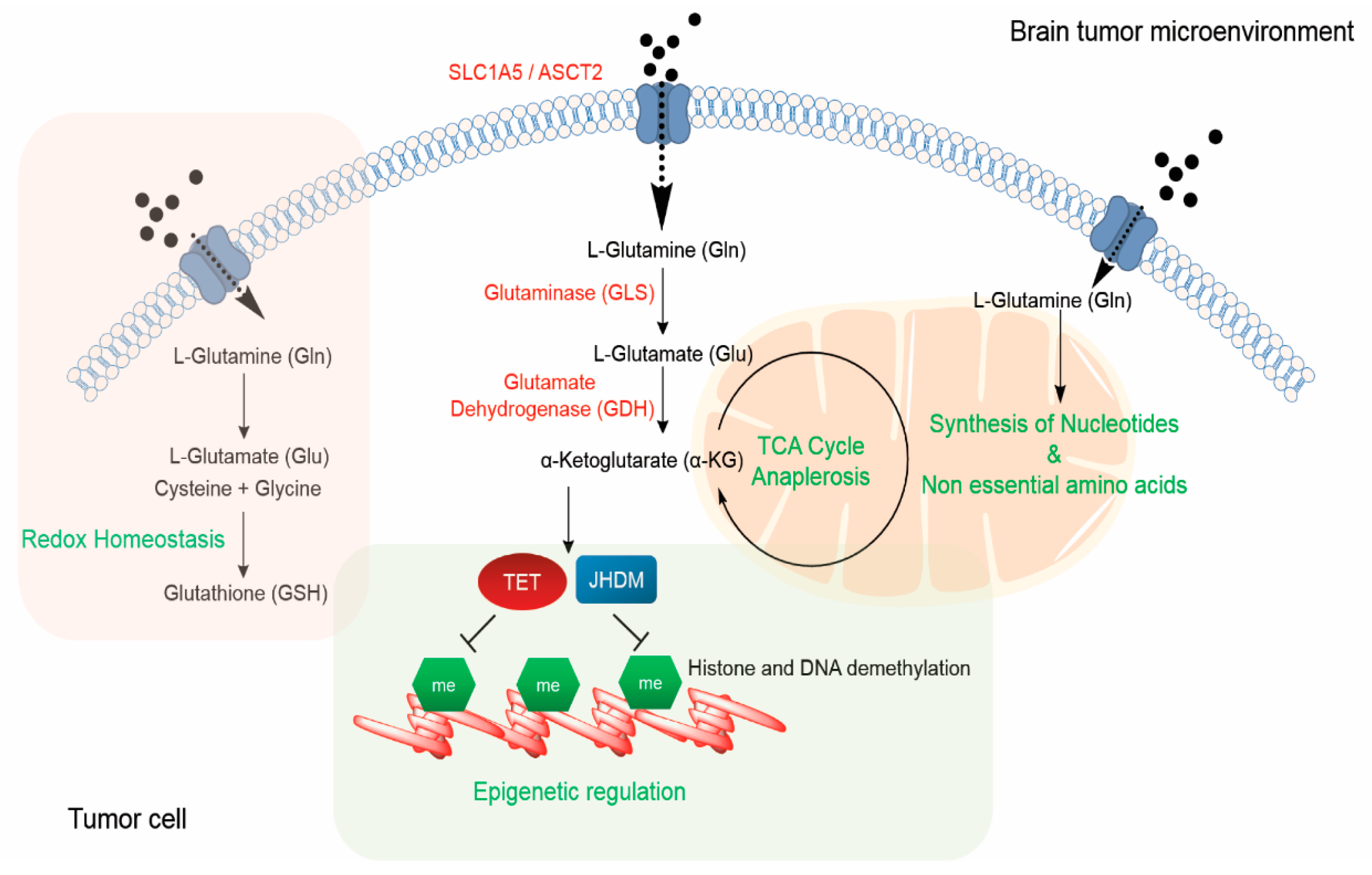
3. Functional Roles of Glutamine
3.1. Glutamine-Derived α-Ketoglutarate can Function as a TCA Cycle Anaplerotic Substrate and Regulate the Epigenome
3.2. Glutamine can Serve as a Nitrogen Donor for Nucleotide Synthesis
3.3. Glutamine as a Source of Non-Essential Amino Acids
3.4. Role of Glutamine in Maintaining Redox Homeostasis
3.5. Understanding Glutamine Metabolic Heterogeneity in Cancers
4. Metabolic Imaging and Therapeutic Targeting of Glutamine Metabolism
4.1. Imaging Glutamine Uptake and Metabolism
4.2. Therapeutic Targeting of Glutamine Metabolism in Cancer
5. Conclusions
Funding
Conflicts of Interest
References
- Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goyal, M.S.; Raichle, M.E. Glucose Requirements of the Developing Human Brain. J. Pediatr. Gastroenterol. Nutr. 2018, 66, S46–S49. [Google Scholar] [CrossRef] [PubMed]
- Hamberger, A.; Nyström, B.; Larsson, S.; Silfvenius, H.; Nordborg, C. Amino acids in the neuronal microenvironment of focal human epileptic lesions. Epilepsy Res. 1991, 9, 32–43. [Google Scholar] [CrossRef]
- Hertz, L. Functional interactions between neurons and astrocytes I. Turnover and metabolism of putative amino acid transmitters. Prog. Neurobiol. 1979, 13, 277–323. [Google Scholar] [CrossRef]
- Daikhin, Y.; Yudkoff, M. Compartmentation of Brain Glutamate Metabolism in Neurons and Glia. J. Nutr. 2000, 130, 1026S–1031S. [Google Scholar] [CrossRef] [PubMed]
- Tani, H.; Dulla, C.G.; Farzampour, Z.; Taylor-Weiner, A.; Huguenard, J.R.; Reimer, R.J. A local glutamate-glutamine cycle sustains synaptic excitatory transmitter release. Neuron 2014, 81, 888–900. [Google Scholar] [CrossRef]
- Choi, D.W. Excitotoxic cell death. J. Neurobiol. 1992, 23, 1261–1276. [Google Scholar] [CrossRef]
- Fujikawa, D.G. Prolonged seizures and cellular injury: Understanding the connection. Epilepsy Behav. 2005, 7, 3–11. [Google Scholar] [CrossRef]
- Mattson, M.P. Glutamate and neurotrophic factors in neuronal plasticity and disease. Ann. N. Y. Acad. Sci. 2008, 1144, 97–112. [Google Scholar] [CrossRef]
- Lieth, E.; LaNoue, K.F.; Berkich, D.A.; Xu, B.; Ratz, M.; Taylor, C.; Hutson, S.M. Nitrogen shuttling between neurons and glial cells during glutamate synthesis. J. Neurochem. 2001, 76, 1712–1723. [Google Scholar] [CrossRef]
- Rothman, D.L.; Behar, K.L.; Hyder, F.; Shulman, R.G. In vivo NMR Studies of the Glutamate Neurotransmitter Flux and Neuroenergetics: Implications for Brain Function. Annu. Rev. Physiol. 2003, 65, 401–427. [Google Scholar] [CrossRef] [PubMed]
- Hollmann, M. Cloned Glutamate Receptors. Annu. Rev. Neurosci. 1994, 17, 31–108. [Google Scholar] [CrossRef] [PubMed]
- Nath, A.; Vetrovec, G.W.; Cowley, M.J.; Newton, M.; DiSciascio, G.; Mukharji, J.; Lewis, S. a Glutamate Receptor Ion CHannels: Structure, Regulation, and Function. Pharmacol. Reveiws 1988, 14, 37–40. [Google Scholar]
- Cull-Candy, S.G.; Leszkiewicz, D.N. Role of distinct NMDA receptor subtypes at central synapses. Sci. STKE 2004, 2004, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Paoletti, P. Molecular basis of NMDA receptor functional diversity. Eur. J. Neurosci. 2011, 33, 1351–1365. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, H.; Singh, S.K.; Mancusso, R.; Gouaux, E. Subunit arrangement and function in NMDA receptors. Nature 2005, 438, 185–192. [Google Scholar] [CrossRef] [PubMed]
- Barria, A.; Malinow, R. NMDA receptor subunit composition controls synaptic plasticity by regulating binding to CaMKII. Neuron 2005, 48, 289–301. [Google Scholar] [CrossRef]
- Wang, C.C.; Held, R.G.; Chang, S.C.; Yang, L.; Delpire, E.; Ghosh, A.; Hall, B.J. A critical role for gluN2B-containing NMDA receptors in cortical development and function. Neuron 2011, 72, 789–805. [Google Scholar] [CrossRef]
- Gambrill, A.C.; Barria, A. NMDA receptor subunit composition controls synaptogenesis and synapse stabilization. Proc. Natl. Acad. Sci. USA 2011, 108, 5855–5860. [Google Scholar] [CrossRef] [Green Version]
- Rojas, A.; Dingledine, R. Ionotropic glutamate receptors: Regulation by G-protein-coupled receptors. Mol. Pharmacol. 2013, 83, 746–752. [Google Scholar] [CrossRef]
- Tang, C.M.; Dichter, M.; Morad, M. Quisqualate activates a rapidly inactivating high conductance ionic channel in hippocampal neurons. Science (80-. ). 1989, 243, 1474–1477. [Google Scholar] [CrossRef] [PubMed]
- Isaac, J.T.R.; Ashby, M.; McBain, C.J. The Role of the GluR2 Subunit in AMPA Receptor Function and Synaptic Plasticity. Neuron 2007, 54, 859–871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakanishi, S. Metabotropic glutamate receptors: Synaptic transmission, modulation, and plasticity. Neuron 1994, 13, 1031–1037. [Google Scholar] [CrossRef]
- Bonsi, P.; Cuomo, D.; De Persis, C.; Centonze, D.; Bernardi, G.; Calabresi, P.; Pisani, A. Modulatory action of metabotropic glutamate receptor (mGluR) 5 on mGluR1 function in striatal cholinergic interneurons. Neuropharmacology 2005, 49, 104–113. [Google Scholar] [CrossRef] [PubMed]
- Gubellini, P.; Pisani, A.; Centonze, D.; Bernardi, G.; Calabresi, P. Metabotropic glutamate receptors and striatal synaptic plasticity: Implications for neurological diseases. Prog. Neurobiol. 2004, 74, 271–300. [Google Scholar] [CrossRef] [PubMed]
- De Vivo, L.; Melone, M.; Bucci, G.; Rothstein, J.D.; Conti, F. Quantitative analysis of EAAT4 promoter activity in neurons and astrocytes of mouse somatic sensory cortex. Neurosci. Lett. 2010, 474, 42–45. [Google Scholar] [CrossRef]
- Lee, A.; Anderson, A.R.; Barnett, N.L.; Stevens, M.G.; Pow, D.V. Alternate splicing and expression of the glutamate transporter EAAT5 in the rat retina. Gene 2012, 506, 283–288. [Google Scholar] [CrossRef]
