Deregulated Lysophosphatidic Acid Metabolism and Signaling in Liver Cancer



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction/Lysophosphatidic Acid (LPA)
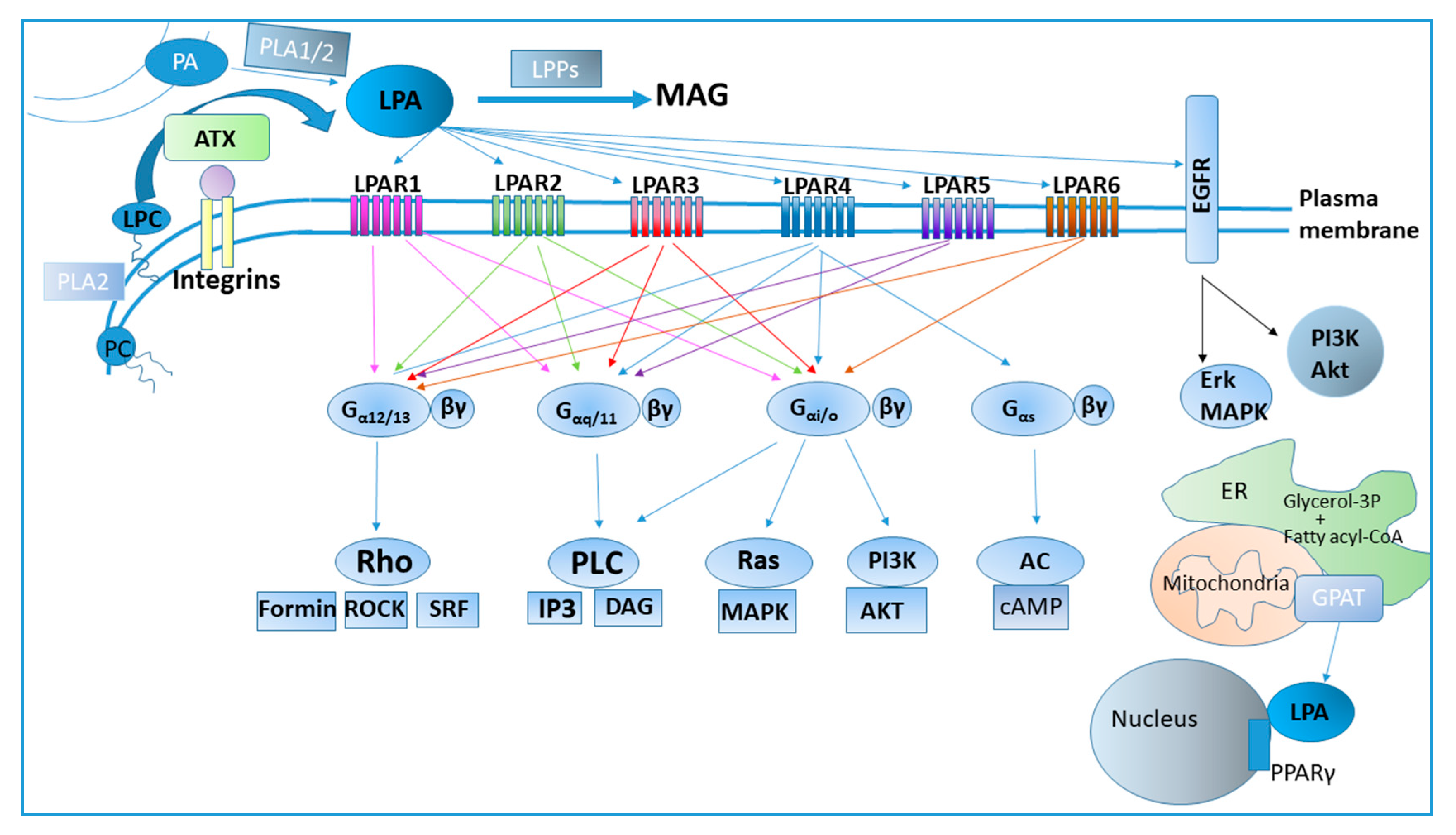
2. LPA Metabolism
3. LPA Receptors and Signaling
4. LPA in Chronic Inflammation
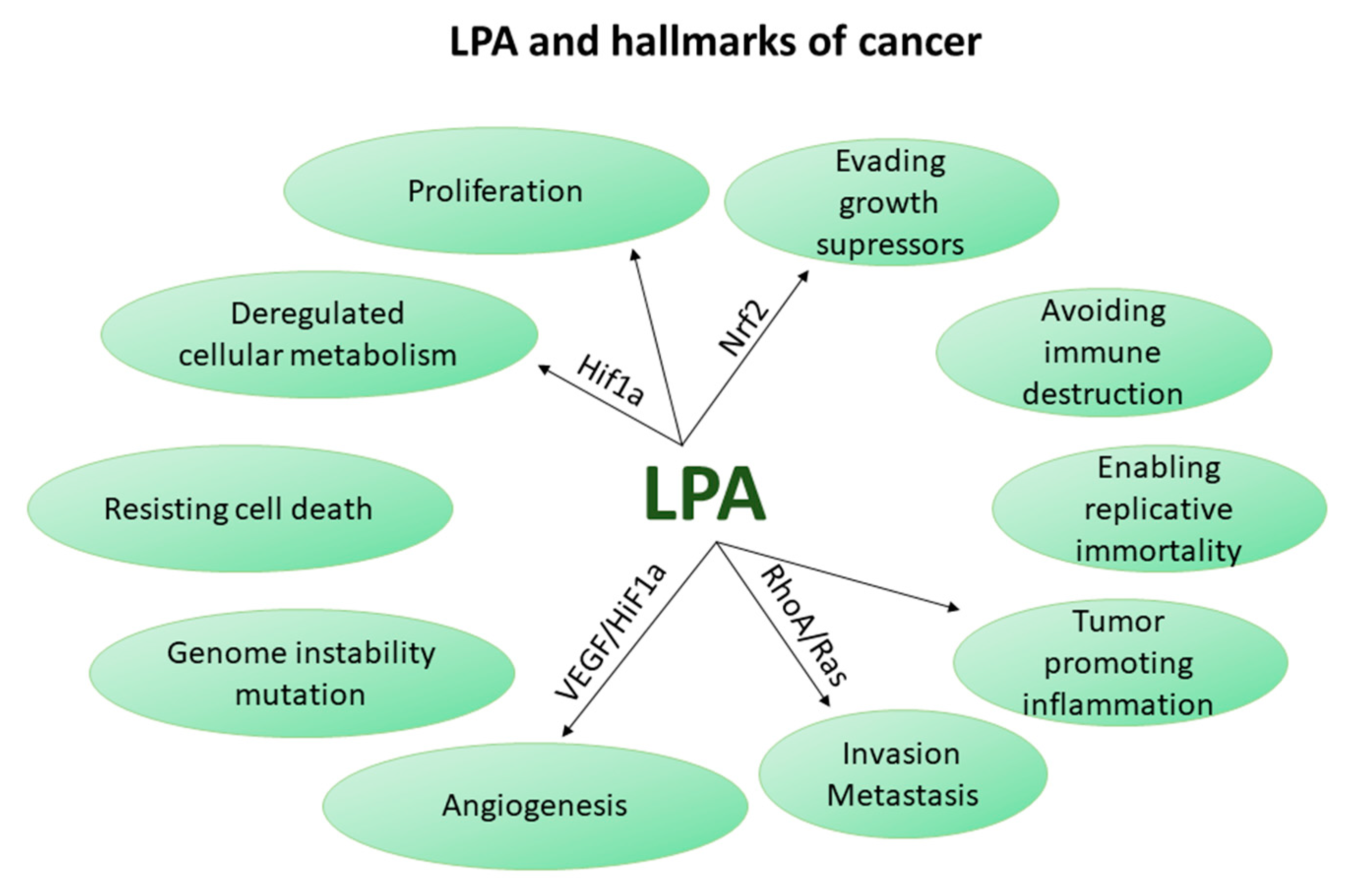
5. LPA Axis in Cancer
6. Liver Cancer
7. Deregulated LPA Metabolism and Risk Factors for Liver Cancer
7.1. Cirrhosis-Related Liver Cancer
7.2. Viral Hepatitis-Related HCC
7.3. Metabolic Diseases-Related HCC
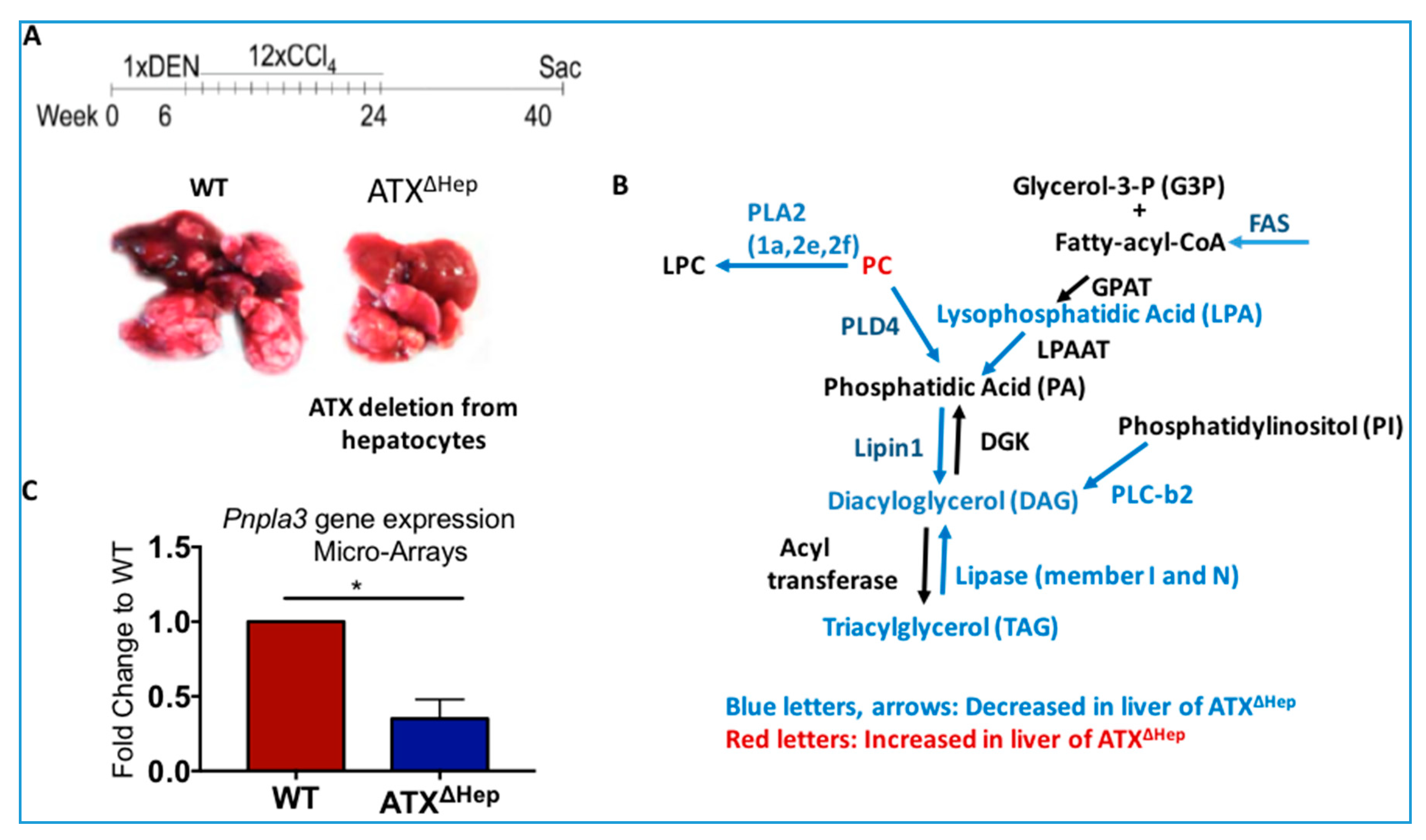
8. Deregulated LPA Signaling and HCC
9. Deregulated LPA Metabolism and CCA
10. Pharmacological Targeting of HCC and Its Risk Factors
11. Conclusions
Funding
Conflicts of Interest
References
- Yung, Y.C.; Stoddard, N.C.; Chun, J. LPA receptor signaling: Pharmacology, physiology, and pathophysiology. J. Lipid Res. 2014, 55, 1192–1214. [Google Scholar] [CrossRef] [PubMed]
- Barbayianni, E.; Kaffe, E.; Aidinis, V.; Kokotos, G. Autotaxin, a secreted lysophospholipase D, as a promising therapeutic target in chronic inflammation and cancer. Prog. Lipid Res. 2015, 58, 76–96. [Google Scholar] [CrossRef] [PubMed]
- Aikawa, S.; Hashimoto, T.; Kano, K.; Aoki, J. Lysophosphatidic acid as a lipid mediator with multiple biological actions. J. Biochem. 2015, 157, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Kazlauskas, A. Lysophosphatidic acid contributes to angiogenic homeostasis. Exp. Cell Res. 2015, 333, 166–170. [Google Scholar] [CrossRef] [PubMed]
- Mueller, P.; Ye, S.; Morris, A.; Smyth, S.S. Lysophospholipid mediators in the vasculature. Exp. Cell Res. 2015, 333, 190–194. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Sardella, A.; Chun, J.; Poubelle, P.E.; Fernandes, M.J.; Bourgoin, S.G. TNF-alpha promotes LPA1- and LPA3-mediated recruitment of leukocytes in vivo through CXCR2 ligand chemokines. J. Lipid Res. 2011, 52, 1307–1318. [Google Scholar] [CrossRef]
- Knowlden, S.; Georas, S.N. The autotaxin-LPA axis emerges as a novel regulator of lymphocyte homing and inflammation. J. Immunol. 2015, 192, 851–857. [Google Scholar] [CrossRef]
- Fotopoulou, S.; Oikonomou, N.; Grigorieva, E.; Nikitopoulou, I.; Paparountas, T.; Thanassopoulou, A.; Zhao, Z.; Xu, Y.; Kontoyiannis, D.L.; Remboutsika, E.; et al. ATX expression and LPA signalling are vital for the development of the nervous system. Dev. Biol. 2010, 339, 451–464. [Google Scholar] [CrossRef]
- Ueda, H.; Matsunaga, H.; Olaposi, O.I.; Nagai, J. Lysophosphatidic acid: Chemical signature of neuropathic pain. Biochim. Biophys. Acta 2013, 1831, 61–73. [Google Scholar] [CrossRef]
- Sims, S.M.; Panupinthu, N.; Lapierre, D.M.; Pereverzev, A.; Dixon, S.J. Lysophosphatidic acid: A potential mediator of osteoblast-osteoclast signaling in bone. Biochim. Biophys. Acta 2013, 1831, 109–116. [Google Scholar] [CrossRef]
- Wu, X.; Ma, Y.; Su, N.; Shen, J.; Zhang, H.; Wang, H. Lysophosphatidic acid: Its role in bone cell biology and potential for use in bone regeneration. Prostaglandins Other Lipid Mediat. 2019, 143, e106335. [Google Scholar] [CrossRef] [PubMed]
- Lidgerwood, G.E.; Pitson, S.M.; Bonder, C.; Pebay, A. Roles of lysophosphatidic acid and sphingosine-1-phosphate in stem cell biology. Prog. Lipid Res. 2018, 72, 42–54. [Google Scholar] [CrossRef] [PubMed]
- Perrakis, A.; Moolenaar, W.H. Autotaxin: Structure-function and signaling. J. Lipid Res. 2014, 55, 1010–1018. [Google Scholar] [CrossRef] [PubMed]
- Tokumura, A.; Majima, E.; Kariya, Y.; Tominaga, K.; Kogure, K.; Yasuda, K.; Fukuzawa, K. Identification of human plasma lysophospholipase D, a lysophosphatidic acid-producing enzyme, as autotaxin, a multifunctional phosphodiesterase. J. Biol. Chem. 2002, 277, 39436–39442. [Google Scholar] [CrossRef]
- Stefan, C.; Jansen, S.; Bollen, M. NPP-type ectophosphodiesterases: Unity in diversity. Trends Biochem. Sci. 2005, 30, 542–550. [Google Scholar] [CrossRef]
- Jansen, S.; Stefan, C.; Creemers, J.W.; Waelkens, E.; Van Eynde, A.; Stalmans, W.; Bollen, M. Proteolytic maturation and activation of autotaxin (NPP2), a secreted metastasis-enhancing lysophospholipase D. J. Cell Sci. 2005, 118, 3081–3089. [Google Scholar] [CrossRef]
- Benesch, M.G.; Tang, X.; Venkatraman, G.; Bekele, R.T.; Brindley, D.N. Recent advances in targeting the autotaxin-lysophosphatidate-lipid phosphate phosphatase axis in vivo. J. Biomed. Res. 2016, 30, 272–284. [Google Scholar] [CrossRef]
- Fulkerson, Z.; Wu, T.; Sunkara, M.; Kooi, C.V.; Morris, A.J.; Smyth, S.S. Binding of autotaxin to integrins localizes lysophosphatidic acid production to platelets and mammalian cells. J. Biol. Chem. 2011, 286, 34654–34663. [Google Scholar] [CrossRef]
- Hausmann, J.; Kamtekar, S.; Christodoulou, E.; Day, J.E.; Wu, T.; Fulkerson, Z.; Albers, H.M.; van Meeteren, L.A.; Houben, A.J.; van Zeijl, L.; et al. Structural basis of substrate discrimination and integrin binding by autotaxin. Nat. Struct. Mol. Biol. 2011, 18, 198–204. [Google Scholar] [CrossRef]
- Leblanc, R.; Lee, S.C.; David, M.; Bordet, J.C.; Norman, D.D.; Patil, R.; Miller, D.; Sahay, D.; Ribeiro, J.; Clezardin, P.; et al. Interaction of platelet-derived autotaxin with tumor integrin alphaVbeta3 controls metastasis of breast cancer cells to bone. Blood 2014, 124, 3141–3150. [Google Scholar] [CrossRef]
- Nishimasu, H.; Okudaira, S.; Hama, K.; Mihara, E.; Dohmae, N.; Inoue, A.; Ishitani, R.; Takagi, J.; Aoki, J.; Nureki, O. Crystal structure of autotaxin and insight into GPCR activation by lipid mediators. Nat. Struct. Mol. Biol. 2011, 18, 205–212. [Google Scholar] [CrossRef] [PubMed]
- Moolenaar, W.H.; Perrakis, A. Insights into autotaxin: How to produce and present a lipid mediator. Nat. Rev. Mol. Cell Biol. 2011, 12, 674–679. [Google Scholar] [CrossRef] [PubMed]
- Giganti, A.; Rodriguez, M.; Fould, B.; Moulharat, N.; Coge, F.; Chomarat, P.; Galizzi, J.P.; Valet, P.; Saulnier-Blache, J.S.; Boutin, J.A.; et al. Murine and human autotaxin {alpha}, {beta}, and {gamma} isoforms: Gene organization, tissue distribution and biochemical characterization. J. Biol. Chem. 2008, 283, 7776–7789. [Google Scholar] [CrossRef] [PubMed]
- Van Meeteren, L.A.; Ruurs, P.; Stortelers, C.; Bouwman, P.; van Rooijen, M.A.; Pradere, J.P.; Pettit, T.R.; Wakelam, M.J.; Saulnier-Blache, J.S.; Mummery, C.L.; et al. Autotaxin, a secreted lysophospholipase D, is essential for blood vessel formation during development. Mol. Cell Biol. 2006, 26, 5015–5022. [Google Scholar] [CrossRef]
- Katsifa, A.; Kaffe, E.; Nikolaidou-Katsaridou, N.; Economides, A.N.; Newbigging, S.; McKerlie, C.; Aidinis, V. The bulk of autotaxin activity is dispensable for adult mouse life. PLoS ONE 2015, 10, e0143083. [Google Scholar] [CrossRef]
- Dusaulcy, R.; Rancoule, C.; Gres, S.; Wanecq, E.; Colom, A.; Guigne, C.; van Meeteren, L.A.; Moolenaar, W.H.; Valet, P.; Saulnier-Blache, J.S. Adipose-specific disruption of autotaxin enhances nutritional fattening and reduces plasma lysophosphatidic acid. J. Lipid Res. 2011, 52, 1247–1255. [Google Scholar] [CrossRef]
- Nishimura, S.; Nagasaki, M.; Okudaira, S.; Aoki, J.; Ohmori, T.; Ohkawa, R.; Nakamura, K.; Igarashi, K.; Yamashita, H.; Eto, K.; et al. ENPP2 contributes to adipose tissue expansion and insulin resistance in diet-induced obesity. Diabetes 2014, 63, 4154–4164. [Google Scholar] [CrossRef]
- Sevastou, I.; Kaffe, E.; Mouratis, M.A.; Aidinis, V. Lysoglycerophospholipids in chronic inflammatory disorders: The PLA(2)/LPC and ATX/LPA axes. Biochim. Biophys. Acta 2013, 1831, 42–60. [Google Scholar] [CrossRef]
- Benesch, M.; MacIntyre, I.; McMullen, T.; Brindley, D. Coming of age for autotaxin and lysophosphatidate signaling: Clinical applications for preventing, detecting and targeting tumor-promoting inflammation. Cancers 2018, 10, 73. [Google Scholar] [CrossRef]
- Aoki, J.; Inoue, A.; Okudaira, S. Two pathways for lysophosphatidic acid production. Biochim. Biophys. Acta 2008, 1781, 513–518. [Google Scholar] [CrossRef]
