Myeloid-Derived Suppressor Cells and Pancreatic Cancer: Implications in Novel Therapeutic Approaches



 and
and 
Abstract
:1. Introduction
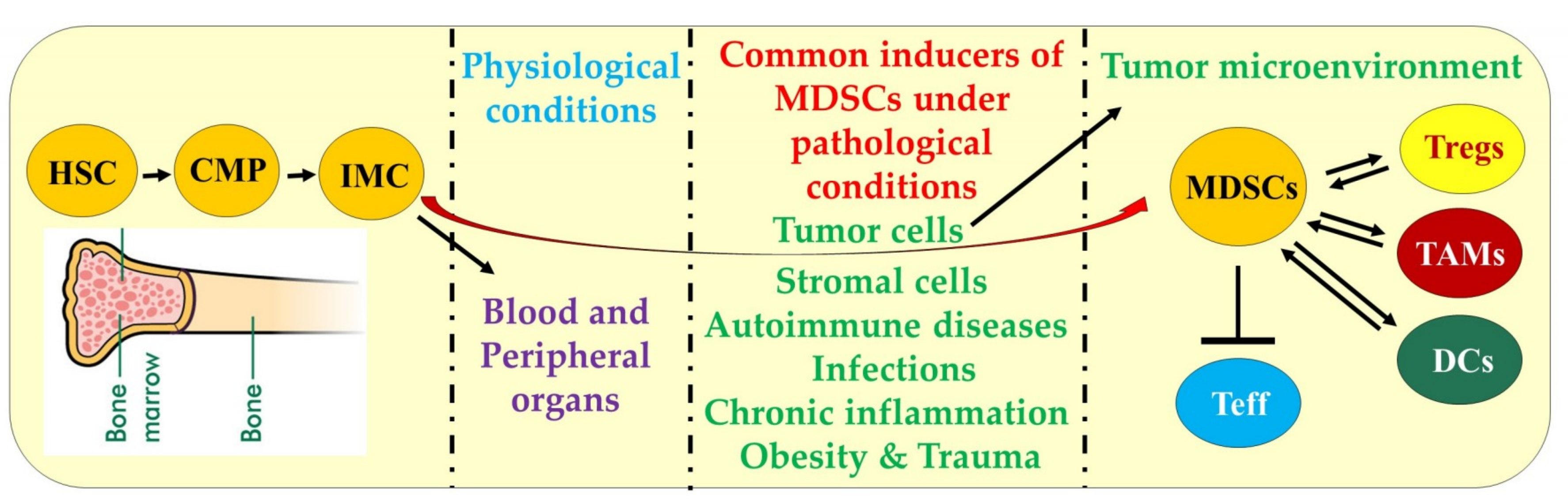
2. Differentiation, Characteristics, and Mechanisms of MDSCs Function
3. Preclinical Studies Delineating the Significance of MDSCs in Pancreatic Cancer and Therapies
3.1. Genetically Engineered Mouse Models of Pancreatic Cancer in MDSCs Studies
3.2. Immune Mediators Affecting MDSCs Function, and Approaches Targeting MDSCs in Mice Models Including GEMM
4. Clinical Studies Delineating the Role of MDSCs in Pancreatic Cancer and Therapies
4.1. Characterization of MDSCs in PDAC Patients
4.2. Effects of Therapeutic Agents on MDSCs Levels in PDAC Patients
5. Conclusions
Funding
Conflicts of Interest
References
- Becker, A.E.; Hernandez, Y.G.; Frucht, H.; Lucas, A.L. Pancreatic ductal adenocarcinoma: Risk factors, screening, and early detection. World J. Gastroenterol. 2014, 20, 11182–11198. [Google Scholar] [CrossRef] [PubMed]
- Pal, S.K.; Miller, M.J.; Agarwal, N.; Chang, S.M.; Chavez-MacGregor, M.; Cohen, E.; Cole, S.; Dale, W.; Magid Diefenbach, C.S.; Disis, M.L.; et al. Clinical cancer advances 2019: Annual report on progress against cancer from the American Society of Clinical Oncology. J. Clin. Oncol. 2019, 37, 834–849. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Zeng, L.; Chen, Y.; Lian, G.; Qian, C.; Chen, S.; Li, J.; Huang, K. Pancreatic cancer epidemiology, detection, and management. Gastroenterol. Res. Pract. 2016, 2016, 8962321. [Google Scholar] [CrossRef] [PubMed]
- McGuigan, A.; Kelly, P.; Turkington, R.C.; Jones, C.; Coleman, H.G.; McCain, R.S. Pancreatic cancer: A review of clinical diagnosis, epidemiology, treatment and outcomes. World J. Gastroenterol. 2018, 24, 4846–4861. [Google Scholar] [CrossRef] [PubMed]
- Cid-Arregui, A.; Juarez, V. Perspectives in the treatment of pancreatic adenocarcinoma. World J. Gastroenterol. 2015, 21, 9297–9316. [Google Scholar] [CrossRef]
- Bullock, A.; Stuart, K.; Jacobus, S.; Abrams, T.; Wadlow, R.; Goldstein, M.; Miksad, R. Capecitabine and oxaliplatin as first and second line treatment for locally advanced and metastatic pancreatic ductal adenocarcinoma. J. Gastrointest. Oncol. 2017, 8, 945–952. [Google Scholar] [CrossRef] [Green Version]
- Chung, K.H.; Ryu, J.K.; Son, J.H.; Lee, J.W.; Jang, D.K.; Lee, S.H.; Kim, Y.T. Efficacy of capecitabine plus oxaliplatin combination chemotherapy for advanced pancreatic cancer after failure of first-line gemcitabine-based therapy. Gut Liver 2017, 11, 298–305. [Google Scholar] [CrossRef]
- Giordano, G.; Pancione, M.; Olivieri, N.; Parcesepe, P.; Velocci, M.; Di Raimo, T.; Coppola, L.; Toffoli, G.; D’Andrea, M.R. Nano albumin bound-paclitaxel in pancreatic cancer: Current evidences and future directions. World J. Gastroenterol. 2017, 23, 5875–5886. [Google Scholar] [CrossRef]
- Glassman, D.C.; Palmaira, R.L.; Covington, C.M.; Desai, A.M.; Ku, G.Y.; Li, J.; Harding, J.J.; Varghese, A.M.; O’Reilly, E.M.; Yu, K.H. Nanoliposomal irinotecan with fluorouracil for the treatment of advanced pancreatic cancer, a single institution experience. BMC Cancer 2018, 18, 693. [Google Scholar] [CrossRef]
- Isacoff, W.H.; Reber, H.A.; Bedford, R.; Hoos, W.; Rahib, L.; Upfill-Brown, A.; Donahue, T.; Hines, O.J. Low-Dose Continuous 5-Fluorouracil Combined with Leucovorin, nab-Paclitaxel, Oxaliplatin, and Bevacizumab for Patients with Advanced Pancreatic Cancer: A Retrospective Analysis. Target. Oncol. 2018, 13, 461–468. [Google Scholar] [CrossRef] [Green Version]