- Danbolt, N.C.; Pines, G.; Kanner, B.I. Purification and Reconstitution of the Sodium- and Potassium-Coupled Glutamate Transport Glycoprotein from Rat Brain. Biochemistry 1990, 29, 6734–6740. [Google Scholar] [CrossRef]
- Haugeto, Ø.; Ullensvang, K.; Levy, L.M.; Chaudhry, F.A.; Honoré, T.; Nielsen, M.; Lehre, K.P.; Danbolt, N.C. Brain glutamate transporter proteins form homomultimers. J. Biol. Chem. 1996, 271, 27715–27722. [Google Scholar] [CrossRef] [PubMed]
- Burdo, J.; Dargusch, R.; Schubert, D. Distribution of the cystine/glutamate antiporter system xc- in the brain, kidney, and duodenum. J. Histochem. Cytochem. 2006, 54, 549–557. [Google Scholar] [CrossRef]
- Piani, D.; Fontana, A. Involvement of the cystine transport system x(c)/- in the macrophage- induced glutamate-dependent cytotoxicity to neurons. J. Immunol. 1994, 152, 3578–3585. [Google Scholar] [PubMed]
- Nakanishi, T.; Sugawara, M.; Huang, W.; Martindale, R.G.; Leibach, F.H.; Ganapathy, M.E.; Prasad, P.D.; Ganapathy, V. Structure, function, and tissue expression pattern of human SN2, a subtype of the amino acid transport system N. Biochem. Biophys. Res. Commun. 2001, 281, 1343–1348. [Google Scholar] [CrossRef] [PubMed]
- Cubelos, B.; González-González, I.M.; Giménez, C.; Zafra, F. Amino acid transporter SNAT5 localizes to glial cells in the rat brain. Glia 2005, 49, 230–244. [Google Scholar] [CrossRef] [PubMed]
- Boulland, J.L.; Rafiki, A.; Levy, L.M.; Storm-Mathisen, J.; Chaudhry, F.A. Highly differential expression of SN1, a bidirectional glutamine transporter, in astroglia and endothelium in the developing rat brain. Glia 2003, 41, 260–275. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Lozada, Z.; Guillem, A.M.; Flores-Méndez, M.; Hernández-Kelly, L.C.; Vela, C.; Meza, E.; Zepeda, R.C.; Caba, M.; Rodríguez, A.; Ortega, A. GLAST/EAAT1-induced Glutamine release via SNAT3 in Bergmann glial cells: Evidence of a functional and physical coupling. J. Neurochem. 2013, 125, 545–554. [Google Scholar] [CrossRef]
- Kobayashi, M.; Mizutani, A.; Nishi, K.; Nakajima, S.; Shikano, N.; Nishii, R.; Fukuchi, K.; Kawai, K. Differences in accumulation and the transport mechanism of L- and D-methionine in high- and low-grade human glioma cells. Nucl. Med. Biol. 2017, 44, 78–82. [Google Scholar] [CrossRef]
- Dolińska, M.; Dybel, A.; Zabłocka, B.; Albrecht, J. Glutamine transport in C6 glioma cells shows ASCT2 system characteristics. Neurochem. Int. 2003, 43, 501–507. [Google Scholar] [CrossRef]
- Sidoryk, M.; Matyja, E.; Dybel, A.; Zielinska, M.; Bogucki, J.; Jaskólski, D.J.; Liberski, P.P.; Kowalczyk, P.; Albrecht, J. Increased expression of a glutamine transporter SNAT3 is a marker of malignant gliomas. Neuroreport 2004, 15, 575–578. [Google Scholar] [CrossRef]
- Medina, M.A.; Sánchez-Jiménez, F.; Márquez, J.; Rodríguez Quesada, A.; de Castro Núñez, I. Relevance of glutamine metabolism to tumor cell growth. Mol. Cell. Biochem. 1992, 113, 1–15. [Google Scholar] [CrossRef]
- De Groot, J.; Sontheimer, H. Glutamate and the biology of gliomas. Glia 2011, 59, 1181–1189. [Google Scholar] [CrossRef]
- Ishiuchi, S.; Tsuzuki, K.; Yoshida, Y.; Yamada, N.; Hagimura, N.; Okado, H.; Miwa, A.; Kurihara, H.; Nakazato, Y.; Sasaki, T.; et al. Blockage of Ca 2+ -permeable AMPA receptors suppresses migration and induces apoptosis in human glioblastoma cells. Nat. Med. 2002, 8, 971–978. [Google Scholar] [CrossRef] [PubMed]
- D’Onofrio, M.; Arcella, A.; Bruno, V.; Ngomba, R.T.; Battaglia, G.; Lombari, V.; Ragona, G.; Calogero, A.; Nicoletti, F. Pharmacological blockade of mGlu2/3 metabotropic glutamate receptors reduces cell proliferation in cultured human glioma cells. J. Neurochem. 2003, 84, 1288–1295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kettenmann, H. Glutamate receptor activation can trigger electrical activity in human glioma cells. Eur. J. Neurosci. 1998, 10, 2153–2162. [Google Scholar]
- Venkatesh, S.; Morishita, W.; Geraghty, A.C.; Silverbush, D.; Gillespie, S.M.; Arzt, M.; Regev, A.; Brang, D.; Vogel, H.; Hervey-jumper, S.; et al. Electrical and synaptic integration of glioma into neural circuits. Nature 2019, 573, 539–545. [Google Scholar] [CrossRef] [PubMed]
- Venkataramani, V.; Tanev, D.I.; Strahle, C.; Studier-fischer, A.; Fankhauser, L.; Kessler, T.; Körber, C.; Kardorff, M.; Ratliff, M.; Xie, R.; et al. Glutamatergic synaptic input to glioma cells drives brain tumour progression. Nature 2019, 573, 532–538. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Q.; Michael, I.P.; Zhang, P.; Saghafinia, S.; Knott, G.; Jiao, W.; McCabe, B.D.; Galván, J.A.; Robinson, H.P.C.; Zlobec, I.; et al. Synaptic proximity enables NMDAR signalling to promote brain metastasis. Nature 2019, 573, 526–531. [Google Scholar] [CrossRef]
- Shih, A.Y.; Erb, H.; Sun, X.; Toda, S.; Kalivas, P.W.; Murphy, T.H. Cystine/glutamate exchange modulates glutathione supply for neuroprotection from oxidative stress and cell proliferation. J. Neurosci. 2006, 26, 10514–10523. [Google Scholar] [CrossRef]
- Lo, M.; Wang, Y.Z.; Gout, P.W. The xc- cystine/glutamate antiporter: A potential target for therapy of cancer and other diseases. J. Cell. Physiol. 2008, 215, 593–602. [Google Scholar] [CrossRef]
- Bansal, A.; Celeste Simon, M. Glutathione metabolism in cancer progression and treatment resistance. J. Cell Biol. 2018, 217, 2291–2298. [Google Scholar] [CrossRef] [Green Version]
- Bump, E.A.; Brown, J.M. Role of glutathione in the radiation response of mammalian cells invitro and in vivo. Pharmacol. Ther. 1990, 47, 117–136. [Google Scholar] [CrossRef]
- Estrela, J.M.; Ortega, A.; Obrador, E. Glutathione in cancer biology and therapy; 2006; Volume 43, ISBN 1040836050052. [Google Scholar]