- Fourcade, O.; Simon, M.F.; Viode, C.; Rugani, N.; Leballe, F.; Ragab, A.; Fournie, B.; Sarda, L.; Chap, H. Secretory phospholipase A2 generates the novel lipid mediator lysophosphatidic acid in membrane microvesicles shed from activated cells. Cell 1995, 80, 919–927. [Google Scholar] [CrossRef]
- Eder, A.M.; Sasagawa, T.; Mao, M.; Aoki, J.; Mills, G.B. Constitutive and lysophosphatidic acid (LPA)-induced LPA production: Role of phospholipase D and phospholipase A2. Clin. Cancer Res. 2000, 6, 2482–2491. [Google Scholar] [PubMed]
- Hiramatsu, T.; Sonoda, H.; Takanezawa, Y.; Morikawa, R.; Ishida, M.; Kasahara, K.; Sanai, Y.; Taguchi, R.; Aoki, J.; Arai, H. Biochemical and molecular characterization of two phosphatidic acid-selective phospholipase A1s, mPA-PLA1alpha and mPA-PLA1beta. J. Biol. Chem. 2003, 278, 49438–49447. [Google Scholar] [CrossRef] [PubMed]
- Scott, S.A.; Mathews, T.P.; Ivanova, P.T.; Lindsley, C.W.; Brown, H.A. Chemical modulation of glycerolipid signaling and metabolic pathways. Biochim. Biophys. Acta 2014, 1841, 1060–1084. [Google Scholar] [CrossRef]
- Tang, X.; Benesch, M.G.; Brindley, D.N. Lipid phosphate phosphatases and their roles in mammalian physiology and pathology. J. Lipid Res. 2015, 56, 2048–2060. [Google Scholar] [CrossRef]
- Tomsig, J.L.; Snyder, A.H.; Berdyshev, E.V.; Skobeleva, A.; Mataya, C.; Natarajan, V.; Brindley, D.N.; Lynch, K.R. Lipid phosphate phosphohydrolase type 1 (LPP1) degrades extracellular lysophosphatidic acid in vivo. Biochem. J. 2009, 419, 611–618. [Google Scholar] [CrossRef]
- Hecht, J.H.; Weiner, J.A.; Post, S.R.; Chun, J. Ventricular zone gene-1 (vzg-1) encodes a lysophosphatidic acid receptor expressed in neurogenic regions of the developing cerebral cortex. J. Cell Biol. 1996, 135, 1071–1083. [Google Scholar] [CrossRef]
- Choi, J.W.; Herr, D.R.; Noguchi, K.; Yung, Y.C.; Lee, C.W.; Mutoh, T.; Lin, M.E.; Teo, S.T.; Park, K.E.; Mosley, A.N.; et al. LPA receptors: Subtypes and biological actions. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 157–186. [Google Scholar] [CrossRef]
- Cai, H.; Xu, Y. The role of LPA and YAP signaling in long-term migration of human ovarian cancer cells. Cell Commun. Signal. 2013, 11, e31. [Google Scholar] [CrossRef]
- Bandoh, K.; Aoki, J.; Taira, A.; Tsujimoto, M.; Arai, H.; Inoue, K. Lysophosphatidic acid (LPA) receptors of the EDG family are differentially activated by LPA species. Structure-activity relationship of cloned LPA receptors. FEBS Lett. 2000, 478, 159–165. [Google Scholar] [CrossRef]
- Noguchi, K.; Ishii, S.; Shimizu, T. Identification of p2y9/GPR23 as a novel G protein-coupled receptor for lysophosphatidic acid, structurally distant from the Edg family. J. Biol. Chem. 2003, 278, 25600–25606. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.W.; Rivera, R.; Dubin, A.E.; Chun, J. LPA(4)/GPR23 is a lysophosphatidic acid (LPA) receptor utilizing G(s)-, G(q)/G(i)-mediated calcium signaling and G(12/13)-mediated Rho activation. J. Biol. Chem. 2007, 282, 4310–4317. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.W.; Rivera, R.; Gardell, S.; Dubin, A.E.; Chun, J. GPR92 as a new G12/13- and Gq-coupled lysophosphatidic acid receptor that increases cAMP, LPA5. J. Biol. Chem. 2006, 281, 23589–23597. [Google Scholar] [CrossRef] [PubMed]
- Yanagida, K.; Masago, K.; Nakanishi, H.; Kihara, Y.; Hamano, F.; Tajima, Y.; Taguchi, R.; Shimizu, T.; Ishii, S. Identification and characterization of a novel lysophosphatidic acid receptor, p2y5/LPA6. J. Biol. Chem. 2009, 284, 17731–17741. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.; Choi, S.; Hallden, G.; Yo, S.J.; Schichnes, D.; Aponte, G.W. P2Y5 is a G(alpha)i, G(alpha)12/13 G protein-coupled receptor activated by lysophosphatidic acid that reduces intestinal cell adhesion. Am. J. Physiol. Gastrointest. Liver Physiol. 2009, 297, 641–654. [Google Scholar] [CrossRef] [PubMed]
- Ward, Y.; Lake, R.; Yin, J.J.; Heger, C.D.; Raffeld, M.; Goldsmith, P.K.; Merino, M.; Kelly, K. LPA receptor heterodimerizes with CD97 to amplify LPA-initiated RHO-dependent signaling and invasion in prostate cancer cells. Cancer Res. 2011, 71, 7301–7311. [Google Scholar] [CrossRef] [PubMed]
- Tabata, K.; Baba, K.; Shiraishi, A.; Ito, M.; Fujita, N. The orphan GPCR GPR87 was deorphanized and shown to be a lysophosphatidic acid receptor. Biochem. Biophys. Res. Commun. 2007, 363, 861–866. [Google Scholar] [CrossRef]
- Murakami, M.; Shiraishi, A.; Tabata, K.; Fujita, N. Identification of the orphan GPCR, P2Y(10) receptor as the sphingosine-1-phosphate and lysophosphatidic acid receptor. Biochem. Biophys. Res. Commun. 2008, 371, 707–712. [Google Scholar] [CrossRef]
- Van Meeteren, L.A.; Moolenaar, W.H. Regulation and biological activities of the autotaxin-LPA axis. Prog. Lipid Res. 2007, 46, 145–160. [Google Scholar] [CrossRef]
- Oldham, W.M.; Hamm, H.E. Heterotrimeric G protein activation by G-protein-coupled receptors. Nat. Rev. Mol. Cell Biol. 2008, 9, 60–71. [Google Scholar] [CrossRef]
- McIntyre, T.M.; Pontsler, A.V.; Silva, A.R.; St Hilaire, A.; Xu, Y.; Hinshaw, J.C.; Zimmerman, G.A.; Hama, K.; Aoki, J.; Arai, H.; et al. Identification of an intracellular receptor for lysophosphatidic acid (LPA): LPA is a transcellular PPARgamma agonist. Proc. Natl. Acad. Sci. USA 2003, 100, 131–136. [Google Scholar] [CrossRef] [PubMed]
- Tsukahara, T.; Tsukahara, R.; Yasuda, S.; Makarova, N.; Valentine, W.J.; Allison, P.; Yuan, H.; Baker, D.L.; Li, Z.; Bittman, R.; et al. Different residues mediate recognition of 1-O-oleyllysophosphatidic acid and rosiglitazone in the ligand binding domain of peroxisome proliferator-activated receptor gamma. J. Biol. Chem. 2006, 281, 3398–3407. [Google Scholar] [CrossRef] [PubMed]
- Gschwind, A.; Prenzel, N.; Ullrich, A. Lysophosphatidic acid-induced squamous cell carcinoma cell proliferation and motility involves epidermal growth factor receptor signal transactivation. Cancer Res. 2002, 62, 6329–6336. [Google Scholar] [PubMed]
- Daub, H.; Weiss, F.U.; Wallasch, C.; Ullrich, A. Role of transactivation of the EGF receptor in signalling by G-protein-coupled receptors. Nature 1996, 379, 557–560. [Google Scholar] [CrossRef] [PubMed]
- Schafer, B.; Marg, B.; Gschwind, A.; Ullrich, A. Distinct ADAM metalloproteinases regulate G protein-coupled receptor-induced cell proliferation and survival. J. Biol. Chem. 2004, 279, 47929–47938. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; He, D.; Saatian, B.; Watkins, T.; Spannhake, E.W.; Pyne, N.J.; Natarajan, V. Regulation of lysophosphatidic acid-induced epidermal growth factor receptor transactivation and interleukin-8 secretion in human bronchial epithelial cells by protein kinase Cdelta, Lyn kinase, and matrix metalloproteinases. J. Biol. Chem. 2006, 281, 19501–19511. [Google Scholar] [CrossRef] [PubMed]
- Ptaszynska, M.M.; Pendrak, M.L.; Stracke, M.L.; Roberts, D.D. Autotaxin signaling via lysophosphatidic acid receptors contributes to vascular endothelial growth factor-induced endothelial cell migration. Mol. Cancer Res. 2010, 8, 309–321. [Google Scholar] [CrossRef]
- Panetti, T.S.; Hannah, D.F.; Avraamides, C.; Gaughan, J.P.; Marcinkiewicz, C.; Huttenlocher, A.; Mosher, D.F. Extracellular matrix molecules regulate endothelial cell migration stimulated by lysophosphatidic acid. J. Thromb. Haemost. 2004, 2, 1645–1656. [Google Scholar] [CrossRef]
- Zhou, Z.; Subramanian, P.; Sevilmis, G.; Globke, B.; Soehnlein, O.; Karshovska, E.; Megens, R.; Heyll, K.; Chun, J.; Saulnier-Blache, J.S.; et al. Lipoprotein-derived lysophosphatidic acid promotes atherosclerosis by releasing CXCL1 from the endothelium. Cell Metab. 2011, 13, 592–600. [Google Scholar] [CrossRef]
- Lin, C.I.; Chen, C.N.; Chen, J.H.; Lee, H. Lysophospholipids increase IL-8 and MCP-1 expressions in human umbilical cord vein endothelial cells through an IL-1-dependent mechanism. J. Cell Biochem. 2006, 99, 1216–1232. [Google Scholar] [CrossRef]