- Kieler, M.; Unseld, M.; Bianconi, D.; Prager, G.W. Cross-over comparison and new chemotherapy regimens in metastatic pancreatic cancer. Memo 2017, 10, 136–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palacio, S.; Hosein, P.J.; Reis, I.; Akunyili, I.I.; Ernani, V.; Pollack, T.; Macintyre, J.; Restrepo, M.H.; Merchan, J.R.; Rocha Lima, C.M. The nab-paclitaxel/gemcitabine regimen for patients with refractory advanced pancreatic adenocarcinoma. J. Gastrointest. Oncol. 2018, 9, 135–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saito, T.; Ishido, K.; Kudo, D.; Kimura, N.; Wakiya, T.; Nakayama, Y.; Hakamada, K. Combination therapy with gemcitabine and nab-paclitaxel for locally advanced unresectable pancreatic cancer. Mol. Clin. Oncol. 2017, 6, 963–967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Liu, Y.; Zhang, Y.; Shang, Y.; Gao, Q. MDSC-decreasing chemotherapy increases the efficacy of cytokine-induced killer cell immunotherapy in metastatic renal cell carcinoma and pancreatic cancer. Oncotarget 2016, 7, 4760–4769. [Google Scholar] [CrossRef]
- Khaled, Y.S.; Ammori, B.J.; Elkord, E. Increased levels of granulocytic myeloid-derived suppressor cells in peripheral blood and tumour tissue of pancreatic cancer patients. J. Immunol. Res. 2014, 2014, 879897. [Google Scholar] [CrossRef]
- Clark, C.E.; Hingorani, S.R.; Mick, R.; Combs, C.; Tuveson, D.A.; Vonderheide, R.H. Dynamics of the immune reaction to pancreatic cancer from inception to invasion. Cancer Res. 2007, 67, 9518–9527. [Google Scholar] [CrossRef]
- Liyanage, U.K.; Moore, T.T.; Joo, H.G.; Tanaka, Y.; Herrmann, V.; Doherty, G.; Drebin, J.A.; Strasberg, S.M.; Eberlein, T.J.; Goedegebuure, P.S.; et al. Prevalence of regulatory T cells is increased in peripheral blood and tumor microenvironment of patients with pancreas or breast adenocarcinoma. J. Immunol. 2002, 169, 2756–2761. [Google Scholar] [CrossRef]
- Ostrand-Rosenberg, S.; Sinham, P.; Beury, D.W.; Clements, V.K. Cross-talk between myeloid-derived suppressor cells (MDSC), macrophages, and dendritic cells enhances tumor-induced immune suppression. Semin. Cancer Biol. 2012, 22, 275–281. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Velez-Delgado, A.; Mathew, E.; Li, D.; Mendez, F.M.; Flannagan, K.; Rhim, A.D.; Simeone, D.M.; Beatty, G.L.; Pasca di Magliano, M. Myeloid cells are required for PD-1/PD-L1 checkpoint activation and the establishment of an immunosuppressive environment in pancreatic cancer. Gut 2017, 66, 124–136. [Google Scholar] [CrossRef]
- Konger, R.L.; Derr-Yellin, E.; Ermatov, N.; Ren, L.; Sahu, R.P. The PPARγ agonist rosiglitazone suppresses syngeneic mouse SCC (Squamous Cell Carcinoma) tumor growth through an immune-mediated mechanism. Molecules 2019, 24, 2192. [Google Scholar] [CrossRef]
- Bruno, A.; Mortara, L.; Baci, D.; Noonan, D.M.; Albini, A. Myeloid derived suppressor cells interactions with natural killer cells and pro-angiogenic activities: Roles in tumor progression. Front. Immunol. 2019, 10, 771. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, E.; Wenthe, J.; Irenaeus, S.; Loskog, A.; Ullenhag, G. Gemcitabine reduces MDSCs, tregs and TGFβ-1 while restoring the teff/treg ratio in patients with pancreatic cancer. J. Transl Med. 2016, 14, 282. [Google Scholar] [CrossRef] [PubMed]
- Faget, J.; Biota, C.; Bachelot, T.; Gobert, M.; Treilleux, I.; Goutagny, N.; Durand, I.; Léon-Goddard, S.; Blay, J.Y.; Caux, C.; et al. Early detection of tumor cells by innate immune cells leads to T(reg) recruitment through CCL22 production by tumor cells. Cancer Res. 2011, 71, 6143–6152. [Google Scholar] [CrossRef] [PubMed]
- Stromnes, I.M.; Brockenbrough, J.S.; Izeradjene, K.; Carlson, M.A.; Cuevas, C.; Simmons, R.M.; Greenberg, P.D.; Hingorani, S.R. Targeted depletion of an MDSC subset unmasks pancreatic ductal adenocarcinoma to adaptive immunity. Gut 2014, 63, 1769–1781. [Google Scholar] [CrossRef] [Green Version]
- Lu, L.C.; Chang, C.J.; Hsu, C.H. Targeting myeloid-derived suppressor cells in the treatment of hepatocellular carcinoma: Current state and future perspectives. J. Hepatocell Carcinoma 2019, 6, 71–84. [Google Scholar] [CrossRef]
- Salminen, A.; Kauppinen, A.; Kaarniranta, K. AMPK activation inhibits the functions of myeloid-derived suppressor cells (MDSC): Impact on cancer and aging. J. Mol. Med. 2019, 97, 1049–1064. [Google Scholar] [CrossRef]
