The Epithelial to Mesenchymal Transition Promotes Glutamine Independence by Suppressing GLS2 Expression


 ,
,  ,
, 
Abstract
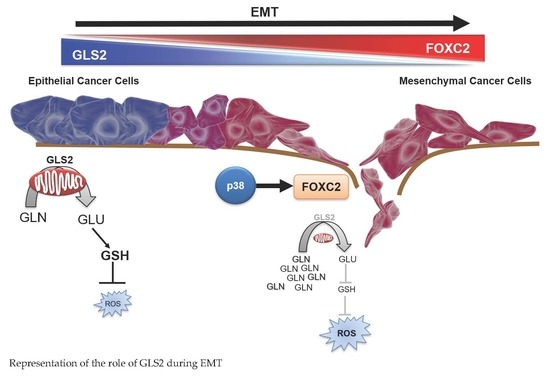
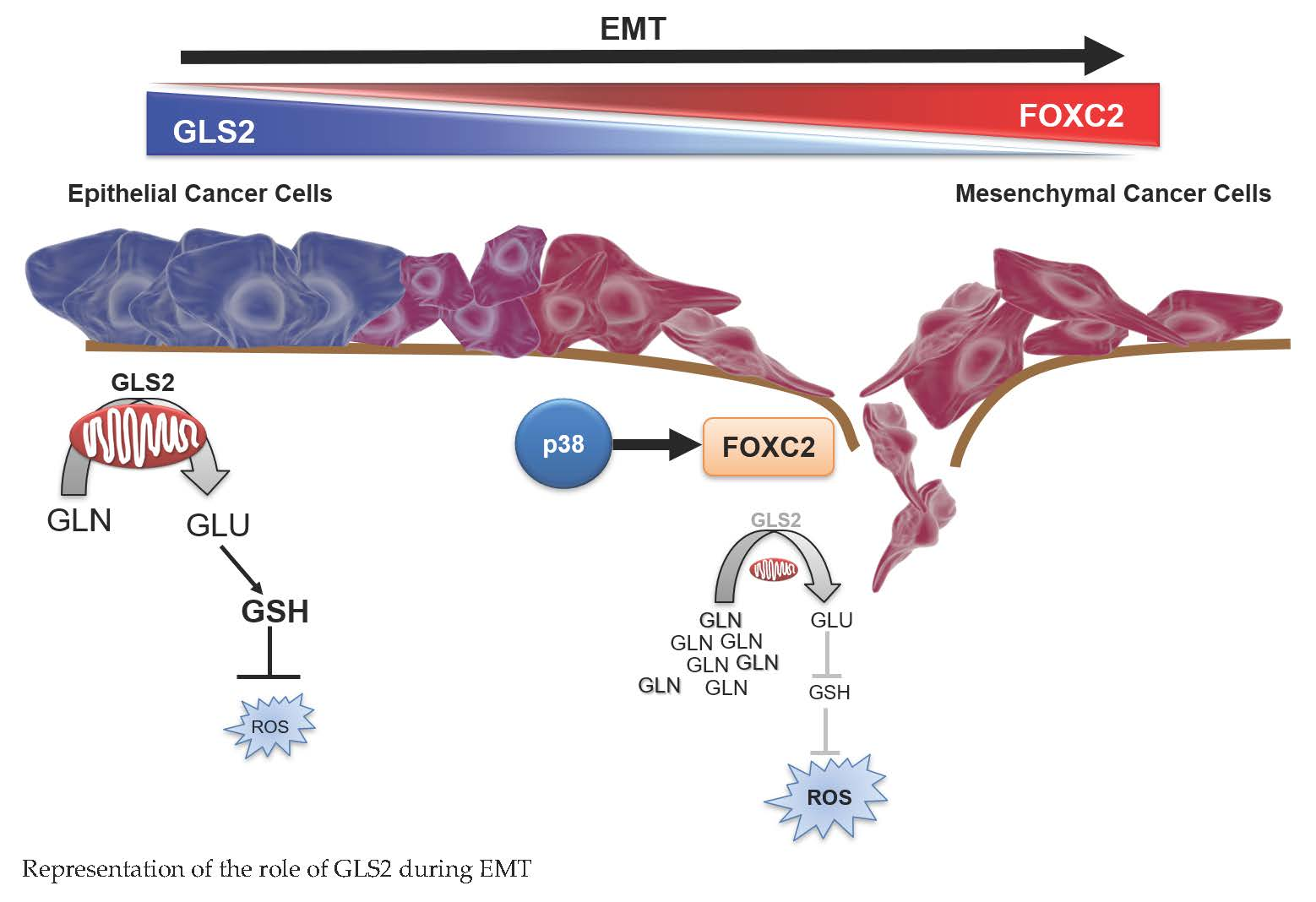{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
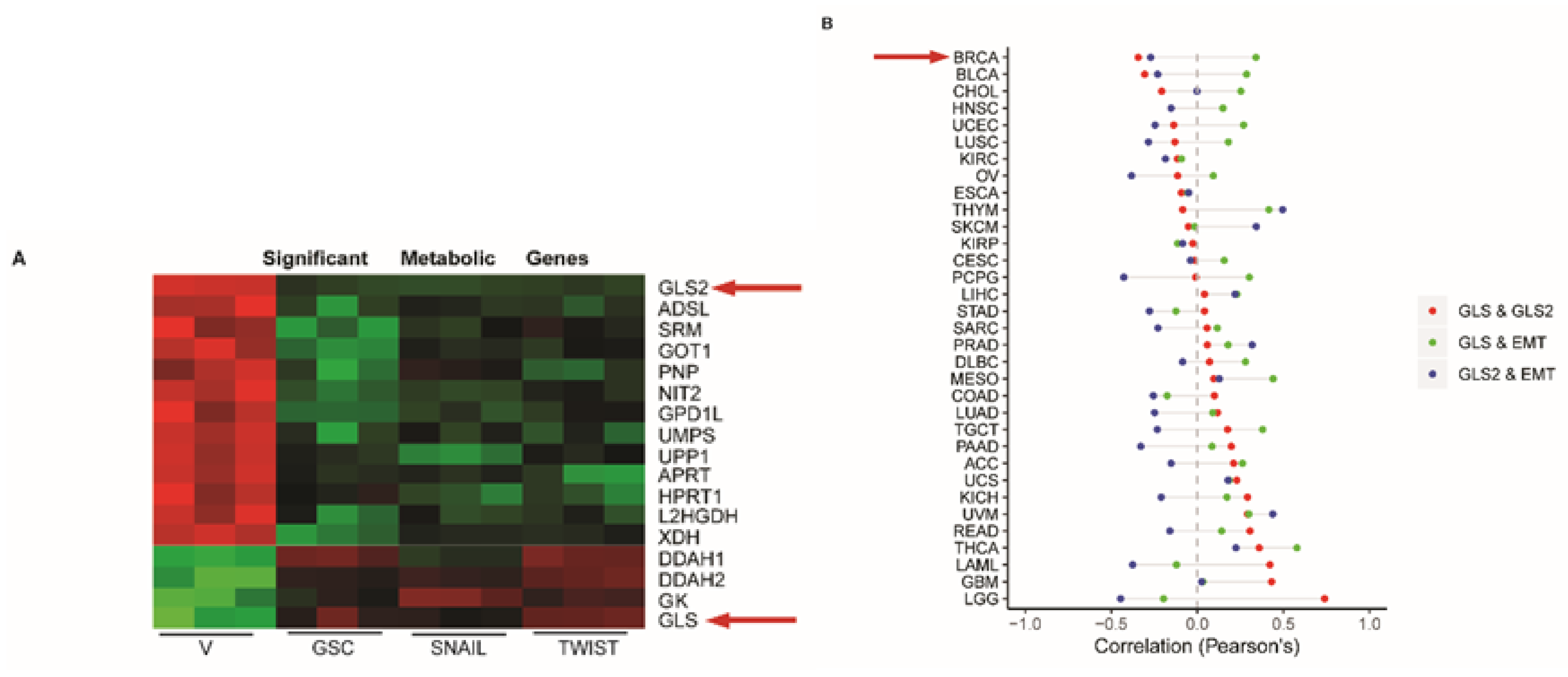
2.1. GLS2 Expression Is Inversely Correlated with EMT in Breast Cancer
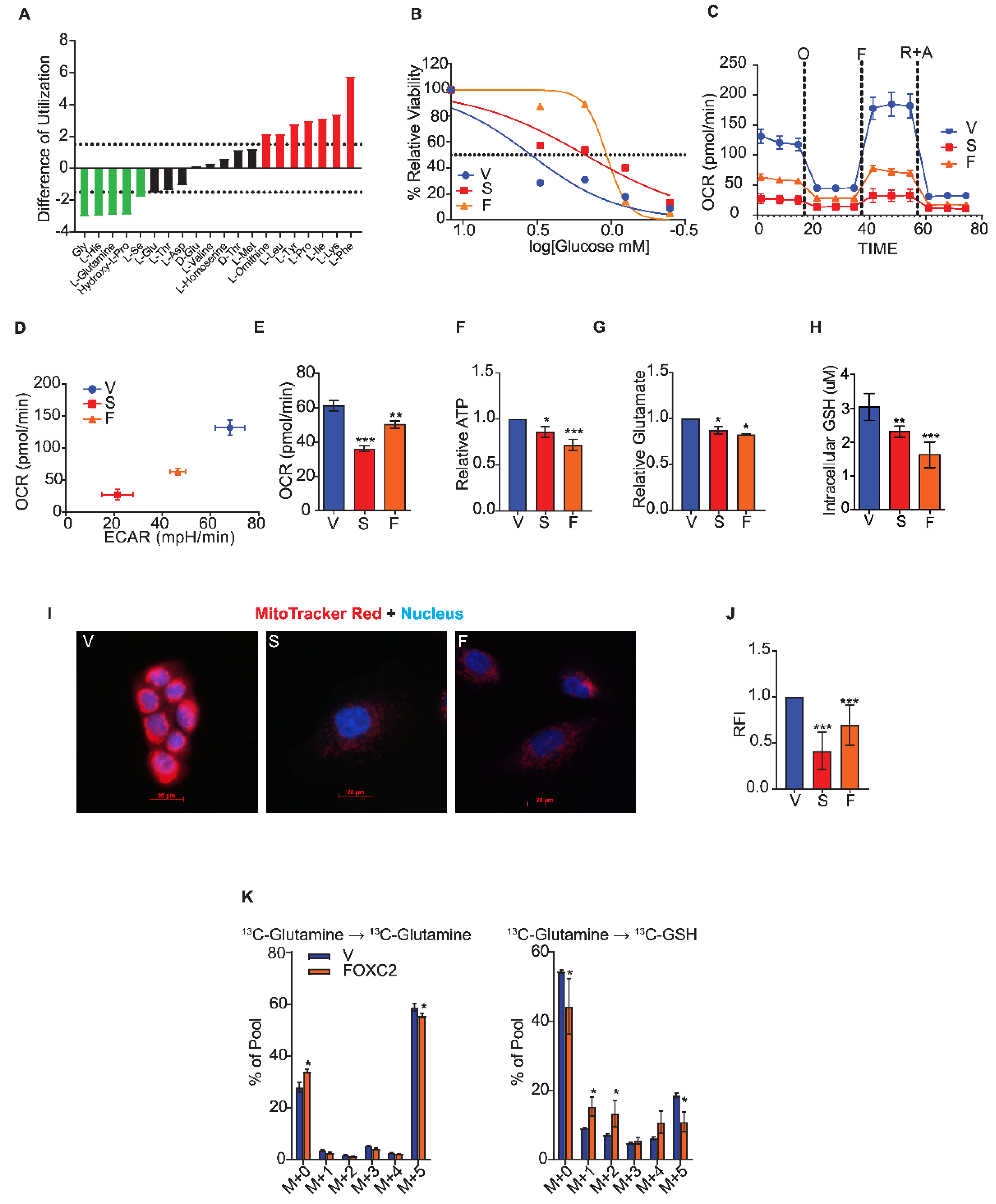
2.2. Cells that Undergo the Epithelial to Mesenchymal Transition Fail to Utilize Glutamine
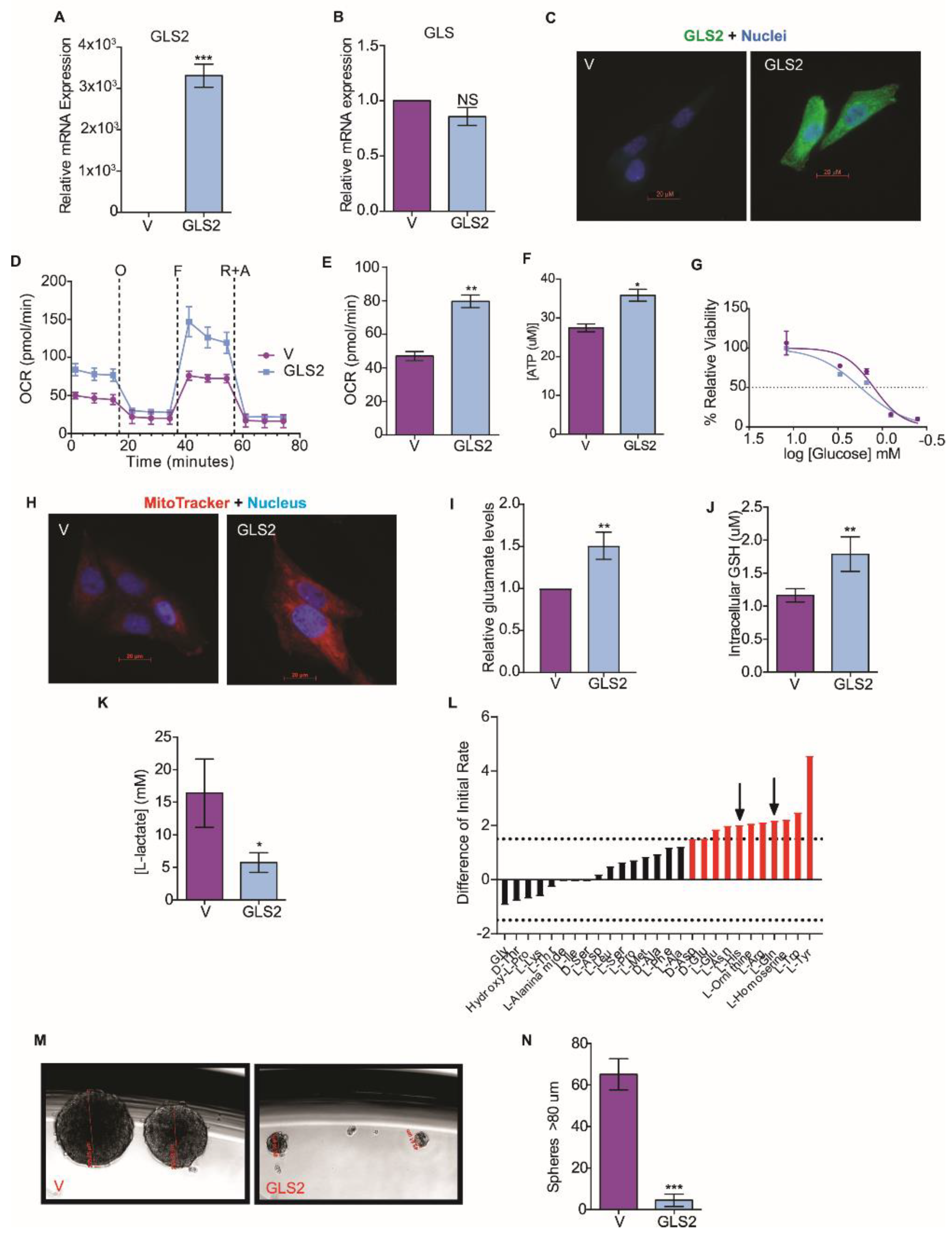
2.3. GLS2 Over-Expression Rescues Glutamine Utilization
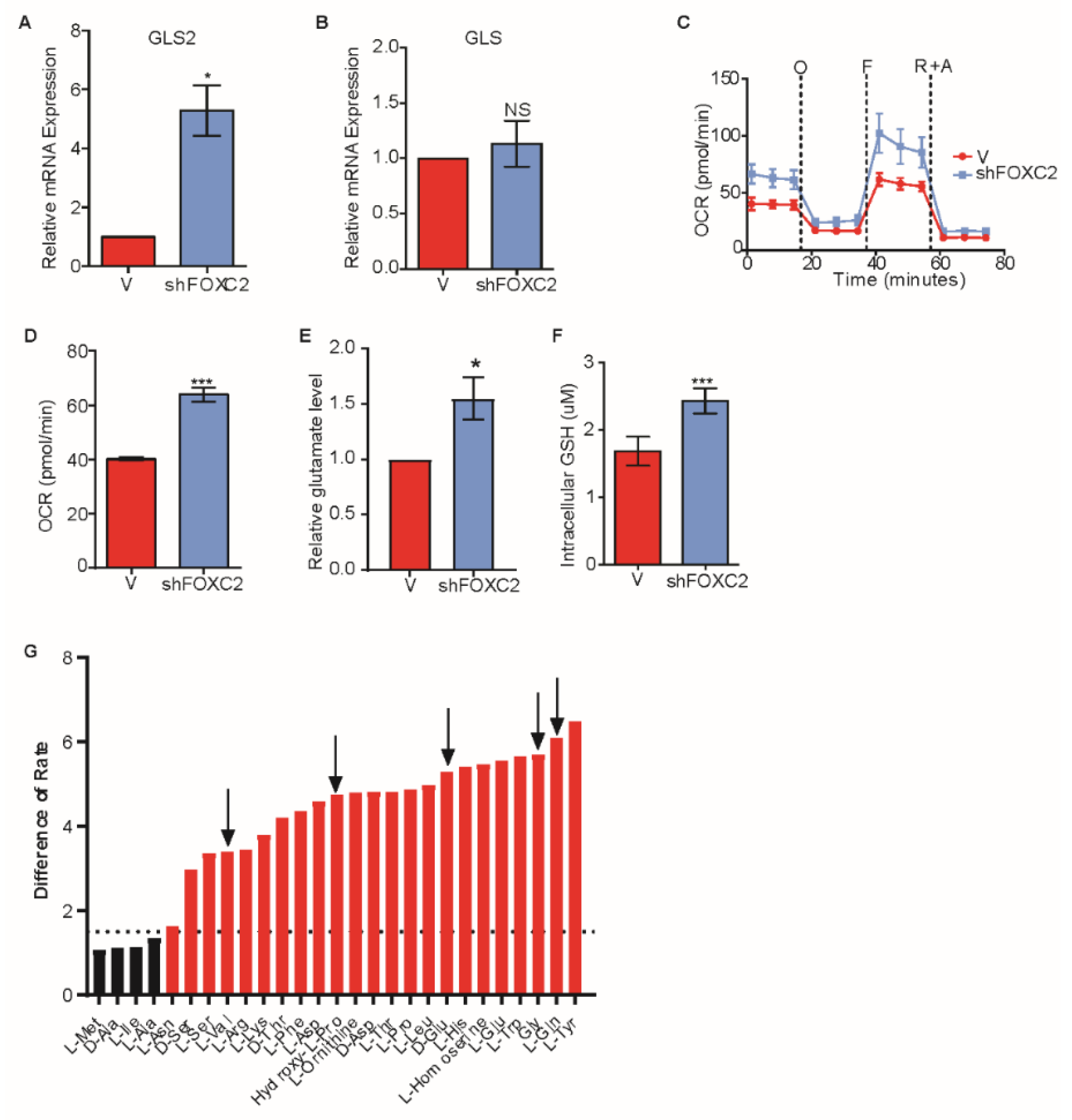
2.4. Inhibition of FOXC2 Enhances GLS2 Expression and Glutamine Utilization
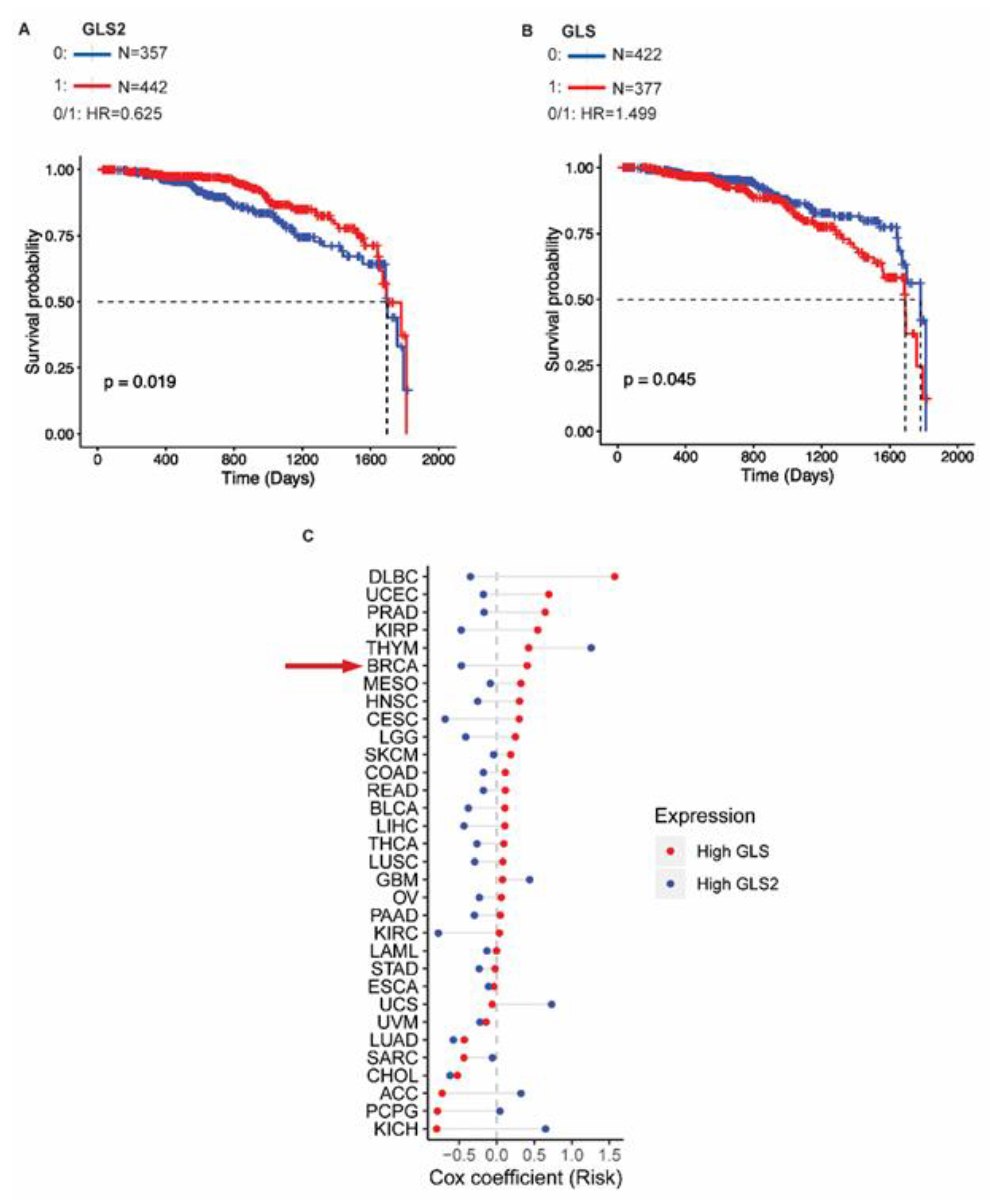