- Shimada, H.; Rajagopalan, L.E. Rho-kinase mediates lysophosphatidic acid-induced IL-8 and MCP-1 production via p38 and JNK pathways in human endothelial cells. FEBS Lett. 2010, 584, 2827–2832. [Google Scholar] [CrossRef] [PubMed]
- Shimada, H.; Rajagopalan, L.E. Rho kinase-2 activation in human endothelial cells drives lysophosphatidic acid-mediated expression of cell adhesion molecules via NF-kappaB p65. J. Biol. Chem. 2010, 285, 12536–12542. [Google Scholar] [CrossRef] [PubMed]
- Sarker, M.H.; Hu, D.E.; Fraser, P.A. Regulation of cerebromicrovascular permeability by lysophosphatidic acid. Microcirculation 2010, 17, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Tager, A.M.; LaCamera, P.; Shea, B.S.; Campanella, G.S.; Selman, M.; Zhao, Z.; Polosukhin, V.; Wain, J.; Karimi-Shah, B.A.; Kim, N.D.; et al. The lysophosphatidic acid receptor LPA1 links pulmonary fibrosis to lung injury by mediating fibroblast recruitment and vascular leak. Nat. Med. 2008, 14, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Bai, Z.; Cai, L.; Umemoto, E.; Takeda, A.; Tohya, K.; Komai, Y.; Veeraveedu, P.T.; Hata, E.; Sugiura, Y.; Kubo, A.; et al. Constitutive lymphocyte transmigration across the basal lamina of high endothelial venules is regulated by the autotaxin/lysophosphatidic acid axis. J. Immunol. 2013, 190, 2036–2048. [Google Scholar] [CrossRef] [PubMed]
- Kanda, H.; Newton, R.; Klein, R.; Morita, Y.; Gunn, M.D.; Rosen, S.D. Autotaxin, an ectoenzyme that produces lysophosphatidic acid, promotes the entry of lymphocytes into secondary lymphoid organs. Nat. Immunol. 2008, 9, 415–423. [Google Scholar] [CrossRef]
- Federico, L.; Ren, H.; Mueller, P.A.; Wu, T.; Liu, S.; Popovic, J.; Blalock, E.M.; Sunkara, M.; Ovaa, H.; Albers, H.M.; et al. Autotaxin and its product lysophosphatidic acid suppress brown adipose differentiation and promote diet-induced obesity in mice. Mol. Endocrinol. 2012, 26, 786–797. [Google Scholar] [CrossRef]
- Brandon, J.A.; Kraemer, M.; Vandra, J.; Halder, S.; Ubele, M.; Morris, A.J.; Smyth, S.S. Adipose-derived autotaxin regulates inflammation and steatosis associated with diet-induced obesity. PLoS ONE 2019, 14, e0208099. [Google Scholar] [CrossRef]
- Lumeng, C.N.; Saltiel, A.R. Inflammatory links between obesity and metabolic disease. J. Clin. Investig. 2011, 121, 2111–2117. [Google Scholar] [CrossRef]
- Oikonomou, N.; Mouratis, M.A.; Tzouvelekis, A.; Kaffe, E.; Valavanis, C.; Vilaras, G.; Karameris, A.; Prestwich, G.D.; Bouros, D.; Aidinis, V. Pulmonary autotaxin expression contributes to the pathogenesis of pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 2012, 47, 566–574. [Google Scholar] [CrossRef]
- Huang, L.S.; Fu, P.; Patel, P.; Harijith, A.; Sun, T.; Zhao, Y.; Garcia, J.G.; Chun, J.; Natarajan, V. Lysophosphatidic acid receptor-2 deficiency confers protection against bleomycin-induced lung injury and fibrosis in mice. Am. J. Respir. Cell Mol. Biol. 2013, 49, 912–922. [Google Scholar] [CrossRef] [PubMed]
- Cummings, R.; Zhao, Y.; Jacoby, D.; Spannhake, E.W.; Ohba, M.; Garcia, J.G.; Watkins, T.; He, D.; Saatian, B.; Natarajan, V. Protein kinase Cdelta mediates lysophosphatidic acid-induced NF-kappaB activation and interleukin-8 secretion in human bronchial epithelial cells. J. Biol. Chem. 2004, 279, 41085–41094. [Google Scholar] [CrossRef] [PubMed]
- Saatian, B.; Zhao, Y.; He, D.; Georas, S.N.; Watkins, T.; Spannhake, E.W.; Natarajan, V. Transcriptional regulation of lysophosphatidic acid-induced interleukin-8 expression and secretion by p38 MAPK and JNK in human bronchial epithelial cells. Biochem. J. 2006, 393, 657–668. [Google Scholar] [CrossRef] [PubMed]
- Ray, R.; Rai, V. Lysophosphatidic acid converts monocytes into macrophages in both mice and humans. Blood 2017, 129, 1177–1183. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.Y.; Porte, J.; Knox, A.J.; Weinreb, P.H.; Maher, T.M.; Violette, S.M.; McAnulty, R.J.; Sheppard, D.; Jenkins, G. Lysophosphatidic acid induces alphavbeta6 integrin-mediated TGF-beta activation via the LPA2 receptor and the small G protein G alpha(q). Am. J. Pathol. 2009, 174, 1264–1279. [Google Scholar] [CrossRef] [PubMed]
- Funke, M.; Zhao, Z.; Xu, Y.; Chun, J.; Tager, A.M. The lysophosphatidic acid receptor LPA1 promotes epithelial cell apoptosis after lung injury. Am. J. Respir. Cell Mol. Biol. 2012, 46, 355–364. [Google Scholar] [CrossRef]
- Maher, T.M.; Kreuter, M.; Lederer, D.J.; Brown, K.K.; Wuyts, W.; Verbruggen, N.; Stutvoet, S.; Fieuw, A.; Ford, P.; Abi-Saab, W.; et al. Rationale, design and objectives of two phase III, randomised, placebo-controlled studies of GLPG1690, a novel autotaxin inhibitor, in idiopathic pulmonary fibrosis (ISABELA 1 and 2). BMJ Open Respir. Res. 2019, 6, e000422. [Google Scholar] [CrossRef]
- Palmer, S.M.; Snyder, L.; Todd, J.L.; Soule, B.; Christian, R.; Anstrom, K.; Luo, Y.; Gagnon, R.; Rosen, G. Randomized, double-blind, placebo-controlled, phase 2 trial of BMS-986020, a lysophosphatidic acid receptor antagonist for the treatment of idiopathic pulmonary fibrosis. Chest 2018, 154, 1061–1069. [Google Scholar] [CrossRef]
- Orosa, B.; Gonzalez, A.; Mera, A.; Gomez-Reino, J.J.; Conde, C. Lysophosphatidic acid receptor 1 suppression sensitizes rheumatoid fibroblast-like synoviocytes to tumor necrosis factor-induced apoptosis. Arthritis Rheum. 2012, 64, 2460–2470. [Google Scholar] [CrossRef]
- Miyabe, Y.; Miyabe, C.; Iwai, Y.; Takayasu, A.; Fukuda, S.; Yokoyama, W.; Nagai, J.; Jona, M.; Tokuhara, Y.; Ohkawa, R.; et al. Necessity of lysophosphatidic acid receptor 1 for development of arthritis. Arthritis Rheum. 2013, 65, 2037–2047. [Google Scholar] [CrossRef]
- Orosa, B.; Garcia, S.; Martinez, P.; Gonzalez, A.; Gomez-Reino, J.J.; Conde, C. Lysophosphatidic acid receptor inhibition as a new multipronged treatment for rheumatoid arthritis. Ann. Rheum. Dis. 2014, 73, 298–305. [Google Scholar] [CrossRef] [PubMed]
- Nikitopoulou, I.; Oikonomou, N.; Karouzakis, E.; Sevastou, I.; Nikolaidou-Katsaridou, N.; Zhao, Z.; Mersinias, V.; Armaka, M.; Xu, Y.; Masu, M.; et al. Autotaxin expression from synovial fibroblasts is essential for the pathogenesis of modeled arthritis. J. Exp. Med. 2012, 209, 925–933. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Fernandes, M.J.; Prestwich, G.D.; Turgeon, M.; Battista, J.; Clair, T. Regulation of lysophosphatidic acid receptor expression and function in human synoviocytes: Implications for rheumatoid arthritis? Mol. Pharmacol. 2008, 73, 587–600. [Google Scholar] [CrossRef]
- Schunkert, H.; Konig, I.R.; Kathiresan, S.; Reilly, M.P.; Assimes, T.L.; Holm, H.; Preuss, M.; Stewart, A.F.; Barbalic, M.; Gieger, C.; et al. Large-scale association analysis identifies 13 new susceptibility loci for coronary artery disease. Nat. Genet. 2011, 43, 333–338. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Yang, X.Y.; Wang, N.D.; Ding, C.; Yang, Y.J.; You, Z.J.; Su, Q.; Chen, J.H. Serum lysophosphatidic acid concentrations measured by dot immunogold filtration assay in patients with acute myocardial infarction. Scand. J. Clin. Lab. Investig. 2003, 63, 497–503. [Google Scholar] [CrossRef]
- Dohi, T.; Miyauchi, K.; Ohkawa, R.; Nakamura, K.; Kurano, M.; Kishimoto, T.; Yanagisawa, N.; Ogita, M.; Miyazaki, T.; Nishino, A.; et al. Increased lysophosphatidic acid levels in culprit coronary arteries of patients with acute coronary syndrome. Atherosclerosis 2013, 229, 192–197. [Google Scholar] [CrossRef]
- Siess, W.; Zangl, K.J.; Essler, M.; Bauer, M.; Brandl, R.; Corrinth, C.; Bittman, R.; Tigyi, G.; Aepfelbacher, M. Lysophosphatidic acid mediates the rapid activation of platelets and endothelial cells by mildly oxidized low density lipoprotein and accumulates in human atherosclerotic lesions. Proc. Natl. Acad. Sci. USA 1999, 96, 6931–6936. [Google Scholar] [CrossRef]
- Bot, M.; Bot, I.; Lopez-Vales, R.; van de Lest, C.H.; Saulnier-Blache, J.S.; Helms, J.B.; David, S.; van Berkel, T.J.; Biessen, E.A. Atherosclerotic lesion progression changes lysophosphatidic acid homeostasis to favor its accumulation. Am. J. Pathol. 2010, 176, 3073–3084. [Google Scholar] [CrossRef]