- Pawelec, G.; Verschoor, C.P.; Ostrand-Rosenberg, S. Myeloid-derived suppressor cells: Not only in tumor Immunity. Front. Immunol. 2019, 10, 1099. [Google Scholar] [CrossRef]
- Consonni, F.M.; Porta, C.; Marino, A.; Pandolfo, C.; Mola, S.; Bleve, A.; Sica, A. Myeloid-derived suppressor Cells: ductile targets in disease. Front. Immunol. 2019, 10, 949. [Google Scholar] [CrossRef]
- Gabrilovich, D.I.; Nagaraj, S. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 2009, 9, 162–174. [Google Scholar] [CrossRef]
- Markowitz, J.; Brooks, T.R.; Duggan, M.C.; Paul, B.K.; Pan, X.; Wei, L.; Abrams, Z.; Luedke, E.; Lesinski, G.B.; Mundy-Bosse, B.; et al. Patients with pancreatic adenocarcinoma exhibit elevated levels of myeloid-derived suppressor cells upon progression of disease. Cancer Immunol. Immunother. 2015, 64, 149–159. [Google Scholar] [CrossRef]
- Solito, S.; Marigo, I.; Pinton, L.; Damuzzo, V.; Mandruzzato, S.; Bronte, V. Myeloid-derived suppressor cell heterogeneity in human cancers. Ann. N. Y. Acad. Sci. 2014, 1319, 47–65. [Google Scholar] [CrossRef] [PubMed]
- Gabrilovich, D.I. Myeloid-derived suppressor cells. Cancer Immunol. Res. 2017, 5, 3–8. [Google Scholar] [CrossRef]
- Veglia, F.; Perego, M.; Gabrilovich, D. Myeloid-derived suppressor cells coming of age. Nat. Immunol. 2018, 19, 108–119. [Google Scholar] [CrossRef] [PubMed]
- Growth, C.; Hu, X.; Weber, R.; Fleming, V.; Altevogt, P.; Utikal, J.; Umansky, V. Immunosuppression mediated by myeloid-derived suppressor cells (MDSCs) during tumour progression. Br. J. Cancer 2019, 120, 16–25. [Google Scholar] [CrossRef] [PubMed]
- Youn, J.I.; Collazo, M.; Shalova, I.N.; Biswas, S.K.; Gabrilovich, D.I. Characterization of the nature of granulocytic myeloid-derived suppressor cells in tumor-bearing mice. J. Leukoc. Biol. 2012, 91, 167–181. [Google Scholar] [CrossRef]
- Bronte, V.; Brandau, S.; Chen, S.H.; Colombo, M.P.; Frey, A.B.; Greten, T.F.; Mandruzzato, S.; Murray, P.J.; Ochoa, A.; Ostrand-Rosenberg, S.; et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat. Commun. 2016, 7, 12150. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez, P.C.; Ernstoff, M.S.; Hernandez, C.; Atkins, M.; Zabaleta, J.; Sierra, R.; Ochoa, A.C. Arginase I-producing myeloid-derived suppressor cells in renal cell carcinoma are a subpopulation of activated granulocytes. Cancer Res. 2009, 69, 1553–1560. [Google Scholar] [CrossRef]
- Bian, Z.; Abdelaal, A.M.; Shi, L.; Liang, H.; Xiong, L.; Kidder, K.; Venkataramani, M.; Culpepper, C.; Zen, K.; Liu, Y. Arginase-1 is neither constitutively expressed in nor required for myeloid-derived suppressor cell-mediated inhibition of T-cell proliferation. Eur. J. Immunol. 2018, 48, 1046–1058. [Google Scholar] [CrossRef]
- Zhao, F.; Obermann, S.; von Wasielewski, R.; Haile, L.; Manns, M.P.; Korangy, F.; Greten, T.F. Increase in frequency of myeloid-derived suppressor cells in mice with spontaneous pancreatic carcinoma. Immunology 2009, 128, 141–149. [Google Scholar] [CrossRef]
- Vernon, P.J.; Loux, T.J.; Schapiro, N.E.; Kang, R.; Muthuswamy, R.; Kalinski, P.; Tang, D.; Lotze, M.T.; Zeh, H.J., 3rd. The receptor for advanced glycation end products promotes pancreatic carcinogenesis and accumulation of myeloid-derived suppressor cells. J. Immunol. 2013, 190, 1372–1379. [Google Scholar] [CrossRef]
- Panni, R.Z.; Sanford, D.E.; Belt, B.A.; Mitchem, J.B.; Worley, L.A.; Goetz, B.D.; Mukherjee, P.; Wang-Gillam, A.; Link, D.C.; Denardo, D.G.; et al. Tumor-induced STAT3 activation in monocytic myeloid-derived suppressor cells enhances stemness and mesenchymal properties in human pancreatic cancer. Cancer Immunol. Immunother. 2014, 63, 513–528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karakhanova, S.; Link, J.; Heinrich, M.; Shevchenko, I.; Yang, Y.; Hassenpflug, M.; Bunge, H.; von Ahn, K.; Brecht, R.; Mathes, A.; et al. Characterization of myeloid leukocytes and soluble mediators in pancreatic cancer: Importance of myeloid-derived suppressor cells. Oncoimmunology 2015, 4, e998519. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Lee, J.; Kim, J.; Jo, S.; Kim, Y.J.; Baek, J.E.; Kwon, E.S.; Lee, K.P.; Yang, S.; Kwon, K.S.; et al. Pancreatic adenocarcinoma up-regulated factor (PAUF) enhances the accumulation and functional activity of myeloid-derived suppressor cells (MDSCs) in pancreatic cancer. Oncotarget 2016, 7, 51840–51853. [Google Scholar] [CrossRef] [PubMed]