2.5. GLS2 Expression Correlates with Survival of Breast Cancer Patients
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Biolog Assay
4.3. Glucose Sensitivity Assay
4.4. Seahorse Assays
4.5. ATP Determination
4.6. Glutamate Determination
4.7. GSH Determination
4.8. MitoTracker Red
4.9. 13C-Glutamine Flux
4.10. Steady-State Mass Spectrometry
4.11. RNA Extraction and qRT-PCR
4.12. Immunofluorescence
4.13. Plasmids and Viral Transduction
4.14. Breast Cancer Data Analysis
4.15. Copy Number Analysis
4.16. L-Lactate Measurement
4.17. Proliferation Assay
4.18. Mammosphere Assay
4.19. Pan-Cancer Correlation and Survival Analysis
4.20. Gene Expression Analysis
4.21. Statistical Analysis
4.22. Western Blot Analysis and Antibodies
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- DeBerardinis, R.J.; Chandel, N.S. Fundamentals of cancer metabolism. Sci. Adv. 2016, 2, e1600200. [Google Scholar] [CrossRef] [PubMed]
- Mates, J.M.; Segura, J.A.; Campos-Sandoval, J.A.; Lobo, C.; Alonso, L.; Alonso, F.J.; Marquez, J. Glutamine homeostasis and mitochondrial dynamics. Int. J. Biochem. Cell Biol. 2009, 41, 2051–2061. [Google Scholar] [CrossRef] [PubMed]
- Marquez, J.; Mates, J.M.; Campos-Sandoval, J.A. Glutaminases. Adv. Neurobiol. 2016, 13, 133–171. [Google Scholar] [CrossRef] [PubMed]
- Son, J.; Lyssiotis, C.A.; Ying, H.; Wang, X.; Hua, S.; Ligorio, M.; Perera, R.M.; Ferrone, C.R.; Mullarky, E.; Shyh-Chang, N.; et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature 2013, 496, 101–105. [Google Scholar] [CrossRef]