- Chang, C.L.; Hsu, H.Y.; Lin, H.Y.; Chiang, W.; Lee, H. Lysophosphatidic acid-induced oxidized low-density lipoprotein uptake is class A scavenger receptor-dependent in macrophages. Prostaglandins Other Lipid Mediat. 2008, 87, 20–25. [Google Scholar] [CrossRef]
- Yang, L.; Kraemer, M.; Fang, X.F.; Angel, P.M.; Drake, R.R.; Morris, A.J.; Smyth, S.S. LPA receptor 4 deficiency attenuates experimental atherosclerosis. J. Lipid Res. 2019, 60, 972–980. [Google Scholar] [CrossRef]
- Mathieu, P.; Boulanger, M.C. Autotaxin and lipoprotein metabolism in calcific aortic valve disease. Front. Cardiovasc. Med. 2019, 6, e18. [Google Scholar] [CrossRef] [PubMed]
- Dennis, J.; Nogaroli, L.; Fuss, B. Phosphodiesterase-Ialpha/autotaxin (PD-Ialpha/ATX): A multifunctional protein involved in central nervous system development and disease. J. Neurosci. Res. 2005, 82, 737–742. [Google Scholar] [CrossRef] [PubMed]
- Ramesh, S.; Govindarajulu, M.; Suppiramaniam, V.; Moore, T.; Dhanasekaran, M. Autotaxin-Lysophosphatidic acid signaling in Alzheimer’s disease. Int. J. Mol. Sci. 2018, 19, 1827. [Google Scholar] [CrossRef] [PubMed]
- Park, F.; Miller, D.D. Role of lysophosphatidic acid and its receptors in the kidney. Physiol. Genomics 2017, 49, 659–666. [Google Scholar] [CrossRef]
- Lee, J.H.; Kim, D.; Oh, Y.S.; Jun, H.S. Lysophosphatidic acid signaling in diabetic nephropathy. Int. J. Mol. Sci. 2019, 20, 2850. [Google Scholar] [CrossRef]
- Yun, C.C. Lysophosphatidic acid and autotaxin-associated effects on the initiation and progression of colorectal cancer. Cancers 2019, 11, 958. [Google Scholar] [CrossRef]
- Xu, Y.; Fang, X.J.; Casey, G.; Mills, G.B. Lysophospholipids activate ovarian and breast cancer cells. Biochem. J. 1995, 309, 933–940. [Google Scholar] [CrossRef]
- Stracke, M.L.; Krutzsch, H.C.; Unsworth, E.J.; Arestad, A.; Cioce, V.; Schiffmann, E.; Liotta, L.A. Identification, purification, and partial sequence analysis of autotaxin, a novel motility-stimulating protein. J. Biol. Chem. 1992, 267, 2524–2529. [Google Scholar]
- Benesch, M.G.; Ko, Y.M.; Tang, X.; Dewald, J.; Lopez-Campistrous, A.; Zhao, Y.Y.; Lai, R.; Curtis, J.M.; Brindley, D.N.; McMullen, T.P. Autotaxin is an inflammatory mediator and therapeutic target in thyroid cancer. Endocr. Relat. Cancer 2015, 22, 593–607. [Google Scholar] [CrossRef]
- Brisbin, A.G.; Asmann, Y.W.; Song, H.; Tsai, Y.Y.; Aakre, J.A.; Yang, P.; Jenkins, R.B.; Pharoah, P.; Schumacher, F.; Conti, D.V.; et al. Meta-analysis of 8q24 for seven cancers reveals a locus between NOV and ENPP2 associated with cancer development. BMC Med. Genet. 2011, 12, e156. [Google Scholar] [CrossRef]
- Willier, S.; Butt, E.; Grunewald, T.G. Lysophosphatidic acid (LPA) signalling in cell migration and cancer invasion: A focussed review and analysis of LPA receptor gene expression on the basis of more than 1700 cancer microarrays. Biol. Cell 2013, 105, 317–333. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Wang, D.; Iyer, S.; Ghaleb, A.M.; Shim, H.; Yang, V.W.; Chun, J.; Yun, C.C. The absence of LPA2 attenuates tumor formation in an experimental model of colitis-associated cancer. Gastroenterology 2009, 136, 1711–1720. [Google Scholar] [CrossRef] [PubMed]
- Brindley, D.N.; Lin, F.T.; Tigyi, G.J. Role of the autotaxin-lysophosphatidate axis in cancer resistance to chemotherapy and radiotherapy. Biochim. Biophys. Acta 2013, 1831, 74–85. [Google Scholar] [CrossRef] [PubMed]
- Popnikolov, N.K.; Dalwadi, B.H.; Thomas, J.D.; Johannes, G.J.; Imagawa, W.T. Association of autotaxin and lysophosphatidic acid receptor 3 with aggressiveness of human breast carcinoma. Tumour Biol. 2012, 33, 2237–2243. [Google Scholar] [CrossRef] [PubMed]
- Minami, K.; Ueda, N.; Maeda, H.; Ishimoto, K.; Otagaki, S.; Tsujiuchi, T. Modulation of chemoresistance by lysophosphatidic acid (LPA) signaling through LPA5 in melanoma cells treated with anticancer drugs. Biochem. Biophys. Res. Commun. 2019, 517, 359–363. [Google Scholar] [CrossRef]
- Tao, K.; Guo, S.; Chen, R.; Yang, C.; Jian, L.; Yu, H.; Liu, S. Lysophosphatidic Acid Receptor 6 (LPAR6) expression and prospective signaling pathway analysis in breast cancer. Mol. Diagn. Ther. 2019, 23, 127–138. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Jeong, K.J.; Park, S.Y.; Cho, K.H.; Sohn, J.S.; Lee, J.; Kim, Y.K.; Kang, J.; Park, C.G.; Han, J.W.; Lee, H.Y. The Rho/ROCK pathway for lysophosphatidic acid-induced proteolytic enzyme expression and ovarian cancer cell invasion. Oncogene 2012, 31, 4279–4289. [Google Scholar] [CrossRef]
- Hoelzinger, D.B.; Nakada, M.; Demuth, T.; Rosensteel, T.; Reavie, L.B.; Berens, M.E. Autotaxin: A secreted autocrine/paracrine factor that promotes glioma invasion. J. Neurooncol. 2008, 86, 297–309. [Google Scholar] [CrossRef]
- Harper, K.; Lavoie, R.R.; Charbonneau, M.; Brochu-Gaudreau, K.; Dubois, C.M. The hypoxic tumor microenvironment promotes invadopodia formation and metastasis through LPA1 receptor and EGFR cooperation. Mol. Cancer Res. 2018, 16, 1601–1613. [Google Scholar] [CrossRef]
- Ren, Z.; Zhang, C.; Ma, L.; Zhang, X.; Shi, S.; Tang, D.; Xu, J.; Hu, Y.; Wang, B.; Zhang, F.; et al. Lysophosphatidic acid induces the migration and invasion of SGC-7901 gastric cancer cells through the LPA2 and Notch signaling pathways. Int. J. Mol. Med. 2019, 44, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Fukushima, K.; Tanaka, K.; Minami, K.; Ishimoto, K.; Otagaki, S.; Fukushima, N.; Honoki, K.; Tsujiuchi, T. Involvement of LPA signaling via LPA receptor-2 in the promotion of malignant properties in osteosarcoma cells. Exp. Cell Res. 2018, 369, 316–324. [Google Scholar] [CrossRef] [PubMed]
- Muinonen-Martin, A.J.; Susanto, O.; Zhang, Q.; Smethurst, E.; Faller, W.J.; Veltman, D.M.; Kalna, G.; Lindsay, C.; Bennett, D.C.; Sansom, O.J.; et al. Melanoma cells break down LPA to establish local gradients that drive chemotactic dispersal. PLoS Biol. 2014, 12, e1001966. [Google Scholar] [CrossRef] [PubMed]
- Susanto, O.; Koh, Y.W.H.; Morrice, N.; Tumanov, S.; Thomason, P.A.; Nielson, M.; Tweedy, L.; Muinonen-Martin, A.J.; Kamphorst, J.J.; Mackay, G.M.; et al. LPP3 mediates self-generation of chemotactic LPA gradients by melanoma cells. J. Cell Sci. 2017, 130, 3455–3466. [Google Scholar] [CrossRef] [PubMed]
- Takara, K.; Eino, D.; Ando, K.; Yasuda, D.; Naito, H.; Tsukada, Y.; Iba, T.; Wakabayashi, T.; Muramatsu, F.; Kidoya, H.; et al. Lysophosphatidic acid receptor 4 activation augments drug delivery in tumors by tightening endothelial cell-cell contact. Cell Rep. 2017, 20, 2072–2086. [Google Scholar] [CrossRef] [PubMed]
- Benesch, M.G.; Tang, X.; Dewald, J.; Dong, W.F.; Mackey, J.R.; Hemmings, D.G.; McMullen, T.P.; Brindley, D.N. Tumor-induced inflammation in mammary adipose tissue stimulates a vicious cycle of autotaxin expression and breast cancer progression. FASEB J. 2015, 29, 3990–4000. [Google Scholar] [CrossRef]
- Reinartz, S.; Lieber, S.; Pesek, J.; Brandt, D.T.; Asafova, A.; Finkernagel, F.; Watzer, B.; Nockher, W.A.; Nist, A.; Stiewe, T.; et al. Cell type-selective pathways and clinical associations of lysophosphatidic acid biosynthesis and signaling in the ovarian cancer microenvironment. Mol. Oncol. 2019, 13, 185–201. [Google Scholar] [CrossRef]
- Ha, J.H.; Radhakrishnan, R.; Jayaraman, M.; Yan, M.; Ward, J.D.; Fung, K.M.; Moxley, K.; Sood, A.K.; Isidoro, C.; Mukherjee, P.; et al. LPA induces metabolic reprogramming in ovarian cancer via a pseudohypoxic response. Cancer Res. 2018, 78, 1923–1934. [Google Scholar] [CrossRef]
- Radhakrishnan, R.; Ha, J.H.; Jayaraman, M.; Liu, J.; Moxley, K.M.; Isidoro, C.; Sood, A.K.; Song, Y.S.; Dhanasekaran, D.N. Ovarian cancer cell-derived lysophosphatidic acid induces glycolytic shift and cancer-associated fibroblast-phenotype in normal and peritumoral fibroblasts. Cancer Lett. 2019, 442, 464–474. [Google Scholar] [CrossRef]