- Vincent, J.; Mignot, G.; Chalmin, F.; Ladoire, S.; Bruchard, M.; Chevriaux, A.; Martin, F.; Apetoh, L.; Rébé, C.; Ghiringhelli, F. 5-Fluorouracil selectively kills tumor-associated myeloid-derived suppressor cells resulting in enhanced T cell-dependent antitumor immunity. Cancer Res. 2010, 70, 3052–3061. [Google Scholar] [CrossRef]
- Hasnis, E.; Alishekevitz, D.; Gingis-Veltski, S.; Bril, R; Fremder, E.; Voloshin, T.; Raviv, Z.; Karban, A.; Shaked, Y. Anti-Bv8 antibody and metronomic gemcitabine improve pancreatic adenocarcinoma treatment outcome following weekly gemcitabine therapy. Neoplasia 2014, 16, 501–510. [Google Scholar] [CrossRef]
- Cappello, P.; Tonoli, E.; Curto, R.; Giordano, D.; Giovarelli, M.; Novelli, F. Anti-α-enolase antibody limits the invasion of myeloid-derived suppressor cells and attenuates their restraining effector T cell response. Oncoimmunology 2015, 5, e1112940. [Google Scholar] [CrossRef]
- Steele, C.W.; Karim, S.A.; Leach, J.D.G.; Bailey, P.; Upstill-Goddard, R.; Rishi, L.; Foth, M.; Bryson, S.; McDaid, K.; Wilson, Z.; et al. CXCR2 inhibition profoundly suppresses metastases and augments immunotherapy in pancreatic ductal adenocarcinoma. Cancer Cell 2016, 29, 832–845. [Google Scholar] [CrossRef]
- Zheng, D.; Chen, H.; Bartee, M.Y.; Williams, J.; Davids, J.A.; Lomas, D.A.; McFadden, G.; Lucas, A.R. Myxomaviral anti-inflammatory serpin reduces myeloid-derived suppressor cells and human pancreatic cancer cell growth in mice. J. Cancer Sci. Ther. 2013, 5, 291–299. [Google Scholar]
- Kang, R.; Tang, D.; Schapiro, N.E.; Livesey, K.M.; Farkas, A.; Loughran, P.; Bierhaus, A.; Lotze, M.T.; Zeh, H.J. The receptor for advanced glycation end products (RAGE) sustains autophagy and limits apoptosis, promoting pancreatic tumor cell survival. Cell Death Differ. 2010, 17, 666–676. [Google Scholar] [CrossRef]
- Kang, R.; Loux, T.; Tang, D.; Schapiro, N.E.; Vernon, P.; Livesey, K.M.; Krasinskas, A.; Lotze, M.T.; Zeh, H.J., 3rd. The expression of the receptor for advanced glycation endproducts (RAGE) is permissive for early pancreatic neoplasia. Proc. Natl. Acad. Sci. USA 2012, 109, 7031–7036. [Google Scholar] [CrossRef] [Green Version]
- Murakami, S.; Shahbazian, D.; Surana, R.; Zhang, W.; Chen, H.; Graham, G.T.; White, S.M.; Weiner, L.M.; Yi, C. Yes-associated protein mediates immune reprogramming in pancreatic ductal adenocarcinoma. Oncogene 2017, 36, 1232–1244. [Google Scholar] [CrossRef] [PubMed]
- Clarke, M.F.; Dick, J.E.; Dirks, P.B.; Eaves, C.J.; Jamieson, C.H.; Jones, D.L.; Visvader, J.; Weissman, I.L.; Wahl, G.M. Cancer stem cells—Perspectives on current status and future directions: AACR Workshop on cancer stem cells. Cancer Res. 2006, 66, 9339–9344. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Heidt, D.G.; Dalerba, P.; Burant, C.F.; Zhang, L.; Adsay, V.; Wicha, M.; Clarke, M.F.; Simeone, DM. Identification of pancreatic cancer stem cells. Cancer Res. 2007, 67, 1030–1037. [Google Scholar] [CrossRef] [PubMed]
- Jinushi, M.; Chiba, S.; Yoshiyama, H.; Masutomi, K.; Kinoshita, I.; Dosaka-Akita, H.; Yagita, H.; Takaoka, A.; Tahara, H. Tumor-associated macrophages regulate tumorigenicity and anticancer drug responses of cancer stem/initiating cells. Proc. Natl. Acad. Sci. USA 2011, 108, 12425–12430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitchem, J.B.; Brennan, D.J.; Knolhoff, B.L.; Belt, B.A.; Zhu, Y.; Sanford, D.E.; Belaygorod, L.; Carpenter, D.; Collins, L.; Piwnica-Worms, D.; et al. Targeting tumor-infiltrating macrophages decreases tumor-initiating cells, relieves immunosuppression, and improves chemotherapeutic responses. Cancer Res. 2013, 73, 1128–1141. [Google Scholar] [CrossRef] [PubMed]