- Wise, D.R.; Deberardinis, R.J.; Mancuso, A.; Sayed, N.; Zhang, X.Y.; Pfeiffer, H.K.; Nissim, I.; Daikhin, E.; Yudkoff, M.; McMahon, S.B.; et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc. Natl. Acad. Sci. USA 2008, 105, 18782–18787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, P.; Tchernyshyov, I.; Chang, T.C.; Lee, Y.S.; Kita, K.; Ochi, T.; Zeller, K.I.; De Marzo, A.M.; Van Eyk, J.E.; Mendell, J.T.; et al. C-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature 2009, 458, 762–765. [Google Scholar] [CrossRef] [PubMed]
- Mannava, S.; Grachtchouk, V.; Wheeler, L.J.; Im, M.; Zhuang, D.; Slavina, E.G.; Mathews, C.K.; Shewach, D.S.; Nikiforov, M.A. Direct role of nucleotide metabolism in C-MYC-dependent proliferation of melanoma cells. Cell Cycle 2008, 7, 2392–2400. [Google Scholar] [CrossRef] [PubMed]
- Han, F.; Hu, R.; Yang, H.; Liu, J.; Sui, J.; Xiang, X.; Wang, F.; Chu, L.; Song, S. PTEN gene mutations correlate to poor prognosis in glioma patients: A meta-analysis. Onco. Targets. Ther. 2016, 9, 3485–3492. [Google Scholar] [PubMed]
- Endersby, R.; Baker, S.J. PTEN signaling in brain: Neuropathology and tumorigenesis. Oncogene 2008, 27, 5416–5430. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Cao, I.; Song, M.S.; Hobbs, R.M.; Laurent, G.; Giorgi, C.; De Boer, V.C.J.; Anastasiou, D.; Ito, K.; Sasaki, A.T.; Rameh, L.; et al. Systemic elevation of PTEN induces a tumor-suppressive metabolic state. Cell 2012, 149, 49–62. [Google Scholar] [CrossRef] [PubMed]
- Juvekar, A.; Hu, H.; Cantley, L.C.; Toker, A.; Lyssiotis, C.A.; Asara, J.M.; Lien, E.C. Glutathione biosynthesis is a metabolic vulnerability in PI(3)K/Akt-driven breast cancer. Nat. Cell Biol. 2016, 18, 572–578. [Google Scholar] [Green Version]
- Mitsuishi, Y.; Taguchi, K.; Kawatani, Y.; Shibata, T.; Nukiwa, T.; Aburatani, H.; Yamamoto, M.; Motohashi, H. Nrf2 Redirects Glucose and Glutamine into Anabolic Pathways in Metabolic Reprogramming. Cancer Cell 2012, 22, 66–79. [Google Scholar] [CrossRef] [Green Version]
- Csibi, A.; Fendt, S.M.; Li, C.; Poulogiannis, G.; Choo, A.Y.; Chapski, D.J.; Jeong, S.M.; Dempsey, J.M.; Parkhitko, A.; Morrison, T.; et al. The mTORC1 pathway stimulates glutamine metabolism and cell proliferation by repressing SIRT4. Cell 2013, 153, 840–854. [Google Scholar] [CrossRef]
- Suzuki, S.; Tanaka, T.; Poyurovsky, M.V.; Nagano, H.; Mayama, T.; Ohkubo, S.; Lokshin, M.; Hosokawa, H.; Nakayama, T.; Suzuki, Y.; et al. Phosphate-activated glutaminase (GLS2), a p53-inducible regulator of glutamine metabolism and reactive oxygen species. Proc. Natl. Acad. Sci. USA 2010, 107, 7461–7466. [Google Scholar] [CrossRef] [Green Version]
- Reynolds, M.R.; Lane, A.N.; Robertson, B.; Kemp, S.; Liu, Y.; Hill, B.G.; Dean, D.C.; Clem, B.F. Control of glutamine metabolism by the tumor suppressor Rb. Oncogene 2014, 33, 556–566. [Google Scholar] [CrossRef] [PubMed]
- Tajan, M.; Hock, A.K.; Blagih, J.; Robertson, N.A.; Labuschagne, C.F.; Kruiswijk, F.; Humpton, T.J.; Adams, P.D.; Vousden, K.H. A Role for p53 in the Adaptation to Glutamine Starvation through the Expression of SLC1A3. Cell Metab. 2018, 28, 721–736.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, J.P.; Yashinskie, J.J.; Koche, R.; Chandwani, R.; Tian, S.; Chen, C.-C.; Baslan, T.; Marinkovic, Z.S.; Sánchez-Rivera, F.J.; Leach, S.D.; et al. α-Ketoglutarate links p53 to cell fate during tumour suppression. Nature 2019. [Google Scholar] [CrossRef] [PubMed]
- Tedjojuwono, K.; Kawase, Y.; Asakura, T. Effect of Particle Density on the Photodetector Signal of Differential-Type Laser Doppler Velocimetry. Opt. 1984, 67, 331–344. [Google Scholar]
- Pizer, E.S.; Wood, F.D.; Heine, H.S.; Romantsev, F.E.; Pasternack, G.R.; Kuhajda, F.P. Inhibition of fatty acid synthesis delays disease progression in a xenograft model of ovarian cancer. Cancer Res. 1996, 56, 1189–1193. [Google Scholar] [PubMed]
- Metallo, C.M.; Gameiro, P.A.; Bell, E.L.; Mattaini, K.R.; Yang, J.; Hiller, K.; Jewell, C.M.; Johnson, Z.R.; Irvine, D.J.; Guarente, L.; et al. Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature 2012, 481, 380–384. [Google Scholar] [CrossRef] [PubMed]
- Dang, L.; White, D.W.; Gross, S.; Bennett, B.D.; Bittinger, M.A.; Driggers, E.M.; Fantin, V.R.; Jang, H.G.; Jin, S.; Keenan, M.C.; et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 2009, 462, 739–744. [Google Scholar] [CrossRef] [Green Version]
- Oizel, K.; Chauvin, C.; Oliver, L.; Gratas, C.; Geraldo, F.; Jarry, U.; Scotet, E.; Rabe, M.; Alves-Guerra, M.C.; Teusan, R.; et al. Efficient mitochondrial glutamine targeting prevails over glioblastoma metabolic plasticity. Clin. Cancer Res. 2017, 23, 6292–6305. [Google Scholar] [CrossRef]
- Wise, D.R.; Ward, P.S.; Shay, J.E.S.; Cross, J.R.; Gruber, J.J.; Sachdeva, U.M.; Platt, J.M.; DeMatteo, R.G.; Simon, M.C.; Thompson, C.B. Hypoxia promotes isocitrate dehydrogenasedependent carboxylation of α-ketoglutarate to citrate to support cell growth and viability. Proc. Natl. Acad. Sci. USA 2011, 108, 19611–19616. [Google Scholar] [CrossRef]
- Mullen, A.R.; Hu, Z.; Shi, X.; Jiang, L.; Boroughs, L.K.; Kovacs, Z.; Boriack, R.; Rakheja, D.; Sullivan, L.B.; Linehan, W.M.; et al. Oxidation of alpha-ketoglutarate is required for reductive carboxylation in cancer cells with mitochondrial defects. Cell Rep. 2014, 7, 1679–1690. [Google Scholar] [CrossRef]
- Fendt, S.M.; Bell, E.L.; Keibler, M.A.; Olenchock, B.A.; Mayers, J.R.; Wasylenko, T.M.; Vokes, N.I.; Guarente, L.; Heiden, M.G.V.; Stephanopoulos, G. Reductive glutamine metabolism is a function of the α-ketoglutarate to citrate ratio in cells. Nat. Commun. 2013, 4, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Carey, B.W.; Finley, L.W.S.; Cross, J.R.; Allis, C.D.; Thompson, C.B. Intracellular α-ketoglutarate maintains the pluripotency of embryonic stem cells. Nature 2015, 518, 413–416. [Google Scholar] [CrossRef] [PubMed]