- Yu, L.; Chen, X.; Sun, X.; Wang, L.; Chen, S. The glycolytic switch in tumors: How many players are involved? J. Cancer 2017, 8, 3430–3440. [Google Scholar] [CrossRef]
- Auciello, F.R.; Bulusu, V.; Oon, C.; Tait-Mulder, J.; Berry, M.; Bhattacharyya, S.; Tumanov, S.; Allen-Petersen, B.L.; Link, J.; Kendsersky, N.D.; et al. A stromal lysolipid-autotaxin signaling axis promotes pancreatic tumor progression. Cancer Discov. 2019, 9, 617–627. [Google Scholar] [CrossRef] [PubMed]
- Samadi, N.; Gaetano, C.; Goping, I.S.; Brindley, D.N. Autotaxin protects MCF-7 breast cancer and MDA-MB-435 melanoma cells against Taxol-induced apoptosis. Oncogene 2009, 28, 1028–1039. [Google Scholar] [CrossRef] [PubMed]
- Shuyu, E.; Lai, Y.J.; Tsukahara, R.; Chen, C.S.; Fujiwara, Y.; Yue, J.; Yu, J.H.; Guo, H.; Kihara, A.; Tigyi, G.; et al. Lysophosphatidic acid 2 receptor-mediated supramolecular complex formation regulates its antiapoptotic effect. J. Biol. Chem. 2009, 284, 14558–14571. [Google Scholar] [CrossRef]
- Venkatraman, G.; Benesch, M.G.; Tang, X.; Dewald, J.; McMullen, T.P.; Brindley, D.N. Lysophosphatidate signaling stabilizes Nrf2 and increases the expression of genes involved in drug resistance and oxidative stress responses: Implications for cancer treatment. FASEB J. 2015, 29, 772–785. [Google Scholar] [CrossRef]
- Su, S.C.; Hu, X.; Kenney, P.A.; Merrill, M.M.; Babaian, K.N.; Zhang, X.Y.; Maity, T.; Yang, S.F.; Lin, X.; Wood, C.G. Autotaxin-lysophosphatidic acid signaling axis mediates tumorigenesis and development of acquired resistance to sunitinib in renal cell carcinoma. Clin. Cancer Res. 2013, 19, 6461–6472. [Google Scholar] [CrossRef]
- Liu, S.; Umezu-Goto, M.; Murph, M.; Lu, Y.; Liu, W.; Zhang, F.; Yu, S.; Stephens, L.C.; Cui, X.; Murrow, G.; et al. Expression of autotaxin and lysophosphatidic acid receptors increases mammary tumorigenesis, invasion, and metastases. Cancer Cell 2009, 15, 539–550. [Google Scholar] [CrossRef]
- Braeuer, R.R.; Zigler, M.; Kamiya, T.; Dobroff, A.S.; Huang, L.; Choi, W.; McConkey, D.J.; Shoshan, E.; Mobley, A.K.; Song, R.; et al. Galectin-3 contributes to melanoma growth and metastasis via regulation of NFAT1 and autotaxin. Cancer Res. 2012, 72, 5757–5766. [Google Scholar] [CrossRef]
- Kmiec, Z. Cooperation of liver cells in health and disease. Adv. Anat. Embryol. Cell Biol. 2001, 161, 1–151. [Google Scholar]
- Heymann, F.; Tacke, F. Immunology in the liver—From homeostasis to disease. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 88–110. [Google Scholar] [CrossRef]
- Yang, J.D.; Hainaut, P.; Gores, G.J.; Amadou, A.; Plymoth, A.; Roberts, L.R. A global view of hepatocellular carcinoma: Trends, risk, prevention and management. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 589–604. [Google Scholar] [CrossRef]
- Mittal, S.; El-Serag, H.B. Epidemiology of hepatocellular carcinoma: Consider the population. J. Clin. Gastroenterol. 2013, 47, 2–6. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Zucman-Rossi, J.; Pikarsky, E.; Sangro, B.; Schwartz, M.; Sherman, M.; Gores, G. Hepatocellular carcinoma. Nat. Rev. Dis. Primers 2016, 2, e16018. [Google Scholar] [CrossRef] [PubMed]
- Gores, G.J. Decade in review-hepatocellular carcinoma: HCC-subtypes, stratification and sorafenib. Nat. Rev. Gastroenterol. Hepatol. 2014, 11, 645–647. [Google Scholar] [CrossRef] [PubMed]
- Razumilava, N.; Gores, G.J. Cholangiocarcinoma. Lancet 2014, 383, 2168–2179. [Google Scholar] [CrossRef]
- Zhang, L.F.; Zhao, H.X. Diabetes mellitus and increased risk of extrahepatic cholangiocarcinoma: A meta-analysis. Hepatogastroenterology 2013, 60, 684–687. [Google Scholar] [CrossRef]
- Petrick, J.L.; Thistle, J.E.; Zeleniuch-Jacquotte, A.; Zhang, X.; Wactawski-Wende, J.; Van Dyke, A.L.; Stampfer, M.J.; Sinha, R.; Sesso, H.D.; Schairer, C.; et al. Body mass index, diabetes and intrahepatic cholangiocarcinoma risk: The liver cancer pooling project and meta-analysis. Am. J. Gastroenterol. 2018, 113, 1494–1505. [Google Scholar] [CrossRef]
- Brivio, S.; Cadamuro, M.; Strazzabosco, M.; Fabris, L. Tumor reactive stroma in cholangiocarcinoma: The fuel behind cancer aggressiveness. World J. Hepatol. 2017, 9, 455–468. [Google Scholar] [CrossRef]
- Brivio, S.; Cadamuro, M.; Fabris, L.; Strazzabosco, M. Epithelial-to-mesenchymal transition and cancer invasiveness: What can we learn from cholangiocarcinoma? J. Clin. Med. 2015, 4, 2028–2041. [Google Scholar] [CrossRef]
- Cadamuro, M.; Stecca, T.; Brivio, S.; Mariotti, V.; Fiorotto, R.; Spirli, C.; Strazzabosco, M.; Fabris, L. The deleterious interplay between tumor epithelia and stroma in cholangiocarcinoma. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 1435–1443. [Google Scholar] [CrossRef]
- Cadamuro, M.; Morton, S.D.; Strazzabosco, M.; Fabris, L. Unveiling the role of tumor reactive stroma in cholangiocarcinoma: An opportunity for new therapeutic strategies. Transl. Gastrointest. Cancer 2013, 2, 130–144. [Google Scholar] [CrossRef]
- Bonato, G.; Cristoferi, L.; Strazzabosco, M.; Fabris, L. Malignancies in primary sclerosing cholangitis—A continuing threat. Dig. Dis. 2015, 33, 140–148. [Google Scholar] [CrossRef] [PubMed]
- Sirica, A.E.; Gores, G.J.; Groopman, J.D.; Selaru, F.M.; Strazzabosco, M.; Wang, X.W.; Zhu, A.X. Intrahepatic cholangiocarcinoma: Continuing challenges and translational advances. Hepatology 2018, 69, 1803–1815. [Google Scholar] [CrossRef] [PubMed]
- Banales, J.M.; Cardinale, V.; Carpino, G.; Marzioni, M.; Andersen, J.B.; Invernizzi, P.; Lind, G.E.; Folseraas, T.; Forbes, S.J.; Fouassier, L.; et al. Expert consensus document: Cholangiocarcinoma: Current knowledge and future perspectives consensus statement from the European Network for the Study of Cholangiocarcinoma (ENS-CCA). Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 261–280. [Google Scholar] [CrossRef] [PubMed]
- Palmer, W.C.; Patel, T. Are common factors involved in the pathogenesis of primary liver cancers? A meta-analysis of risk factors for intrahepatic cholangiocarcinoma. J. Hepatol. 2012, 57, 69–76. [Google Scholar] [CrossRef]
- Nakagawa, H.; Ikeda, H.; Nakamura, K.; Ohkawa, R.; Masuzaki, R.; Tateishi, R.; Yoshida, H.; Watanabe, N.; Tejima, K.; Kume, Y.; et al. Autotaxin as a novel serum marker of liver fibrosis. Clin. Chim. Acta 2011, 412, 1201–1206. [Google Scholar] [CrossRef]
- Wunsch, E.; Krawczyk, M.; Milkiewicz, M.; Trottier, J.; Barbier, O.; Neurath, M.F.; Lammert, F.; Kremer, A.E.; Milkiewicz, P. Serum autotaxin is a marker of the severity of liver injury and overall survival in patients with cholestatic liver diseases. Sci. Rep. 2016, 6, e30847. [Google Scholar] [CrossRef]
- Pleli, T.; Martin, D.; Kronenberger, B.; Brunner, F.; Koberle, V.; Grammatikos, G.; Farnik, H.; Martinez, Y.; Finkelmeier, F.; Labocha, S.; et al. Serum autotaxin is a parameter for the severity of liver cirrhosis and overall survival in patients with liver cirrhosis—A prospective cohort study. PLoS ONE 2014, 9, e103532. [Google Scholar] [CrossRef]
- Honda, Y.; Imajo, K.; Kobayashi, T.; Kessoku, T.; Ogawa, Y.; Tomeno, W.; Yoneda, M.; Kobayashi, N.; Saito, S.; Nakajima, A. Autotaxin is a valuable biomarker for the prediction of liver fibrosis in patients with non-alcoholic fatty liver disease. Hepatol. Res. 2019. [Google Scholar] [CrossRef]
- Joshita, S.; Umemura, T.; Usami, Y.; Yamashita, Y.; Norman, G.L.; Sugiura, A.; Yamazaki, T.; Fujimori, N.; Kimura, T.; Matsumoto, A.; et al. Serum autotaxin is a useful disease progression marker in patients with primary biliary cholangitis. Sci. Rep. 2018, 8, e8159. [Google Scholar] [CrossRef]
- Wu, J.M.; Xu, Y.; Skill, N.J.; Sheng, H.; Zhao, Z.; Yu, M.; Saxena, R.; Maluccio, M.A. Autotaxin expression and its connection with the TNF-alpha-NF-kappaB axis in human hepatocellular carcinoma. Mol. Cancer 2010, 9, e71. [Google Scholar] [CrossRef]