- Rasheed, Z.A.; Yang, J.; Wang, Q.; Kowalski, J.; Freed, I.; Murter, C.; Hong, S.M.; Koorstra, J.B.; Rajeshkumar, N.V.; He, X.; et al. Prognostic significance of tumorigenic cells with mesenchymal features in pancreatic adenocarcinoma. J. Natl. Cancer Inst. 2010, 102, 340–351. [Google Scholar] [CrossRef]
- Cappello, P.; Tomaino, B.; Chiarle, R.; Ceruti, P.; Novarino, A.; Castagnoli, C.; Migliorini, P.; Perconti, G.; Giallongo, A.; Milella, M.; et al. An integrated humoral and cellular response is elicited in pancreatic cancer by alpha-enolase, a novel pancreatic ductal adenocarcinoma-associated antigen. Int. J. Cancer 2009, 125, 639–648. [Google Scholar] [CrossRef]
- Cappello, P.; Rolla, S.; Chiarle, R.; Principe, M.; Cavallo, F.; Perconti, G.; Feo, S.; Giovarelli, M.; Novelli, F. Vaccination with ENO1 DNA prolongs survival of genetically engineered mice with pancreatic cancer. Gastroenterology 2013, 144, 1098–10106. [Google Scholar] [CrossRef]
- Eash, K.J.; Greenbaum, A.M.; Gopalan, P.K.; Link, D.C. CXCR2 and CXCR4 antagonistically regulate neutrophil trafficking from murine bone marrow. J. Clin. Investig. 2010, 120, 2423–2431. [Google Scholar] [CrossRef] [Green Version]
- Highfill, S.L.; Cui, Y.; Giles, A.J.; Smith, J.P.; Zhang, H.; Morse, E.; Kaplan, R.N.; Mackall, C.L. Disruption of CXCR2-mediated MDSC tumor trafficking enhances anti-PD1 efficacy. Sci. Transl. Med. 2014, 6, 237ra267. [Google Scholar] [CrossRef]
- Jamieson, T.; Clarke, M.; Steele, C.W; Samuel, M.S.; Neumann, J.; Jung, A.; Huels, D.; Olson, M.F.; Das, S.; Nibbs, R.J.; et al. Inhibition of CXCR2 profoundly suppresses inflammation-driven and spontaneous tumorigenesis. J. Clin. Investig. 2012, 122, 3127–3144. [Google Scholar] [CrossRef] [PubMed]
- Saintigny, P.; Massarelli, E.; Lin, S.; Ahn, Y.H.; Chen, Y.; Goswami, S.; Erez, B.; O’Reilly, M.S.; Liu, D.; Lee, J.J. CXCR2 expression in tumor cells is a poor prognostic factor and promotes invasion and metastasis in lung adenocarcinoma. Cancer Res. 2013, 73, 571–582. [Google Scholar] [CrossRef] [PubMed]
- Ijichi, H.; Chytil, A.; Gorska, A.E.; Aakre, M.E.; Bierie, B.; Tada, M.; Mohri, D.; Miyabayashi, K.; Asaoka, Y.; Maeda, S. Inhibiting Cxcr2 disrupts tumor-stromal interactions and improves survival in a mouse model of pancreatic ductal adenocarcinoma. J. Clin. Investig. 2011, 121, 4106–4117. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; King, J.; Moro, A.; Sugi, M.D.; Dawson, D.W.; Kaplan, J.; Li, G.; Lu, X.; Strieter, R.M.; Burdick, M. Overexpression of CXCL5 is associated with poor survival in patients with pancreatic cancer. Am. J. Pathol. 2011, 178, 1340–1349. [Google Scholar] [CrossRef]
- Dass, K.; Ahmad, A.; Azmi, A.S.; Sarkar, S.H.; Sarkar, F.H. Evolving role of uPA/uPAR system in human cancers. Cancer Treat. Rev. 2008, 34, 122–136. [Google Scholar] [CrossRef] [PubMed]
- Porembka, M.R.; Mitchem, J.B.; Belt, B.A.; Hsieh, C.S.; Lee, H.M.; Herndon, J.; Gillanders, W.E.; Linehan, D.C.; Goedegebuure, P. Pancreatic adenocarcinoma induces bone marrow mobilization of myeloid-derived suppressor cells which promote primary tumor growth. Cancer Immunol. Immunother. 2012, 61, 1373–1385. [Google Scholar] [CrossRef] [Green Version]
- Trovato, R.; Fiore, A.; Sartori, S.; Canè, S.; Giugno, R.; Cascione, L.; Paiella, S.; Salvia, R.; De Sanctis, F.; Poffe, O.; et al. Immunosuppression by monocytic myeloid-derived suppressor cells in patients with pancreatic ductal carcinoma is orchestrated by STAT3. J. Immunother. Cancer 2019, 7, 255. [Google Scholar] [CrossRef]