- Parsons, D.W.; Jones, S.; Zhang, X.; Lin, J.C.H.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Siu, I.M.; Gallia, G.L.; et al. An integrated genomic analysis of human glioblastoma multiforme. Science (80-. ). 2008, 321, 1807–1812. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Parsons, D.W.; Jin, G.; McLendon, R.; Rasheed, B.A.; Yuan, W.; Kos, I.; Batinic-Haberle, I.; Jones, S.; Riggins, G.J.; et al. Mutations in Gliomas. N. Engl. J. Med. 2009, 360, 765–773. [Google Scholar] [CrossRef] [PubMed]
- Ward, P.S.; Patel, J.; Wise, D.R.; Abdel-Wahab, O.; Bennett, B.D.; Coller, H.A.; Cross, J.R.; Fantin, V.R.; Hedvat, C.V.; Perl, A.E.; et al. The Common Feature of Leukemia-Associated IDH1 and IDH2 Mutations Is a Neomorphic Enzyme Activity Converting α-Ketoglutarate to 2-Hydroxyglutarate. Cancer Cell 2010, 17, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Salamanca-Cardona, L.; Shah, H.; Poot, A.J.; Correa, F.M.; Di Gialleonardo, V.; Lui, H.; Miloushev, V.Z.; Granlund, K.L.; Tee, S.S.; Cross, J.R.; et al. In Vivo Imaging of Glutamine Metabolism to the Oncometabolite 2-Hydroxyglutarate in IDH1/2 Mutant Tumors. Cell Metab. 2017, 26, 830–841.e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chowdhury, R.; Yeoh, K.K.; Tian, Y.M.; Hillringhaus, L.; Bagg, E.A.; Rose, N.R.; Leung, I.K.H.; Li, X.S.; Woon, E.C.Y.; Yang, M.; et al. The oncometabolite 2-hydroxyglutarate inhibits histone lysine demethylases. EMBO Rep. 2011, 12, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Yang, H.; Liu, Y.; Yang, Y.; Wang, P.; Kim, S.H.; Ito, S.; Yang, C.; Wang, P.; Xiao, M.T.; et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of α-ketoglutarate-dependent dioxygenases. Cancer Cell 2011, 19, 17–30. [Google Scholar] [CrossRef] [PubMed]
- Turcan, S.; Rohle, D.; Goenka, A.; Walsh, L.A.; Fang, F.; Yilmaz, E.; Campos, C.; Fabius, A.W.M.; Lu, C.; Ward, P.S.; et al. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature 2012, 483, 479–483. [Google Scholar] [CrossRef]
- Turcan, S.; Makarov, V.; Taranda, J.; Wang, Y.; Fabius, A.W.M.; Wu, W.; Zheng, Y.; El-Amine, N.; Haddock, S.; Nanjangud, G.; et al. Mutant-IDH1-dependent chromatin state reprogramming, reversibility, and persistence. Nat. Genet. 2018, 50, 62–72. [Google Scholar] [CrossRef]
- Lu, C.; Ward, P.S.; Kapoor, G.S.; Rohle, D.; Turcan, S.; Abdel-Wahab, O.; Edwards, C.R.; Khanin, R.; Figueroa, M.E.; Melnick, A.; et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature 2012, 483, 474–478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wise, D.R.; Thompson, C.B. Glutamine addiction: a new therapeutic target in cancer. Trends Biochem. Sci. 2010, 35, 427–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cory, J.G.; Cory, A.N.N.H. Critical Roles of Glutamine as Nitrogen Donors in Purine and Pyrimidine Nucleotide Synthesis. In Vivo 2006, 590, 587–589. [Google Scholar]
- Goswami, M.T.; Chen, G.; Chakravarthi, B.V.S.K.; Pathi, S.S.; Anand, S.K.; Carskadon, S.L.; Giordano, T.J.; Chinnaiyan, A.M.; Thomas, D.G.; Palanisamy, N.; et al. Role and regulation of coordinately expressed de novo purine biosynthetic enzymes PPAT and PAICS in lung cancer. Oncotarget 2015, 6, 23445–23461. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, J.T.; Moreno, M.V.; Lodi, A.; Ronen, S.M.; Ruggero, D. Protein and nucleotide biosynthesis are coupled by a single rate-limiting enzyme, PRPS2, to drive cancer. Cell 2014, 157, 1088–1103. [Google Scholar] [CrossRef] [PubMed]
- Robitaille, A.M.; Christen, S.; Shimobayashi, M.; Cornu, M.; Fava, L.L.; Moes, S.; Prescianotto-Baschong, C.; Sauer, U.; Jenoe, P.; Hall, M.N. Quantitative phosphoproteomics reveal mTORC1 activates de novo pyrimidine synthesis. Science (80-. ). 2013, 339, 1320–1323. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Moss, T.; Mangala, L.S.; Marini, J.; Zhao, H.; Wahlig, S.; Armaiz-Pena, G.; Jiang, D.; Achreja, A.; Win, J.; et al. Metabolic shifts toward glutamine regulate tumor growth, invasion and bioenergetics in ovarian cancer. Mol. Syst. Biol. 2014, 10, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Gaglio, D.; Soldati, C.; Vanoni, M.; Alberghina, L.; Chiaradonna, F. Glutamine deprivation induces abortive S-phase rescued by deoxyribonucleotides in K-ras transformed fibroblasts. PLoS ONE 2009, 4. [Google Scholar] [CrossRef]
- Lane, A.N.; Fan, T.W.M. Regulation of mammalian nucleotide metabolism and biosynthesis. Nucleic Acids Res. 2015, 43, 2466–2485. [Google Scholar] [CrossRef] [Green Version]
- Patel, D.; Menon, D.; Bernfeld, E.; Mroz, V.; Kalan, S.; Loayza, D.; Foster, D.A. Aspartate rescues S-phase arrest caused by suppression of glutamine utilization in KRas-driven cancer cells. J. Biol. Chem. 2016, 291, 9322–9329. [Google Scholar] [CrossRef]
- Davidson, S.M.; Papagiannakopoulos, T.; Olenchock, B.A.; Heyman, J.E.; Keibler, M.A.; Luengo, A.; Bauer, M.R.; Jha, A.K.; O’Brien, J.P.; Pierce, K.A.; et al. Environment impacts the metabolic dependencies of ras-driven non-small cell lung cancer. Cell Metab. 2016, 23, 517–528. [Google Scholar] [CrossRef] [PubMed]
- Choi, B.H.; Coloff, J.L. The diverse functions of non-essential amino acids in cancer. Cancers (Basel) 2019, 11, 675. [Google Scholar] [CrossRef] [PubMed]
- Hao, Y.; Samuels, Y.; Li, Q.; Krokowski, D.; Guan, B.-J.; Wang, C.; Jin, Z.; Dong, B.; Cao, B.; Feng, X.; et al. Oncogenic PIK3CA mutations reprogram glutamine metabolism in colorectal cancer. Nat. Commun. 2016, 7, 11971. [Google Scholar] [CrossRef] [PubMed]
- Coloff, J.L.; Murphy, J.P.; Braun, C.R.; Harris, I.S.; Shelton, L.M.; Kami, K.; Gygi, S.P.; Selfors, L.M.; Brugge, J.S. Differential Glutamate Metabolism in Proliferating and Quiescent Mammary Epithelial Cells. Cell Metab. 2016, 23, 867–880. [Google Scholar] [CrossRef] [PubMed]