- Gao, P.; Tchernyshyov, I.; Chang, T.C.; Lee, Y.S.; Kita, K.; Ochi, T.; Zeller, K.I.; De Marzo, A.M.; Van Eyk, J.E.; Mendell, J.T.; et al. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature 2009, 458, 762–765. [Google Scholar] [CrossRef]
- Suzuki, S.; Tanaka, T.; Poyurovsky, M.V.; Nagano, H.; Mayama, T.; Ohkubo, S.; Lokshin, M.; Hosokawa, H.; Nakayama, T.; Suzuki, Y.; et al. Phosphate-activated glutaminase (GLS2), a p53-inducible regulator of glutamine metabolism and reactive oxygen species. Proc. Natl. Acad. Sci. USA 2010, 107, 7461–7466. [Google Scholar] [CrossRef]
- Giacobbe, A.; Bongiorno-Borbone, L.; Bernassola, F.; Terrinoni, A.; Markert, E.K.; Levine, A.J.; Feng, Z.; Agostini, M.; Zolla, L.; Agro, A.F.; et al. p63 regulates glutaminase 2 expression. Cell Cycle 2013, 12, 1395–1405. [Google Scholar] [CrossRef]
- Velletri, T.; Romeo, F.; Tucci, P.; Peschiaroli, A.; Annicchiarico-Petruzzelli, M.; Niklison-Chirou, M.V.; Amelio, I.; Knight, R.A.; Mak, T.W.; Melino, G.; et al. GLS2 is transcriptionally regulated by p73 and contributes to neuronal differentiation. Cell Cycle 2013, 12, 3564–3573. [Google Scholar] [CrossRef]
- Szeliga, M.; Bogacinska-Karas, M.; Kuzmicz, K.; Rola, R.; Albrecht, J. Downregulation of GLS2 in glioblastoma cells is related to DNA hypermethylation but not to the p53 status. Mol. Carcinog. 2016, 55, 1309–1316. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, C.; Chen, M.; Cao, J.; Zhong, Y.; Chen, L.; Shen, H.M.; Xia, D. Epigenetic silencing of glutaminase 2 in human liver and colon cancers. BMC Cancer 2013, 13, 601. [Google Scholar] [CrossRef]