- Sanford, D.E.; Porembka, M.R.; Panni, R.Z.; Mitchem, J.B.; Belt, B.A.; Plambeck-Suess, S.M.; Lin, G.; Denardo, D.G.; Fields, R.C.; Hawkins, W.G.; et al. A study of zoledronic acid as neo-adjuvant, perioperative therapy in patients with resectable pancreatic ductal adenocarcinoma. J. Cancer Ther. 2013, 4, 797–803. [Google Scholar] [CrossRef]
- Annels, N.E.; Shaw, V.E.; Gabitass, R.F.; Billingham, L.; Corrie, P.; Eatock, M.; Valle, J.; Smith, D.; Wadsley, J.; Cunningham, D.; et al. The effects of gemcitabine and capecitabine combination chemotherapy and of low-dose adjuvant GM-CSF on the levels of myeloid-derived suppressor cells in patients with advanced pancreatic cancer. Cancer Immunol. Immunother. 2014, 63, 175–183. [Google Scholar] [CrossRef]
- Nagaraj, S.; Youn, J.I.; Weber, H.; Iclozan, C.; Lu, L.; Cotter, M.J.; Meyer, C.; Becerra, C.R.; Fishman, M.; Antonia, S.; et al. Anti-inflammatory triterpenoid blocks immune suppressive function of MDSCs and improves immune response in cancer. Clin. Cancer Res. 2010, 16, 1812–1823. [Google Scholar] [CrossRef]
- Dominguez, G.A.; Condamine, T.; Mony, S.; Hashimoto, A.; Wang, F.; Liu, Q.; Forero, A.; Bendell, J.; Witt, R.; Hockstein, N.; et al. Selective targeting of myeloid-derived suppressor cells in cancer patients using DS-8273a, an agonistic TRAIL-R2 antibody. Clin. Cancer Res. 2017, 23, 2942–2950. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, S.A.; Lotze, M.T.; Muul, L.M.; Leitman, S.; Chang, A.E.; Ettinghausen, S.E.; Matory, Y.L.; Skibber, J.M.; Shiloni, E.; Vetto, J.T. Observations on the systemic administration of autologous lymphokine-activated killer cells and recombinant interleukin-2 to patients with metastatic cancer. N. Engl. J. Med. 1985, 313, 1485–1492. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, S.A.; Yang, J.C.; Sherry, R.M.; Kammula, U.S.; Hughes, M.S.; Phan, G.Q.; Citrin, D.E.; Restifo, N.P.; Robbins, P.F.; Wunderlich, J.R.; et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin. Cancer Res. 2011, 17, 4550–4557. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Zhang, W.; Qi, X.; Li, H.; Yu, J.; Wei, S.; Hao, X.; Ren, X. Randomized study of autologous cytokine-induced killer cell immunotherapy in metastatic renal carcinoma. Clin. Cancer Res. 2012, 18, 1751–1759. [Google Scholar] [CrossRef]
- Schmeel, L.C.; Schmeel, F.C.; Coch, C.; Schmidt-Wolf, I.G. Cytokine-induced killer (CIK) cells in cancer immunotherapy: Report of the international registry on CIK cells (IRCC). J. Cancer Res. Clin. Oncol 2015, 141, 839–849. [Google Scholar] [CrossRef]
- Wang, Z.; Zhang, Y.; Liu, Y.; Wang, L.; Zhao, L.; Yang, T.; He, C.; Song, Y.; Gao, Q. Association of myeloid-derived suppressor cells and efficacy of cytokine-induced killer cell immunotherapy in metastatic renal cell carcinoma patients. J. Immunother. 2014, 37, 43–50. [Google Scholar] [CrossRef]
- Ling, X.; Konopleva, M.; Zeng, Z.; Ruvolo, V.; Stephens, L.C.; Schober, W.; McQueen, T.; Dietrich, M.; Madden, T.L.; Andreeff, M. The novel triterpenoid C-28 methyl ester of 2-cyano-3, 12-dioxoolen-1, 9-dien-28-oic acid inhibits metastatic murine breast tumor growth through inactivation of STAT3 signaling. Cancer Res. 2007, 67, 4210–4218. [Google Scholar] [CrossRef]
- Johnstone, R.W.; Frew, A.J.; Smyth, M.J. The TRAIL apoptotic pathway in cancer onset, progression and therapy. Nat. Rev. Cancer. 2008, 8, 782–798. [Google Scholar] [CrossRef]
- Wiezorek, J.; Holland, P.; Graves, J. Death receptor agonists as a targeted therapy for cancer. Clin. Cancer Res. 2010, 16, 1701–1708. [Google Scholar] [CrossRef]
- Condamine, T.; Kumar, V.; Ramachandran, I.R.; Youn, J.I.; Celis, E.; Finnberg, N.; El-Deiry, W.S.; Winograd, R.; Vonderheide, R.H.; English, N.R.; et al. ER stress regulates myeloid-derived suppressor cell fate through TRAIL-R-mediated apoptosis. J. Clin. Investig. 2014, 124, 2626–2639. [Google Scholar] [CrossRef]
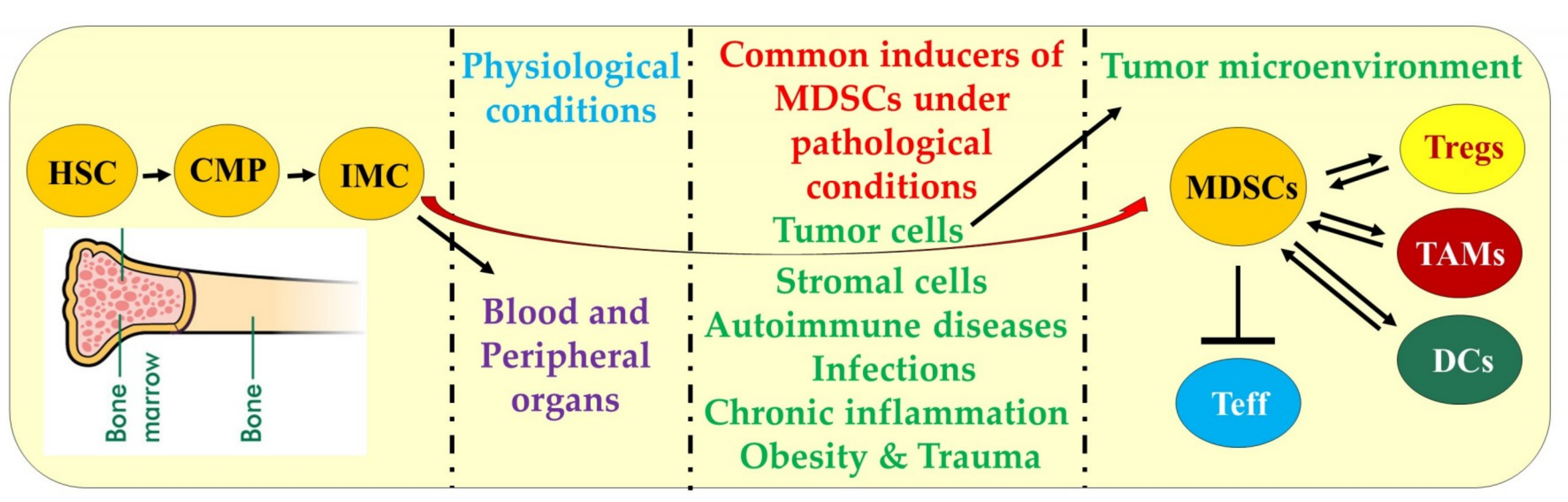
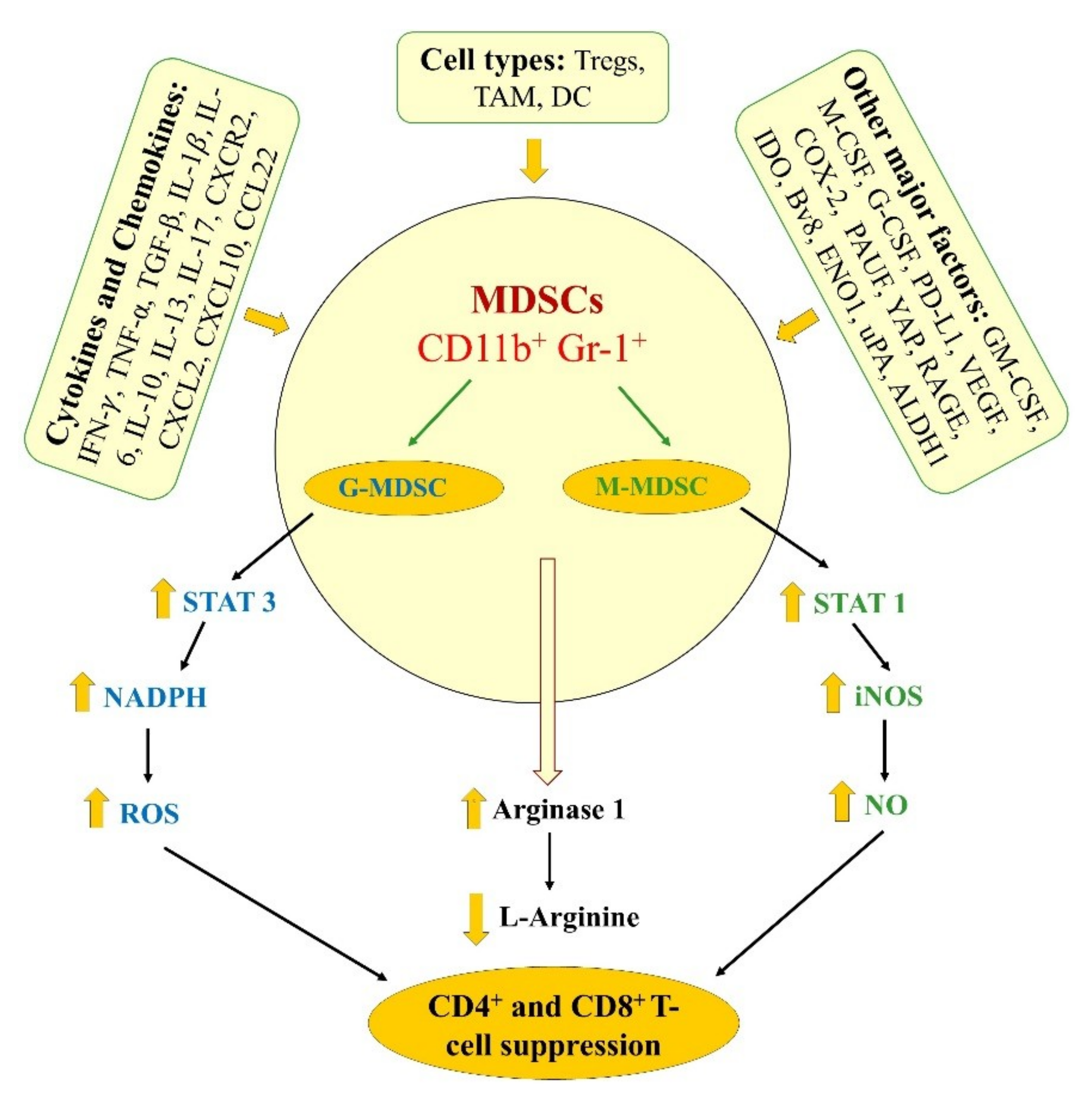
 inhibition and
inhibition and  increase or upregulation.