- Son, J.; Lyssiotis, C.A.; Ying, H.; Wang, X.; Hua, S.; Ligorio, M.; Perera, R.M.; Ferrone, C.R.; Mullarky, E.; Shyh-Chang, N.; et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature 2013, 496, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, L.B.; Luengo, A.; Danai, L.V.; Bush, L.N.; Diehl, F.F.; Hosios, A.M.; Lau, A.N.; Elmiligy, S.; Malstrom, S.; Lewis, C.A.; et al. Aspartate is an endogenous metabolic limitation for tumour growth. Nat. Cell Biol. 2018, 20, 782–788. [Google Scholar] [CrossRef]
- Garcia-Bermudez, J.; Baudrier, L.; La, K.; Zhu, X.G.; Fidelin, J.; Sviderskiy, V.O.; Papagiannakopoulos, T.; Molina, H.; Snuderl, M.; Lewis, C.A.; et al. Aspartate is a limiting metabolite for cancer cell proliferation under hypoxia and in tumours. Nat. Cell Biol. 2018, 20, 775–781. [Google Scholar] [CrossRef]
- Alkan, H.F.; Walter, K.E.; Luengo, A.; Madreiter-Sokolowski, C.T.; Stryeck, S.; Lau, A.N.; Al-Zoughbi, W.; Lewis, C.A.; Thomas, C.J.; Hoefler, G.; et al. Cytosolic Aspartate Availability Determines Cell Survival When Glutamine Is Limiting. Cell Metab. 2018, 28, 706–720.e6. [Google Scholar] [CrossRef] [Green Version]
- Birsoy, K.; Wang, T.; Chen, W.W.; Freinkman, E.; Abu-Remaileh, M.; Sabatini, D.M. An Essential Role of the Mitochondrial Electron Transport Chain in Cell Proliferation Is to Enable Aspartate Synthesis. Cell 2015, 162, 540–551. [Google Scholar] [CrossRef] [Green Version]
- Bertero, T.; Oldham, W.M.; Grasset, E.M.; Bourget, I.; Boulter, E.; Pisano, S.; Hofman, P.; Bellvert, F.; Meneguzzi, G.; Bulavin, D.V.; et al. Tumor-Stroma Mechanics Coordinate Amino Acid Availability to Sustain Tumor Growth and Malignancy. Cell Metab. 2019, 29, 124–140.e10. [Google Scholar] [CrossRef]
- Tessem, M.B.; Swanson, M.G.; Keshari, K.R.; Albers, M.J.; Joun, D.; Tabatabai, Z.L.; Simko, J.P.; Shinohara, K.; Nelson, S.J.; Vigneron, D.B.; et al. Evaluation of lactate and alanine as metabolic biomarkers of prostate cancer using 1H HR-MAS spectroscopy of biopsy tissues. Magn. Reson. Med. 2008, 60, 510–516. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Stampouloglou, E.; Kingston, N.M.; Zhang, L.; Monti, S.; Varelas, X. Glutamine-utilizing transaminases are a metabolic vulnerability of TAZ/YAP-activated cancer cells. EMBO Rep. 2018, 19, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Li, B.S.; Gu, L.J.; Luo, C.Y.; Li, W.S.; Jiang, L.M.; Shen, S.H.; Jiang, H.; Shen, S.H.; Zhang, B.; Chen, J.; et al. The downregulation of asparagine synthetase expression can increase the sensitivity of cells resistant to L-asparaginase [13]. Leukemia 2006, 20, 2199–2201. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Fan, J.; Venneti, S.; Cross, J.R.; Takagi, T.; Bhinder, B.; Djaballah, H.; Kanai, M.; Cheng, E.H.; Judkins, A.R.; et al. Asparagine plays a critical role in regulating cellular adaptation to glutamine depletion. Mol. Cell 2014, 56, 205–218. [Google Scholar] [CrossRef] [PubMed]
- Wojcik, M.; Seidle, H.F.; Bieganowski, P.; Brenner, C. Glutamine-dependent NAD+ Synthetase: How a two-domain, three-substrate enzyme avoids waste. J. Biol. Chem. 2006, 281, 33395–33402. [Google Scholar] [CrossRef] [PubMed]
- Jones, M.E. Conversion of glutamate to ornithine and proline: Pyrroline-5-carboxylate, a possible modulator of arginine requirements. J. Nutr. 1985, 115, 509–515. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, F.; Hamanaka, R.; Wheaton, W.W.; Weinberg, S.; Joseph, J.; Lopez, M.; Kalyanaraman, B.; Mutlu, G.M.; Budinger, G.R.S.; Chandel, N.S. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc. Natl. Acad. Sci. USA 2010, 107, 8788–8793. [Google Scholar] [CrossRef] [Green Version]
- Hamanaka, R.B.; Chandel, N.S. Mitochondrial reactive oxygen species regulate cellular signaling and dictate biological outcomes. Trends Biochem. Sci. 2010, 35, 505–513. [Google Scholar] [CrossRef] [Green Version]
- Badur, M.G.; Muthusamy, T.; Parker, S.J.; Ma, S.; McBrayer, S.K.; Cordes, T.; Magana, J.H.; Guan, K.L.; Metallo, C.M. Oncogenic R132 IDH1 Mutations Limit NADPH for De Novo Lipogenesis through (D)2-Hydroxyglutarate Production in Fibrosarcoma Sells. Cell Rep. 2018, 25, 1018–1026.e4. [Google Scholar] [CrossRef]
- Fack, F.; Tardito, S.; Hochart, G.; Oudin, A.; Zheng, L.; Fritah, S.; Golebiewska, A.; Nazarov, P.V.; Bernard, A.; Hau, A.; et al. Altered metabolic landscape in IDH -mutant gliomas affects phospholipid, energy, and oxidative stress pathways. EMBO Mol. Med. 2017, 9, 1681–1695. [Google Scholar] [CrossRef]
- Godwin, A.K.; Meister, A.; O’Dwyer, P.J.; Huang, C.S.; Hamilton, T.C.; Anderson, M.E. High resistance to cisplatin in human ovarian cancer cell lines is associated with marked increase of glutathione synthesis. Proc. Natl. Acad. Sci. USA 1992, 89, 3070–3074. [Google Scholar] [CrossRef] [PubMed]
- Boysen, G. The Glutathione Conundrum: Stoichiometric Disconnect between Its Formation and Oxidative Stress. Chem. Res. Toxicol. 2017, 30, 1113–1116. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Shestov, A.A.; Swain, P.; Yang, C.; Parker, S.J.; Wang, Q.A.; Terada, L.S.; Adams, N.D.; McCabe, M.T.; Pietrak, B.; et al. Reductive carboxylation supports redox homeostasis during anchorage-independent growth. Nature 2016, 532, 255–258. [Google Scholar] [CrossRef] [PubMed]
- Yuneva, M.O.; Fan, T.W.M.; Allen, T.D.; Higashi, R.M.; Ferraris, D.V.; Tsukamoto, T.; Matés, J.M.; Alonso, F.J.; Wang, C.; Seo, Y.; et al. The metabolic profile of tumors depends on both the responsible genetic lesion and tissue type. Cell Metab. 2012, 15, 157–170. [Google Scholar] [CrossRef] [PubMed]
- Cheng, T.; Sudderth, J.; Yang, C.; Mullen, A.R.; Jin, E.S.; Matés, J.M.; DeBerardinis, R.J. Pyruvate carboxylase is required for glutamine-independent growth of tumor cells. Proc. Natl. Acad. Sci. USA 2011, 108, 8674–8679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sellers, K.; Fox, M.P.; Bousamra, M.; Slone, S.P.; Higashi, R.M.; Miller, D.M.; Wang, Y.; Yan, J.; Yuneva, M.O.; Deshpande, R. Pyruvate carboxylase is critical for non-small-cell lung cancer proliferation. J Clin Invest 2015, 125, 687–698. [Google Scholar] [CrossRef] [PubMed]