- Kondo, M.; Ishizawa, T.; Enooku, K.; Tokuhara, Y.; Ohkawa, R.; Uranbileg, B.; Nakagawa, H.; Tateishi, R.; Yoshida, H.; Kokudo, N.; et al. Increased serum autotaxin levels in hepatocellular carcinoma patients were caused by background liver fibrosis but not by carcinoma. Clin. Chim. Acta 2014, 433, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Kaffe, E.; Katsifa, A.; Xylourgidis, N.; Ninou, I.; Zannikou, M.; Harokopos, V.; Foka, P.; Dimitriadis, A.; Evangelou, K.; Moulas, A.N.; et al. Hepatocyte autotaxin expression promotes liver fibrosis and cancer. Hepatology 2017, 65, 1369–1383. [Google Scholar] [CrossRef]
- Simo, K.A.; Niemeyer, D.J.; Hanna, E.M.; Swet, J.H.; Thompson, K.J.; Sindram, D.; Iannitti, D.A.; Eheim, A.L.; Sokolov, E.; Zuckerman, V.; et al. Altered lysophosphatidic acid (LPA) receptor expression during hepatic regeneration in a mouse model of partial hepatectomy. HPB (Oxford) 2014, 16, 534–542. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, H.; Yatomi, Y.; Yanase, M.; Satoh, H.; Nishihara, A.; Kawabata, M.; Fujiwara, K. Effects of lysophosphatidic acid on proliferation of stellate cells and hepatocytes in culture. Biochem. Biophys. Res. Commun. 1998, 248, 436–440. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, S.; Wei, L.; Song, W.M.; Higashi, T.; Ghoshal, S.; Kim, R.S.; Bian, C.B.; Yamada, S.; Sun, X.; Venkatesh, A.; et al. Molecular liver cancer prevention in cirrhosis by organ transcriptome analysis and lysophosphatidic acid pathway inhibition. Cancer Cell 2016, 30, 879–890. [Google Scholar] [CrossRef] [PubMed]
- Coulouarn, C.; Clement, B. Stellate cells and the development of liver cancer: Therapeutic potential of targeting the stroma. J. Hepatol. 2014, 60, 1306–1309. [Google Scholar] [CrossRef] [PubMed]
- Boucharaba, A.; Guillet, B.; Menaa, F.; Hneino, M.; van Wijnen, A.J.; Clezardin, P.; Peyruchaud, O. Bioactive lipids lysophosphatidic acid and sphingosine 1-phosphate mediate breast cancer cell biological functions through distinct mechanisms. Oncol. Res. 2009, 18, 173–184. [Google Scholar] [CrossRef]
- Castelino, F.V.; Bain, G.; Pace, V.A.; Black, K.E.; George, L.; Probst, C.K.; Goulet, L.; Lafyatis, R.; Tager, A.M. An autotaxin/Lysophosphatidic acid/Interleukin-6 amplification loop drives scleroderma fibrosis. Arthritis Rheumatol. 2016, 68, 2964–2974. [Google Scholar] [CrossRef]
- Mu, H.; Calderone, T.L.; Davies, M.A.; Prieto, V.G.; Wang, H.; Mills, G.B.; Bar-Eli, M.; Gershenwald, J.E. Lysophosphatidic acid induces lymphangiogenesis and IL-8 production in vitro in human lymphatic endothelial cells. Am. J. Pathol. 2012, 180, 2170–2181. [Google Scholar] [CrossRef]
- Tsukahara, T.; Haniu, H. Lysophosphatidic acid stimulates MCP-1 secretion from C2C12 myoblast. ISRN Inflamm. 2012, 2012, 1–6. [Google Scholar] [CrossRef]
- Hu, X.; Mendoza, F.J.; Sun, J.; Banerji, V.; Johnston, J.B.; Gibson, S.B. Lysophosphatidic acid (LPA) induces the expression of VEGF leading to protection against apoptosis in B-cell derived malignancies. Cell Signal. 2008, 20, 1198–1208. [Google Scholar] [CrossRef] [PubMed]
- Anzola, M. Hepatocellular carcinoma: Role of hepatitis B and hepatitis C viruses proteins in hepatocarcinogenesis. J. Viral Hepat. 2004, 11, 383–393. [Google Scholar] [CrossRef] [PubMed]
- Levrero, M.; Zucman-Rossi, J. Mechanisms of HBV-induced hepatocellular carcinoma. J. Hepatol. 2016, 64, 84–101. [Google Scholar] [CrossRef] [PubMed]
- Zemer, R.; Kitay Cohen, Y.; Naftaly, T.; Klein, A. Presence of hepatitis C virus DNA sequences in the DNA of infected patients. Eur. J. Clin. Investig. 2008, 38, 845–848. [Google Scholar] [CrossRef]
- Dejean, A.; de The, H. Hepatitis B virus as an insertional mutagene in a human hepatocellular carcinoma. Mol. Biol. Med. 1990, 7, 213–222. [Google Scholar]
- Watanabe, N.; Ikeda, H.; Nakamura, K.; Ohkawa, R.; Kume, Y.; Aoki, J.; Hama, K.; Okudaira, S.; Tanaka, M.; Tomiya, T.; et al. Both plasma lysophosphatidic acid and serum autotaxin levels are increased in chronic hepatitis C. J. Clin. Gastroenterol. 2007, 41, 616–623. [Google Scholar] [CrossRef]
- Yamazaki, T.; Joshita, S.; Umemura, T.; Usami, Y.; Sugiura, A.; Fujimori, N.; Shibata, S.; Ichikawa, Y.; Komatsu, M.; Matsumoto, A.; et al. Association of serum autotaxin levels with liver fibrosis in patients with chronic hepatitis, C. Sci. Rep. 2017, 7, e46705. [Google Scholar] [CrossRef]
- Joshita, S.; Ichikawa, Y.; Umemura, T.; Usami, Y.; Sugiura, A.; Shibata, S.; Yamazaki, T.; Fujimori, N.; Komatsu, M.; Matsumoto, A.; et al. Serum autotaxin is a useful liver fibrosis marker in patients with chronic hepatitis B virus infection. Hepatol. Res. 2018, 48, 275–285. [Google Scholar] [CrossRef]
- Cooper, A.B.; Wu, J.; Lu, D.; Maluccio, M.A. Is autotaxin (ENPP2) the link between hepatitis C and hepatocellular cancer? J. Gastrointest. Surg. 2007, 11, 1628–1635. [Google Scholar] [CrossRef]
- Farquhar, M.J.; Humphreys, I.S.; Rudge, S.A.; Wilson, G.K.; Bhattacharya, B.; Ciaccia, M.; Hu, K.; Zhang, Q.; Mailly, L.; Reynolds, G.M.; et al. Autotaxin-lysophosphatidic acid receptor signalling regulates hepatitis C virus replication. J. Hepatol. 2017, 66, 919–929. [Google Scholar] [CrossRef]
- Yamazaki, T.; Joshita, S.; Umemura, T.; Usami, Y.; Sugiura, A.; Fujimori, N.; Kimura, T.; Matsumoto, A.; Igarashi, K.; Ota, M.; et al. Changes in serum levels of autotaxin with direct-acting antiviral therapy in patients with chronic hepatitis C. PLoS ONE 2018, 13, e0195632. [Google Scholar] [CrossRef] [PubMed]
- Schlatzer, D.M.; Sugalski, J.M.; Chen, Y.; Barnholtz-Sloan, J.; Davitkov, P.; Hazlett, F.E.; Funderburg, N.; Rodriguez, B.; Lederman, M.M.; Sieg, S.F.; et al. Plasma proteome analysis reveals overlapping, yet distinct mechanisms of immune activation in chronic HCV and HIV infections. J. Acquir. Immune Defic. Syndr. 2013, 63, 563–571. [Google Scholar] [CrossRef] [PubMed]
- Kostadinova, L.; Shive, C.L.; Judge, C.; Zebrowski, E.; Compan, A.; Rife, K.; Hirsch, A.; Falck-Ytter, Y.; Schlatzer, D.M.; Li, X.; et al. During Hepatitis C Virus (HCV) infection and HCV-HIV coinfection, an elevated plasma level of autotaxin is associated with lysophosphatidic acid and markers of immune activation that normalize during interferon-free HCV therapy. J. Infect. Dis. 2016, 214, 1438–1448. [Google Scholar] [CrossRef] [PubMed]
- Sokolov, E.; Eheim, A.L.; Ahrens, W.A.; Walling, T.L.; Swet, J.H.; McMillan, M.T.; Simo, K.A.; Thompson, K.J.; Sindram, D.; McKillop, I.H. Lysophosphatidic acid receptor expression and function in human hepatocellular carcinoma. J. Surg. Res. 2013, 180, 104–113. [Google Scholar] [CrossRef]
- Ratziu, V.; Bellentani, S.; Cortez-Pinto, H.; Day, C.; Marchesini, G. A position statement on NAFLD/NASH based on the EASL 2009 special conference. J. Hepatol. 2010, 53, 372–384. [Google Scholar] [CrossRef]
- Kawada, N.; Imanaka, K.; Kawaguchi, T.; Tamai, C.; Ishihara, R.; Matsunaga, T.; Gotoh, K.; Yamada, T.; Tomita, Y. Hepatocellular carcinoma arising from non-cirrhotic nonalcoholic steatohepatitis. J. Gastroenterol. 2009, 44, 1190–1194. [Google Scholar] [CrossRef]
- Alexander, J.; Torbenson, M.; Wu, T.T.; Yeh, M.M. Non-alcoholic fatty liver disease contributes to hepatocarcinogenesis in non-cirrhotic liver: A clinical and pathological study. J. Gastroenterol. Hepatol. 2013, 28, 848–854. [Google Scholar] [CrossRef]
- Mohamad, B.; Shah, V.; Onyshchenko, M.; Elshamy, M.; Aucejo, F.; Lopez, R.; Hanouneh, I.A.; Alhaddad, R.; Alkhouri, N. Characterization of hepatocellular carcinoma (HCC) in non-alcoholic fatty liver disease (NAFLD) patients without cirrhosis. Hepatol. Int. 2016, 10, 632–639. [Google Scholar] [CrossRef]
- Michelotti, G.A.; Machado, M.V.; Diehl, A.M. NAFLD, NASH and liver cancer. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 656–665. [Google Scholar] [CrossRef]
- Perumpail, R.B.; Liu, A.; Wong, R.J.; Ahmed, A.; Harrison, S.A. Pathogenesis of hepatocarcinogenesis in non-cirrhotic nonalcoholic fatty liver disease: Potential mechanistic pathways. World J. Hepatol. 2015, 7, 2384–2388. [Google Scholar] [CrossRef]
- Hu, F.B. Globalization of diabetes: The role of diet, lifestyle, and genes. Diabetes Care 2011, 34, 1249–1257. [Google Scholar] [CrossRef] [PubMed]