inhibition and increase or upregulation.
increase or upregulation.
inhibition and increase or upregulation.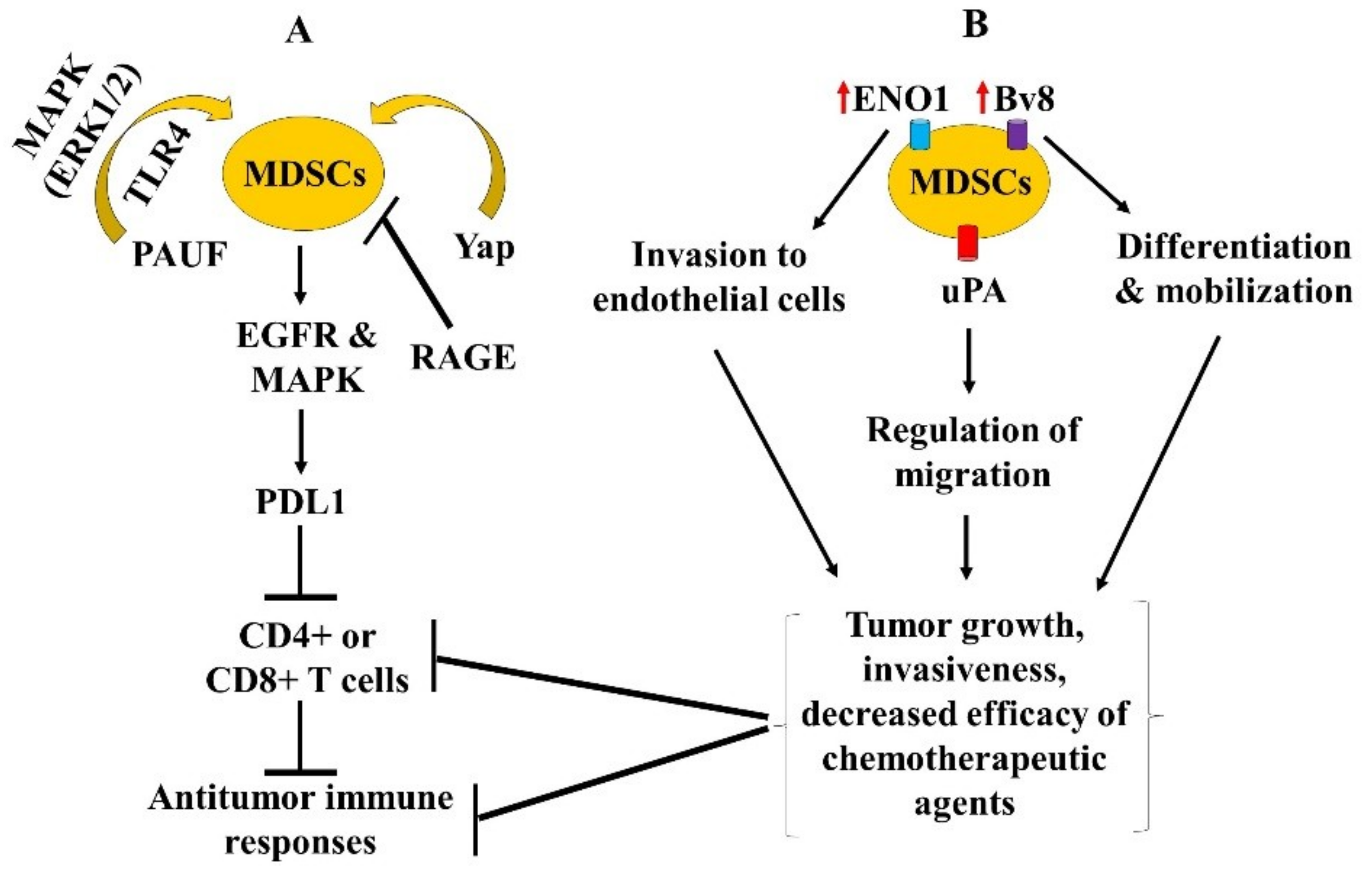

{kind=link}
{kind=link}
{kind=link}
| MDSCs | Common PMN-MDSCs Markers | Common M-MDSCs Markers | eMDSCs |
|---|---|---|---|
| Human | HLA-DR-CD33+CD11b+CD15+CD14− | HLA-DRlow-CD11b+ CD14+ CD15− | Lin−(CD3/14/15/19/56)/HLA-DR−/CD33+ |
| Murine | CD11b+Gr-1+Ly6GhighLy6Clow | CD11b+Gr-1+Ly6GlowLy6Chigh | — |
| Mouse Models | Cells Lines | Therapeutic Agent(s) | Findings | Target(s) | Ref. |
|---|---|---|---|---|---|
| CD11b-DTR, iKras*, iKras*P53*, iKras*; CD11b-DTR, iKras*; p53*; CD11b-DTR mice | Primary human (1319, UM2, UM5, UM18 and UM19) and primary mouse (iKras*1, iKRAS*2, iKras*3, 65 671, 7940B) cell lines | – | Myeloid cell subsets favor tumor immunoevasion in EGFR-MAPK-dependent regulation of tumoral PD-L1 expression and inhibition of CD8+ T cell antitumor immunity | KrasG12D, PD-L1 | [19] |
| KrasLSL-G12D/+; Cre (KC) and KrasLSL-G12D/+; Trp53LSL-R172H/+; Cre (KPC) mice | – | – | G-MDSCs inhibit T cell proliferation and induce T cell death and its depletion unmask PDAC to adaptive immune response. | – | [24] |
| EL-TGF-α/p53−/− double transgenic, EL-TGF-α/p53+/− heterozygous, C57BL/6 and BALB/c mice | Murine mPAC cell line | – | Increased frequency of MDSCs was detected at early stages of tumor development with further increased during tumor progression. | – | [39] |
| KCR, KC, RAGE-null, and C57BL/6- wild type mice | – | – | RAGE ablation resulted in the accumulation of MDSCs. | RAGE | [40] |
| GCSFR−/−, NU/J and C57BL/6-WT mice | KCM, KCKO, Panc-1, BxPC3 and Pan02 cell lines | – | STAT3 signaling in M-MDSCs promotes CSCs stemness in pancreatic cancer. | STAT3 | [41] |
| C57BL/6 mice | Murine Panc02 cell line | Sildenafil | Sildenafil treatment reduced MDSCs frequency and VEGF levels and increased the survival of tumor-bearing female mice. | VEGF | [42] |
| NOD/SCID mice | PANC-1, CFPAC-1 and EL4 cell lines | Neutralizing antibodies against PAUF and TLR4, and inhibitor of the MAPK pathway | PAUF regulates the functional activation of MDSCs via TLR4 and the MAPK-dependent pathways. | PAUF | [43] |
| TLR4−/− C57BL/6-WT and Nude mice | EL4, MSC-1 and MSC-2 cell lines | Cyclophosphamide, doxorubicin, oxaliplatin, paclitaxel, gemcitabine, 5FU, and raltitrexed | 5FU-mediated depletion of MDSCs promoted CD8+T-cell-dependent anti-tumor responses. | – | [44] |
| Severe combined immunodeficiency disease (SCID) and C57BL/6-WT mice | Human Panc-1 and Murine Panc-02 cell lines | Gemcitabine, metronomic chemotherapy (MC) with gemcitabine and anti-Bv8 antibody | MC with gemcitabine or anti-Bv8 antibody enhanced gemcitabine efficacy. | – | [45] |
| LSL-KrasG12D; Pdx-1/Cre (KC) and pdx-1/Cre (Cre) mice | Murine PDA cell line | Monoclonal antibody targeting ENO1 | Anti-ENO1 impaired MDSCs invasion and elicited sustained effector T-cell function. | ENO1 | [46] |
| KPC Cxcr2−/− and KPC mice | – | CXCR2 inhibitor, gemcitabine, and anti-PD1 immunotherapy | CXCR2 inhibition suppressed metastasis and enhanced therapeutic responses of chemotherapy and immunotherapy to prolong mice survival. | CXCR2 | [47] |