- Christen, S.; Lorendeau, D.; Schmieder, R.; Broekaert, D.; Metzger, K.; Veys, K.; Elia, I.; Buescher, J.M.; Orth, M.F.; Davidson, S.M.; et al. Breast Cancer-Derived Lung Metastases Show Increased Pyruvate Carboxylase-Dependent Anaplerosis. Cell Rep. 2016, 17, 837–848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marin-Valencia, I.; Yang, C.; Mashimo, T.; Cho, S.; Baek, H.; Yang, X.L.; Rajagopalan, K.N.; Maddie, M.; Vemireddy, V.; Zhao, Z.; et al. Analysis of tumor metabolism reveals mitochondrial glucose oxidation in genetically diverse human glioblastomas in the mouse brain in vivo. Cell Metab. 2012, 15, 827–837. [Google Scholar] [CrossRef]
- Tardito, S.; Oudin, A.; Ahmed, S.U.; Fack, F.; Keunen, O.; Zheng, L.; Miletic, H.; Sakariassen, P.Ø.; Weinstock, A.; Wagner, A.; et al. Glutamine synthetase activity fuels nucleotide biosynthesis and supports growth of glutamine-restricted glioblastoma. Nat. Cell Biol. 2015, 17. [Google Scholar] [CrossRef]
- Dranoff, G.; Elion, G.B.; Friedman, H.S.; Le, G.; Campbell, M.; Bigner, D.D. Influence of Glutamine on the Growth of Human Glioma and Medulloblastoma in Culture. Cancer Res. 1985, 45, 4077–4081. [Google Scholar]
- Stumvoll, M.; Perriello, G.; Meyer, C.; Gerich, J. Role of glutamine in human carbohydrate metabolism in kidney and other tissues. Kidney Int. 1999, 55, 778–792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tischler, M.E.; Goldberg, A.L. Leucine degradation and release of glutamine and alanine by adipose tissue. J. Biol. Chem. 1980, 255, 8074–8081. [Google Scholar] [PubMed]
- Muir, A.; Danai, L.V.; Gui, D.Y.; Waingarten, C.Y.; Lewis, C.A.; Vander Heiden, M.G. Environmental cystine drives glutamine anaplerosis and sensitizes cancer cells to glutaminase inhibition. Elife 2017, 6, 1–27. [Google Scholar] [CrossRef] [PubMed]
- Timmerman, L.A.; Holton, T.; Yuneva, M.; Louie, R.J.; Padró, M.; Daemen, A.; Hu, M.; Chan, D.A.; Ethier, S.P.; van’tVeer, L.J.; et al. Glutamine Sensitivity Analysis Identifies the xCT Antiporter as a Common Triple-Negative Breast Tumor Therapeutic Target. Cancer Cell 2013, 24, 450–465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, C.S.; Mishra, P.; Watrous, J.D.; Carelli, V.; D’Aurelio, M.; Jain, M.; Chan, D.C. The glutamate/cystine xCT antiporter antagonizes glutamine metabolism and reduces nutrient flexibility. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, M.R.; Danai, L.V.; Lewis, C.A.; Chan, S.H.; Gui, D.Y.; Kunchok, T.; Dennstedt, E.A.; Vander Heiden, M.G.; Muir, A. Quantification of microenvironmental metabolites in murine cancers reveals determinants of tumor nutrient availability. Elife 2019, 8, 1–27. [Google Scholar] [CrossRef]
- García-Canaveras, J.C.; Chen, L.; Rabinowitz, J.D. The tumor metabolic microenvironment: Lessons from lactate. Cancer Res. 2019, 79, 3155–3162. [Google Scholar] [CrossRef] [PubMed]
- Graeber, M.B.; Scheithauer, B.W.; Kreutzberg, G.W. Microglia in brain tumors. Glia 2002, 40, 252–259. [Google Scholar] [CrossRef]
- Venmar, K.T.; Kimmel, D.W.; Cliffel, D.E.; Fingleton, B. IL4 receptor α mediates enhanced glucose and glutamine metabolism to support breast cancer growth. Biochim. Biophys. Acta - Mol. Cell Res. 2015, 1853, 1219–1228. [Google Scholar] [CrossRef]
- Jha, A.K.; Huang, S.C.C.; Sergushichev, A.; Lampropoulou, V.; Ivanova, Y.; Loginicheva, E.; Chmielewski, K.; Stewart, K.M.; Ashall, J.; Everts, B.; et al. Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity 2015, 42, 419–430. [Google Scholar] [CrossRef]
- Palmieri, E.M.; Menga, A.; Martín-Pérez, R.; Quinto, A.; Riera-Domingo, C.; De Tullio, G.; Hooper, D.C.; Lamers, W.H.; Ghesquière, B.; McVicar, D.W.; et al. Pharmacologic or Genetic Targeting of Glutamine Synthetase Skews Macrophages toward an M1-like Phenotype and Inhibits Tumor Metastasis. Cell Rep. 2017, 20, 1654–1666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakaya, M.; Xiao, Y.; Zhou, X.; Chang, J.H.; Chang, M.; Cheng, X.; Blonska, M.; Lin, X.; Sun, S.C. Inflammatory T cell responses rely on amino acid transporter ASCT2 facilitation of glutamine uptake and mTORC1 kinase activation. Immunity 2014, 40, 692–705. [Google Scholar] [CrossRef] [PubMed]
- Johnson, M.O.; Wolf, M.M.; Madden, M.Z.; Andrejeva, G.; Sugiura, A.; Contreras, D.C.; Maseda, D.; Liberti, M.V.; Paz, K.; Kishton, R.J.; et al. Distinct Regulation of Th17 and Th1 Cell Differentiation by Glutaminase-Dependent Metabolism. Cell 2018, 175, 1780–1795.e19. [Google Scholar] [CrossRef] [PubMed]
- Kelloff, G.J.; Hoffman, J.M.; Johnson, B.; Scher, H.I.; Siegel, B.A.; Cheng, E.Y.; Cheson, B.D.; O’Shaughnessy, J.; Guyton, K.Z.; Mankoff, D.A.; et al. Progress and promise of FDG-PET imaging for cancer patient management and oncologic drug development. Clin. Cancer Res. 2005, 11, 2785–2808. [Google Scholar] [CrossRef] [PubMed]
- Petrirena, G.J.; Goldman, S.; Delattre, J.Y. Advances in PET imaging of brain tumors: A referring physician’s perspective. Curr. Opin. Oncol. 2011, 23, 617–623. [Google Scholar] [CrossRef] [PubMed]
- Kläsner, B.D.; Krause, B.J.; Beer, A.J.; Drzezga, A. PET imaging of gliomas using novel tracers: A sleeping beauty waiting to be kissed. Expert Rev. Anticancer Ther. 2010, 10, 609–613. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Zha, Z.; Li, G.; Lieberman, B.P.; Choi, S.R.; Ploessl, K.; Kung, H.F. [18 F](2S,4S)-4-(3-Fluoropropyl)glutamine as a tumor imaging agent. Mol. Pharm. 2014, 11, 3852–3866. [Google Scholar] [CrossRef] [PubMed]
- Qu, W.; Zha, Z.; Ploessl, K.; Lieberman, B.P.; Zhu, L.; Wise, D.R.; Thompson, C.B.; Kung, H.F. Synthesis of optically pure 4-fluoro-glutamines as potential metabolic imaging agents for tumors. J. Am. Chem. Soc. 2011, 133, 1122–1133. [Google Scholar] [CrossRef] [PubMed]