- Imperatore, G.; Boyle, J.P.; Thompson, T.J.; Case, D.; Dabelea, D.; Hamman, R.F.; Lawrence, J.M.; Liese, A.D.; Liu, L.L.; Mayer-Davis, E.J.; et al. Projections of type 1 and type 2 diabetes burden in the U.S. population aged <20 years through 2050: Dynamic modeling of incidence, mortality, and population growth. Diabetes Care 2012, 35, 2515–2520. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Choi, S.; Park, S.M. Association of fasting serum glucose level and type 2 diabetes with hepatocellular carcinoma in men with chronic hepatitis B infection: A large cohort study. Eur. J. Cancer 2018, 102, 103–113. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Deng, Y.L.; Liu, C.; Huang, L.H.; Shang, L.; Chen, X.G.; Wang, L.T.; Du, J.Z.; Wang, Y.; Wang, P.X.; et al. Diabetes mellitus may affect the long-term survival of hepatitis B virus-related hepatocellular carcinoma patients after liver transplantation. World J. Gastroenterol. 2016, 22, 9571–9585. [Google Scholar] [CrossRef]
- Bechmann, L.P.; Hannivoort, R.A.; Gerken, G.; Hotamisligil, G.S.; Trauner, M.; Canbay, A. The interaction of hepatic lipid and glucose metabolism in liver diseases. J. Hepatol. 2012, 56, 952–964. [Google Scholar] [CrossRef]
- D’Souza, K.; Paramel, G.V.; Kienesberger, P.C. Lysophosphatidic acid signaling in obesity and insulin resistance. Nutrients 2018, 10, 399. [Google Scholar] [CrossRef]
- Rachakonda, V.P.; Reeves, V.L.; Aljammal, J.; Wills, R.C.; Trybula, J.S.; DeLany, J.P.; Kienesberger, P.C.; Kershaw, E.E. Serum autotaxin is independently associated with hepatic steatosis in women with severe obesity. Obesity (Silver Spring) 2015, 23, 965–972. [Google Scholar] [CrossRef]
- Reeves, V.L.; Trybula, J.S.; Wills, R.C.; Goodpaster, B.H.; Dube, J.J.; Kienesberger, P.C.; Kershaw, E.E. Serum Autotaxin/ENPP2 correlates with insulin resistance in older humans with obesity. Obesity (Silver Spring) 2015, 23, 2371–2376. [Google Scholar] [CrossRef]
- Fayyaz, S.; Japtok, L.; Schumacher, F.; Wigger, D.; Schulz, T.J.; Haubold, K.; Gulbins, E.; Voller, H.; Kleuser, B. Lysophosphatidic acid inhibits insulin signaling in primary rat hepatocytes via the LPA3 receptor subtype and is increased in obesity. Cell. Physiol. Biochem. 2017, 43, 445–456. [Google Scholar] [CrossRef]
- Han, M.S.; Park, S.Y.; Shinzawa, K.; Kim, S.; Chung, K.W.; Lee, J.H.; Kwon, C.H.; Lee, K.W.; Lee, J.H.; Park, C.K.; et al. Lysophosphatidylcholine as a death effector in the lipoapoptosis of hepatocytes. J. Lipid Res. 2008, 49, 84–97. [Google Scholar] [CrossRef]
- Kolodziejczyk, A.A.; Zheng, D.; Shibolet, O.; Elinav, E. The role of the microbiome in NAFLD and NASH. EMBO Mol. Med. 2019, 11, e9302. [Google Scholar] [CrossRef] [PubMed]
- Goedeke, L.; Perry, R.J.; Shulman, G.I. Emerging pharmacological targets for the treatment of nonalcoholic fatty liver disease, insulin resistance, and type 2 diabetes. Annu. Rev. Pharmacol. Toxicol. 2019, 59, 65–87. [Google Scholar] [CrossRef] [PubMed]
- Petersen, M.C.; Shulman, G.I. Mechanisms of insulin action and insulin resistance. Physiol. Rev. 2018, 98, 2133–2223. [Google Scholar] [CrossRef] [PubMed]
- Samuel, V.T.; Shulman, G.I. The pathogenesis of insulin resistance: Integrating signaling pathways and substrate flux. J. Clin. Investig. 2016, 126, 12–22. [Google Scholar] [CrossRef] [PubMed]
- Shulman, G.I. Ectopic fat in insulin resistance, dyslipidemia, and cardiometabolic disease. N. Engl. J. Med. 2014, 371, 1131–1141. [Google Scholar] [CrossRef]
- Perry, R.J.; Samuel, V.T.; Petersen, K.F.; Shulman, G.I. The role of hepatic lipids in hepatic insulin resistance and type 2 diabetes. Nature 2014, 510, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Perry, R.J.; Zhang, D.; Zhang, X.M.; Boyer, J.L.; Shulman, G.I. Controlled-release mitochondrial protonophore reverses diabetes and steatohepatitis in rats. Science 2015, 347, 1253–1256. [Google Scholar] [CrossRef]
- D’Souza, K.; Nzirorera, C.; Cowie, A.M.; Varghese, G.P.; Trivedi, P.; Eichmann, T.O.; Biswas, D.; Touaibia, M.; Morris, A.J.; Aidinis, V.; et al. Autotaxin-LPA signaling contributes to obesity-induced insulin resistance in muscle and impairs mitochondrial metabolism. J. Lipid Res. 2018, 59, 1805–1817. [Google Scholar] [CrossRef]
- Dusaulcy, R.; Daviaud, D.; Pradere, J.P.; Gres, S.; Valet, P.; Saulnier-Blache, J.S. Altered food consumption in mice lacking lysophosphatidic acid receptor-1. J. Physiol. Biochem. 2009, 65, 345–350. [Google Scholar] [CrossRef]
- Im, D.S.; Fujioka, T.; Katada, T.; Kondo, Y.; Ui, M.; Okajima, F. Characterization of sphingosine 1-phosphate-induced actions and its signaling pathways in rat hepatocytes. Am. J. Physiol. 1997, 272, 1091–1099. [Google Scholar] [CrossRef]
- Rancoule, C.; Attane, C.; Gres, S.; Fournel, A.; Dusaulcy, R.; Bertrand, C.; Vinel, C.; Treguer, K.; Prentki, M.; Valet, P.; et al. Lysophosphatidic acid impairs glucose homeostasis and inhibits insulin secretion in high-fat diet obese mice. Diabetologia 2013, 56, 1394–1402. [Google Scholar] [CrossRef] [PubMed]
- Yea, K.; Kim, J.; Lim, S.; Park, H.S.; Park, K.S.; Suh, P.G.; Ryu, S.H. Lysophosphatidic acid regulates blood glucose by stimulating myotube and adipocyte glucose uptake. J. Mol. Med. (Berl.) 2008, 86, 211–220. [Google Scholar] [CrossRef] [PubMed]
- Cao, H.; Gerhold, K.; Mayers, J.R.; Wiest, M.M.; Watkins, S.M.; Hotamisligil, G.S. Identification of a lipokine, a lipid hormone linking adipose tissue to systemic metabolism. Cell 2008, 134, 933–944. [Google Scholar] [CrossRef] [PubMed]
- Parimisetty, A.; Dorsemans, A.C.; Awada, R.; Ravanan, P.; Diotel, N.; Lefebvre d’Hellencourt, C. Secret talk between adipose tissue and central nervous system via secreted factors-an emerging frontier in the neurodegenerative research. J. Neuroinflamm. 2016, 13, e67. [Google Scholar] [CrossRef]
- Gluchowski, N.L.; Becuwe, M.; Walther, T.C.; Farese, R.V., Jr. Lipid droplets and liver disease: From basic biology to clinical implications. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 343–355. [Google Scholar] [CrossRef]
- Calvisi, D.F. De novo lipogenesis: Role in hepatocellular carcinoma. Pathologe 2011, 32, 174–180. [Google Scholar] [CrossRef]
- Ameer, F.; Scandiuzzi, L.; Hasnain, S.; Kalbacher, H.; Zaidi, N. De novo lipogenesis in health and disease. Metabolism 2014, 63, 895–902. [Google Scholar] [CrossRef]
- Mukherjee, A.; Wu, J.; Barbour, S.; Fang, X. Lysophosphatidic acid activates lipogenic pathways and de novo lipid synthesis in ovarian cancer cells. J. Biol. Chem. 2012, 287, 24990–25000. [Google Scholar] [CrossRef]
- Aragones, G.; Auguet, T.; Armengol, S.; Berlanga, A.; Guiu-Jurado, E.; Aguilar, C.; Martinez, S.; Sabench, F.; Porras, J.A.; Ruiz, M.D.; et al. PNPLA3 expression is related to liver steatosis in morbidly obese women with non-alcoholic fatty liver disease. Int. J. Mol. Sci. 2016, 17, 630. [Google Scholar] [CrossRef]
- Li, J.F.; Zheng, E.Q.; Xie, M. Association between rs738409 polymorphism in patatin-like phospholipase domain-containing protein 3 (PNPLA3) gene and hepatocellular carcinoma susceptibility: Evidence from case-control studies. Gene 2019, 685, 143–148. [Google Scholar] [CrossRef]
- Liu, Y.L.; Patman, G.L.; Leathart, J.B.; Piguet, A.C.; Burt, A.D.; Dufour, J.F.; Day, C.P.; Daly, A.K.; Reeves, H.L.; Anstee, Q.M. Carriage of the PNPLA3 rs738409 C >G polymorphism confers an increased risk of non-alcoholic fatty liver disease associated hepatocellular carcinoma. J. Hepatol. 2014, 61, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Sanders, F.W.; Griffin, J.L. De novo lipogenesis in the liver in health and disease: More than just a shunting yard for glucose. Biol. Rev. Camb. Philos. Soc. 2016, 91, 452–468. [Google Scholar] [CrossRef] [PubMed]
- Skill, N.J.; Scott, R.E.; Wu, J.; Maluccio, M.A. Hepatocellular carcinoma associated lipid metabolism reprogramming. J. Surg. Res. 2011, 169, 51–56. [Google Scholar] [CrossRef]