| NOD/SCID mice | Human Hs766t, and MIA PaCa-2 cell lines | Serp-1, neuroserpin, and M-T7 | Serp-1 and neuroserpin treatment reduced pancreatic tumor growth via decreasing splenic and tumoral MDSCs as well as tumor infiltration of macrophage. | uPA | [48] |
| Therapeutic Agent(s) | Findings | Ref. |
|---|---|---|
| – | MDSCs play an essential role in pancreatic cancer but were not correlated with tumor stage. | [15] |
| – | MDSCs play importance role in pancreatic cancer progression | [66] |
| – | M-MDSCs can be characterized as circulating STAT3/arginase-1-expressing CD14+ cells in pancreatic cancer patients. | [67] |
| Chemotherapy + Cytokine-induced killer cell (CIK) immunotherapy | MDSCs-targeting chemotherapy improved the survival response of CIK immunotherapy. | [14] |
| Chemotherapy | Analysis of MDSCs in peripheral blood may represent a predictive biomarker for chemotherapy failure in pancreatic cancer patients. | [30] |
| Zoledronic Acid (ZA) | No differences were observed in the prevalence of G-MDSCs in the blood and bone marrow of PDAC patients treated (pre- and post) with ZA. | [68] |
| Gemcitabine + Capecitabine alone versus GV1001 vaccine with gemcitabine + capecitabine along with GM-CSF as adjuvant | Gemcitabine and capecitabine combination did not result in a consistent reduction in MDSCs levels. High levels of MDSCs pre-vaccination do not prevent the development of an immune response to tumor antigens. | [69] |
| CDDO-Me alone and CDDO-Me combination with gemcitabine | CDDO-Me abrogated the immune suppressive effects of MDSCs and improved immune response. | [70] |
| DS-82373a, an agonistic TRAIL-R2 antibody | DS-82373a selectively reduced MDSCs subsets in peripheral blood and tumor tissues of cancer patients, including pancreatic cancer. | [71] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thyagarajan, A.; Alshehri, M.S.A.; Miller, K.L.R.; Sherwin, C.M.; Travers, J.B.; Sahu, R.P. Myeloid-Derived Suppressor Cells and Pancreatic Cancer: Implications in Novel Therapeutic Approaches. Cancers 2019, 11, 1627. https://doi.org/10.3390/cancers11111627
Thyagarajan A, Alshehri MSA, Miller KLR, Sherwin CM, Travers JB, Sahu RP. Myeloid-Derived Suppressor Cells and Pancreatic Cancer: Implications in Novel Therapeutic Approaches. Cancers. 2019; 11(11):1627. https://doi.org/10.3390/cancers11111627
Chicago/Turabian StyleThyagarajan, Anita, Mamdouh Salman A. Alshehri, Kelly L.R. Miller, Catherine M. Sherwin, Jeffrey B. Travers, and Ravi P. Sahu. 2019. "Myeloid-Derived Suppressor Cells and Pancreatic Cancer: Implications in Novel Therapeutic Approaches" Cancers 11, no. 11: 1627. https://doi.org/10.3390/cancers11111627
APA StyleThyagarajan, A., Alshehri, M. S. A., Miller, K. L. R., Sherwin, C. M., Travers, J. B., & Sahu, R. P. (2019). Myeloid-Derived Suppressor Cells and Pancreatic Cancer: Implications in Novel Therapeutic Approaches. Cancers, 11(11), 1627. https://doi.org/10.3390/cancers11111627