- Lieberman, B.P.; Ploessl, K.; Wang, L.; Qu, W.; Zha, Z.; Wise, D.R.; Chodosh, L.A.; Belka, G.; Thompson, C.B.; Kung, H.F. PET imaging of glutaminolysis in tumors by 18F-(2S,4R)4- fluoroglutamine. J. Nucl. Med. 2011, 52, 1947–1955. [Google Scholar] [CrossRef]
- Qu, W.; Oya, S.; Lieberman, B.P.; Ploessl, K.; Wang, L.; Wise, D.R.; Divgi, C.R.; Chodosh, L.P.; Thompson, C.B.; Kung, H.F. Preparation and characterization of L-[5- 11C]-glutamine for metabolic imaging of tumors. J. Nucl. Med. 2012, 53, 98–105. [Google Scholar] [CrossRef]
- Ploessl, K.; Wang, L.; Lieberman, B.P.; Qu, W.; Kung, H.F. Comparative evaluation of 18F-labeled glutamic acid and glutamine as tumor metabolic imaging agents. J. Nucl. Med. 2012, 53, 1616–1624. [Google Scholar] [CrossRef] [PubMed]
- Venneti, S.; Dunphy, M.P.; Zhang, H.; Pitter, K.L.; Zanzonico, P.; Campos, C.; Carlin, S.D.; La Rocca, G.; Lyashchenko, S.; Ploessl, K.; et al. Glutamine-based PET imaging facilitates enhanced metabolic evaluation of gliomas in vivo. Sci. Transl. Med. 2015, 7, 274ra17. [Google Scholar] [CrossRef] [PubMed]
- Dunphy, M.P.S.; Harding, J.J.; Venneti, S.; Zhang, H.; Burnazi, E.M.; Bromberg, J.; Omuro, A.M.; Hsieh, J.J.; Mellinghoff, I.K.; Staton, K.; et al. In Vivo PET Assay of Tumor Glutamine Flux and Metabolism: In-Human Trial of 18 F-(2 S,4 R )-4-Fluoroglutamine. Radiology 2018, 287, 667–675. [Google Scholar] [CrossRef] [PubMed]
- Mittra, E.S.; Koglin, N.; Mosci, C.; Kumar, M.; Hoehne, A.; Keu, K.V.; Iagaru, A.H.; Mueller, A.; Berndt, M.; Bullich, S.; et al. Pilot preclinical and clinical evaluation of (4S)-4-(3-[18F]Fluoropropyl)-L-Glutamate (18F-FSPG) for PET/CT imaging of intracranial malignancies. PLoS ONE 2016, 11, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Koglin, N.; Mueller, A.; Berndt, M.; Schmitt-Willich, H.; Toschi, L.; Stephens, A.W.; Gekeler, V.; Friebe, M.; Dinkelborg, L.M. Specific PET imaging of x C- transporter activity using a 18 F-labeled glutamate derivative reveals a dominant pathway in tumor metabolism. Clin. Cancer Res. 2011, 17, 6000–6011. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Ploessl, K.; Zhou, R.; Mankoff, D.; Kung, H.F. Metabolic Imaging of Glutamine in Cancer. J. Nucl. Med. 2017, 58, 533–537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glunde, K.; Bhujwalla, Z.M. Metabolic tumor imaging using magnetic resonance spectroscopy. Semin. Oncol. 2011, 38, 26–41. [Google Scholar] [CrossRef]
- Griffin, J.L.; Shockcor, J.P. Metabolic profiles of cancer cells. Nat. Rev. Cancer 2004, 4, 551–561. [Google Scholar] [CrossRef]
- Majós, C.; Julià-Sapé, M.; Alonso, J.; Serrallonga, M.; Aguilera, C.; Acebes, J.J.; Arús, C.; Gili, J. Brain tumor classification by proton MR spectroscopy: Comparison of diagnostic accuracy at short and long TE. Am. J. Neuroradiol. 2004, 25, 1696–1704. [Google Scholar]
- Wilson, M.; Gill, S.K.; MacPherson, L.; English, M.; Arvanitis, T.N.; Peet, A.C. Noninvasive detection of glutamate predicts survival in pediatric medulloblastoma. Clin. Cancer Res. 2014, 20, 4532–4539. [Google Scholar] [CrossRef]
- Chawla, S.; Oleaga, L.; Wang, S.; Krejza, J.; Wolf, R.L.; Woo, J.H.; O’Rourke, D.M.; Judy, K.D.; Grady, M.S.; Melhem, E.R.; et al. Role of proton magnetic resonance spectroscopy in differentiating oligodendrogliomas from astrocytomas: Clinical investigative study. J. Neuroimaging 2010, 20, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Chawla, S.; Wang, S.; Wolf, R.L.; Woo, J.H.; Wang, J.; O’Rourke, D.M.; Judy, K.D.; Grady, M.S.; Melhem, E.R.; Poptani, H. Arterial spin-labeling and MR spectroscopy in the differentiation of gliomas. Am. J. Neuroradiol. 2007, 28, 1683–1689. [Google Scholar] [CrossRef] [PubMed]
- Buescher, J.M.; Antoniewicz, M.R.; Boros, L.G.; Burgess, S.C.; Brunengraber, H.; Clish, C.B.; DeBerardinis, R.J.; Feron, O.; Frezza, C.; Ghesquiere, B.; et al. A roadmap for interpreting 13 C metabolite labeling patterns from cells. Curr. Opin. Biotechnol. 2015, 34, 189–201. [Google Scholar] [CrossRef] [PubMed]
- Ardenkjaer-Larsen, J.H.; Fridlund, B.; Gram, A.; Hansson, G.; Hansson, L.; Lerche, M.H.; Servin, R.; Thaning, M.; Golman, K. Increase in signal-to-noise ratio of > 10,000 times in liquid-state NMR. Proc. Natl. Acad. Sci. USA 2003, 100, 10158–10163. [Google Scholar] [CrossRef] [PubMed]
- Nelson, S.J.; Kurhanewicz, J.; Vigneron, D.B.; Larson, P.E.Z.; Harzstark, A.L.; Ferrone, M.; Van Criekinge, M.; Chang, J.W.; Bok, R.; Park, I.; et al. Metabolic imaging of patients with prostate cancer using hyperpolarized [1-13C]pyruvate. Sci. Transl. Med. 2013, 5. [Google Scholar] [CrossRef] [PubMed]
- Wilson, D.M.; Kurhanewicz, J. Hyperpolarized 13 C MR for molecular imaging of prostate cancer. J. Nucl. Med. 2014, 55, 1567–1572. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, F.A.; Kettunen, M.I.; Day, S.E.; Lerche, M.; Brindle, K.M. 13C MR spectroscopy measurements of glutaminase activity in human hepatocellular carcinoma cells using hyperpolarized 13C-labeled glutamine. Magn. Reson. Med. 2008, 60, 253–257. [Google Scholar] [CrossRef] [PubMed]
- Cabella, C.; Karlsson, M.; Canapè, C.; Catanzaro, G.; Serra, S.C.; Miragoli, L.; Poggi, L.; Uggeri, F.; Venturi, L.; Jensen, P.R.; et al. In vivo and in vitro liver cancer metabolism observed with hyperpolarized [5-13C]glutamine. J. Magn. Reson. 2013, 232, 45–52. [Google Scholar] [CrossRef]
- Brindle, K.M.; Bohndiek, S.E.; Gallagher, F.A.; Kettunen, M.I. Tumor imaging using hyperpolarized 13C magnetic resonance spectroscopy. Magn. Reson. Med. 2011, 66, 505–519. [Google Scholar] [CrossRef]