- Skill, N.J.; Jianmin, W.; Yan, X.; Zhao, Z.; Tector, A.J.; Maluccio, M.A. Lysophospholipid variants in hepatocellular carcinoma. J. Surg. Res. 2013, 182, 241–249. [Google Scholar] [CrossRef] [PubMed]
- Wendel, A.A.; Lewin, T.M.; Coleman, R.A. Glycerol-3-phosphate acyltransferases: Rate limiting enzymes of triacylglycerol biosynthesis. Biochim. Biophys. Acta 2009, 1791, 501–506. [Google Scholar] [CrossRef]
- Enooku, K.; Uranbileg, B.; Ikeda, H.; Kurano, M.; Sato, M.; Kudo, H.; Maki, H.; Koike, K.; Hasegawa, K.; Kokudo, N.; et al. Higher LPA2 and LPA6 mRNA levels in hepatocellular carcinoma are associated with poorer differentiation, microvascular invasion and earlier recurrence with higher serum autotaxin levels. PLoS ONE 2016, 11, e0161825. [Google Scholar] [CrossRef]
- Frau, F.; Crowther, D.; Ruetten, H.; Allebrandt, K.V. Type-2 diabetes-associated variants with cross-trait relevance: Post-GWAs strategies for biological function interpretation. Mol. Genet. Metab. 2017, 121, 43–50. [Google Scholar] [CrossRef]
- Mazzocca, A.; Dituri, F.; De Santis, F.; Filannino, A.; Lopane, C.; Betz, R.C.; Li, Y.Y.; Mukaida, N.; Winter, P.; Tortorella, C.; et al. Lysophosphatidic acid receptor LPAR6 supports the tumorigenicity of hepatocellular carcinoma. Cancer Res. 2015, 75, 532–543. [Google Scholar] [CrossRef]
- Zuckerman, V.; Sokolov, E.; Swet, J.H.; Ahrens, W.A.; Showlater, V.; Iannitti, D.A.; McKillop, I.H. Expression and function of lysophosphatidic acid receptors (LPARs) 1 and 3 in human hepatic cancer progenitor cells. Oncotarget 2016, 7, 2951–2967. [Google Scholar] [CrossRef]
- Park, S.Y.; Jeong, K.J.; Panupinthu, N.; Yu, S.; Lee, J.; Han, J.W.; Kim, J.M.; Lee, J.S.; Kang, J.; Park, C.G.; et al. Lysophosphatidic acid augments human hepatocellular carcinoma cell invasion through LPA1 receptor and MMP-9 expression. Oncogene 2011, 30, 1351–1359. [Google Scholar] [CrossRef]
- Xu, M.; Liu, Z.; Wang, C.; Yao, B.; Zheng, X. EDG2 enhanced the progression of hepatocellular carcinoma by LPA/PI3K/AKT/ mTOR signaling. Oncotarget 2017, 8, 66154–66168. [Google Scholar] [CrossRef] [PubMed]
- Mukai, M.; Nakamura, H.; Tatsuta, M.; Iwasaki, T.; Togawa, A.; Imamura, F.; Akedo, H. Hepatoma cell migration through a mesothelial cell monolayer is inhibited by cyclic AMP-elevating agents via a Rho-dependent pathway. FEBS Lett. 2000, 484, 69–73. [Google Scholar] [CrossRef]
- Fosslien, E. Cancer morphogenesis: Role of mitochondrial failure. Ann. Clin. Lab. Sci. 2008, 38, 307–329. [Google Scholar] [PubMed]
- Guerra, F.; Arbini, A.A.; Moro, L. Mitochondria and cancer chemoresistance. Biochim. Biophys. Acta Bioenerg. 2017, 1858, 686–699. [Google Scholar] [CrossRef] [PubMed]
- Rangarajan, S.; Bernard, K.; Thannickal, V.J. Mitochondrial dysfunction in pulmonary fibrosis. Ann. Am. Thorac. Soc. 2017, 14, 383–388. [Google Scholar] [CrossRef] [PubMed]
- Gong, Z.; Pan, J.; Shen, Q.; Li, M.; Peng, Y. Mitochondrial dysfunction induces NLRP3 inflammasome activation during cerebral ischemia/reperfusion injury. J. Neuroinflamm. 2018, 15, e242. [Google Scholar] [CrossRef]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef]
- Ghouri, Y.A.; Mian, I.; Blechacz, B. Cancer review: Cholangiocarcinoma. J. Carcinog. 2015, 14, e1. [Google Scholar] [CrossRef]
- Kremer, A.E.; Martens, J.J.; Kulik, W.; Rueff, F.; Kuiper, E.M.; van Buuren, H.R.; van Erpecum, K.J.; Kondrackiene, J.; Prieto, J.; Rust, C.; et al. Lysophosphatidic acid is a potential mediator of cholestatic pruritus. Gastroenterology 2010, 139, 1008–1018.e1. [Google Scholar] [CrossRef]
- Kremer, A.E.; van Dijk, R.; Leckie, P.; Schaap, F.G.; Kuiper, E.M.; Mettang, T.; Reiners, K.S.; Raap, U.; van Buuren, H.R.; van Erpecum, K.J.; et al. Serum autotaxin is increased in pruritus of cholestasis, but not of other origin, and responds to therapeutic interventions. Hepatology 2012, 56, 1391–1400. [Google Scholar] [CrossRef]
- Sun, Y.; Zhang, W.; Evans, J.F.; Floreani, A.; Zou, Z.; Nishio, Y.; Qi, R.; Leung, P.S.; Bowlus, C.L.; Gershwin, M.E. Autotaxin, pruritus and Primary Biliary Cholangitis (PBC). Autoimmun. Rev. 2016, 15, 795–800. [Google Scholar] [CrossRef] [PubMed]
- Bolier, R.; Tolenaars, D.; Kremer, A.E.; Saris, J.; Pares, A.; Verheij, J.; Bosma, P.J.; Beuers, U.; Oude Elferink, R.P.J. Enteroendocrine cells are a potential source of serum autotaxin in men. Biochim. Biophys. Acta 2016, 1862, 696–704. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.X.; Zhao, B.; Panupinthu, N.; Jewell, J.L.; Lian, I.; Wang, L.H.; Zhao, J.; Yuan, H.; Tumaneng, K.; Li, H.; et al. Regulation of the Hippo-YAP pathway by G-protein-coupled receptor signaling. Cell 2012, 150, 780–791. [Google Scholar] [CrossRef] [PubMed]
- Marti, P.; Stein, C.; Blumer, T.; Abraham, Y.; Dill, M.T.; Pikiolek, M.; Orsini, V.; Jurisic, G.; Megel, P.; Makowska, Z.; et al. YAP promotes proliferation, chemoresistance, and angiogenesis in human cholangiocarcinoma through TEAD transcription factors. Hepatology 2015, 62, 1497–1510. [Google Scholar] [CrossRef] [PubMed]
- Rizvi, S.; Fischbach, S.R.; Bronk, S.F.; Hirsova, P.; Krishnan, A.; Dhanasekaran, R.; Smadbeck, J.B.; Smoot, R.L.; Vasmatzis, G.; Gores, G.J. YAP-associated chromosomal instability and cholangiocarcinoma in mice. Oncotarget 2018, 9, 5892–5905. [Google Scholar] [CrossRef] [PubMed]
- Pei, T.; Li, Y.; Wang, J.; Wang, H.; Liang, Y.; Shi, H.; Sun, B.; Yin, D.; Sun, J.; Song, R.; et al. YAP is a critical oncogene in human cholangiocarcinoma. Oncotarget 2015, 6, 17206–17220. [Google Scholar] [CrossRef]
- Huang, Y.L.; Lin, Y.C.; Lin, C.C.; Chen, W.M.; Chen, B.P.C.; Lee, H. High glucose induces VEGF-C expression via the LPA1/3-Akt-ROS-LEDGF signaling axis in human prostate cancer PC-3 cells. Cell. Physiol. Biochem. 2018, 50, 597–611. [Google Scholar] [CrossRef]
- Mariotti, V.; Strazzabosco, M.; Fabris, L.; Calvisi, D.F. Animal models of biliary injury and altered bile acid metabolism. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 1254–1261. [Google Scholar] [CrossRef]
- Skill, N.; Wu, J.; Xu, Y.; Zhao, Z.; Maluccio, M. Lysophosphatidic acid aberrancies and hepatocellular carcinoma: Studies in the MDR2 gene knockout mouse. Cancer Investig. 2013, 31, 145–155. [Google Scholar] [CrossRef]
- Lozano, E.; Sanchez-Vicente, L.; Monte, M.J.; Herraez, E.; Briz, O.; Banales, J.M.; Marin, J.J.; Macias, R.I. Cocarcinogenic effects of intrahepatic bile acid accumulation in cholangiocarcinoma development. Mol. Cancer Res. 2014, 12, 91–100. [Google Scholar] [CrossRef]
- Bain, G.; Shannon, K.E.; Huang, F.; Darlington, J.; Goulet, L.; Prodanovich, P.; Ma, G.L.; Santini, A.M.; Stein, A.J.; Lonergan, D.; et al. Selective inhibition of autotaxin is efficacious in mouse models of liver fibrosis. J. Pharmacol. Exp. Ther. 2017, 360, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Allanore, Y.; Distler, O.; Jagerschmidt, A.; Illiano, S.; Ledein, L.; Boitier, E.; Agueusop, I.; Denton, C.P.; Khanna, D. Lysophosphatidic acid receptor 1 antagonist SAR100842 for patients with diffuse cutaneous systemic sclerosis: A double-blind, randomized, eight-week placebo-controlled study followed by a sixteen-week open-label extension study. Arthritis Rheumatol. 2018, 70, 1634–1643. [Google Scholar] [CrossRef] [PubMed]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kaffe, E.; Magkrioti, C.; Aidinis, V. Deregulated Lysophosphatidic Acid Metabolism and Signaling in Liver Cancer. Cancers 2019, 11, 1626. https://doi.org/10.3390/cancers11111626
Kaffe E, Magkrioti C, Aidinis V. Deregulated Lysophosphatidic Acid Metabolism and Signaling in Liver Cancer. Cancers. 2019; 11(11):1626. https://doi.org/10.3390/cancers11111626
Chicago/Turabian StyleKaffe, Eleanna, Christiana Magkrioti, and Vassilis Aidinis. 2019. "Deregulated Lysophosphatidic Acid Metabolism and Signaling in Liver Cancer" Cancers 11, no. 11: 1626. https://doi.org/10.3390/cancers11111626
APA StyleKaffe, E., Magkrioti, C., & Aidinis, V. (2019). Deregulated Lysophosphatidic Acid Metabolism and Signaling in Liver Cancer. Cancers, 11(11), 1626. https://doi.org/10.3390/cancers11111626