- Hassanein, M.; Hoeksema, M.D.; Shiota, M.; Qian, J.; Harris, B.K.; Chen, H.; Clark, J.E.; Alborn, W.E.; Eisenberg, R.; Massion, P.P. SLC1A5 mediates glutamine transport required for lung cancer cell growth and survival. Clin. Cancer Res. 2013, 19, 560–570. [Google Scholar] [CrossRef]
- Hassanein, M.; Qian, J.; Hoeksema, M.D.; Wang, J.; Jacobovitz, M.; Ji, X.; Harris, F.T.; Harris, B.K.; Boyd, K.L.; Chen, H.; et al. Targeting SLC1a5-mediated glutamine dependence in non-small cell lung cancer. Int. J. Cancer 2015, 137, 1587–1597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schulte, M.L.; Fu, A.; Zhao, P.; Li, J.; Geng, L.; Smith, S.T.; Kondo, J.; Coffey, R.J.; Johnson, M.O.; Rathmell, J.C.; et al. Pharmacological blockade of ASCT2-dependent glutamine transport leads to antitumor efficacy in preclinical models. Nat. Med. 2018, 24, 194–202. [Google Scholar] [CrossRef] [PubMed]
- Hanaford, A.R.; Alt, J.; Rais, R.; Wang, S.Z.; Kaur, H.; Thorek, D.L.J.; Eberhart, C.G.; Slusher, B.S.; Martin, A.M.; Raabe, E.H. Orally bioavailable glutamine antagonist prodrug JHU-083 penetrates mouse brain and suppresses the growth of MYC-driven medulloblastoma. Transl. Oncol. 2019, 12, 1314–1322. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.M.; McBryant, S.J.; Tsukamoto, T.; Rojas, C.; Ferraris, D.V.; Hamilton, S.K.; Hansen, J.C.; Curthoys, N.P. Novel mechanism of inhibition of rat kidney-type glutaminase by bis-2-(5-phenylacetamido-1,2,4-thiadiazol-2-yl)ethyl sulfide (BPTES). Biochem. J. 2007, 406, 407–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.B.; Erickson, J.W.; Fuji, R.; Ramachandran, S.; Gao, P.; Dinavahi, R.; Wilson, K.F.; Ambrosio, A.L.B.; Dias, S.M.G.; Dang, C.V.; et al. Targeting mitochondrial glutaminase activity inhibits oncogenic transformation. Cancer Cell 2010, 18, 207–219. [Google Scholar] [CrossRef]
- Gross, M.I.; Demo, S.D.; Dennison, J.B.; Chen, L.; Chernov-Rogan, T.; Goyal, B.; Janes, J.R.; Laidig, G.J.; Lewis, E.R.; Li, J.; et al. Antitumor activity of the glutaminase inhibitor CB-839 in triple-negative breast cancer. Mol. Cancer Ther. 2014, 13, 890–901. [Google Scholar] [CrossRef] [PubMed]
- Delabarre, B.; Gross, S.; Fang, C.; Gao, Y.; Jha, A.; Jiang, F.; Song J., J.; Wei, W.; Hurov, J.B. Full-length human glutaminase in complex with an allosteric inhibitor. Biochemistry 2011, 50, 10764–10770. [Google Scholar] [CrossRef] [PubMed]
- Jacque, N.; Ronchetti, A.M.; Larrue, C.; Meunier, G.; Birsen, R.; Willems, L.; Saland, E.; Decroocq, J.; Maciel, T.T.; Lambert, M.; et al. Targeting glutaminolysis has antileukemic activity in acute myeloid leukemia and synergizes with BCL-2 inhibition. Blood 2015, 126, 1346–1356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meric-Bernstam, F.; DeMichele, A.; Telli, M.L.; Munster, P.; Orford, K.W.; Demitri, G.D.; Schwartz, G.K.; Iliopoulos, O.; Mier, J.W.; Owonikoko, T.K.; et al. Abstract C49: Phase 1 study of CB-839, a first-in-class, orally administered small molecule inhibitor of glutaminase in patients with refractory solid tumors. AACR J. 2015, 14, C49. [Google Scholar]
- Biancur, D.E.; Paulo, J.A.; Małachowska, B.; Del Rey, M.Q.; Sousa, C.M.; Wang, X.; Sohn, A.S.W.; Chu, G.C.; Gygi, S.P.; Harper, J.W.; et al. Compensatory metabolic networks in pancreatic cancers upon perturbation of glutamine metabolism. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef]
- Li, C.; Allen, A.; Kwagh, J.; Doliba, N.M.; Qin, W.; Najafi, H.; Collins, H.W.; Matschinsky, F.M.; Stanley, C.A.; Smith, T.J. Green tea polyphenols modulate insulin secretion by inhibiting glutamate dehydrogenase. J. Biol. Chem. 2006, 281, 10214–10221. [Google Scholar] [CrossRef] [PubMed]
- Korangath, P.; Teo, W.W.; Sadik, H.; Han, L.; Mori, N.; Huijts, C.M.; Wildes, F.; Bharti, S.; Zhang, Z.; Santa-Maria, C.A.; et al. Targeting glutamine metabolism in breast cancer with aminooxyacetate. Clin. Cancer Res. 2015, 21, 3263–3273. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, E.L.; Carroll, P.A.; Thalhofer, A.B.; Lagunoff, M. Latent KSHV Infected Endothelial Cells Are Glutamine Addicted and Require Glutaminolysis for Survival. PLoS Pathog. 2015, 11, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Vandekeere, S.; Kalucka, J.; Bierhansl, L.; Zecchin, A.; Brüning, U.; Visnagri, A.; Yuldasheva, N.; Goveia, J.; Cruys, B.; et al. Role of glutamine and interlinked asparagine metabolism in vessel formation. EMBO J. 2017, 36, 2334–2352. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| Class | Drug | Status |
|---|---|---|
| Glutamine mimics | DON JHU–083 [164] Azaserine Acivicin | Limited by off target toxicity |
| Glutaminase (GLS) inhibitors | Compound 968 [166] CB-839 [167] BPTES [165,168] | Preclinical tool Successful in Phase I clinical trials [NCT02071927, NCT02071888] Preclinical tool |
| SLC1A5 inhibition | V-9302 [163] Benzylserine GPNA γ-FBP | Effective, preclinical tools |
| Glutamine depletion | L–Asparginase | Clinically used for hematological malignancies Limited by toxicity |
| Glutamate dehydrogenase (GLUD) inhibitors | EGCG R162 | Preclinical tool compounds |
| Aminotransferase inhibitors | AOA | Used for tinnitus treatment Limited by toxicity |
| SLC7A11 or xCT inhibitors | Sulfasalazine Erastin | Pre-clinical tools Potent inducers of ferroptosis |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Natarajan, S.K.; Venneti, S. Glutamine Metabolism in Brain Tumors. Cancers 2019, 11, 1628. https://doi.org/10.3390/cancers11111628
Natarajan SK, Venneti S. Glutamine Metabolism in Brain Tumors. Cancers. 2019; 11(11):1628. https://doi.org/10.3390/cancers11111628
Chicago/Turabian StyleNatarajan, Siva Kumar, and Sriram Venneti. 2019. "Glutamine Metabolism in Brain Tumors" Cancers 11, no. 11: 1628. https://doi.org/10.3390/cancers11111628
APA StyleNatarajan, S. K., & Venneti, S. (2019). Glutamine Metabolism in Brain Tumors. Cancers, 11(11), 1628. https://doi.org/10.3390/cancers11111628