- Commisso, C.; Davidson, S.M.; Soydaner-Azeloglu, R.G.; Parker, S.J.; Kamphorst, J.J.; Hackett, S.; Grabocka, E.; Nofal, M.; Drebin, J.A.; Thompson, C.B.; et al. Macropinocytosis of protein is an amino acid supply route in Ras-transformed cells. Nature 2013, 497, 633–637. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Kepp, O.; Vander Heiden, M.G.; Kroemer, G. Metabolic targets for cancer therapy. Nat. Rev. Drug Discov. 2013, 12, 829–846. [Google Scholar] [CrossRef] [PubMed]
- Venneti, S.; Dunphy, M.P.; Zhang, H.; Pitter, K.L.; Zanzonico, P.; Campos, C.; Carlin, S.D.; La Rocca, G.; Lyashchenko, S.; Ploessl, K.; et al. Glutamine-based PET imaging facilitates enhanced metabolic evaluation of gliomas in vivo. Sci. Transl. Med. 2015, 7, 274ra217. [Google Scholar] [CrossRef] [PubMed]
- Altman, B.J.; Stine, Z.E.; Dang, C.V. From Krebs to clinic: Glutamine metabolism to cancer therapy. Nat. Rev. Cancer 2016, 16, 749. [Google Scholar] [CrossRef] [PubMed]
- Mani, S.A.; Yang, J.; Brooks, M.; Schwaninger, G.; Zhou, A.; Miura, N.; Kutok, J.L.; Hartwell, K.; Richardson, A.L.; Weinberg, R.A. Mesenchyme Forkhead 1 (FOXC2) plays a key role in metastasis and is associated with aggressive basal-like breast cancers. Proc. Natl. Acad. Sci. USA 2007, 104, 10069–10074. [Google Scholar] [CrossRef] [PubMed]
- Creighton, C.J.; Li, X.; Landis, M.; Dixon, J.M.; Neumeister, V.M.; Sjolund, A.; Rimm, D.L.; Wong, H.; Rodriguez, A.; Herschkowitz, J.I.; et al. Residual breast cancers after conventional therapy display mesenchymal as well as tumor-initiating features. Proc. Natl. Acad. Sci. USA 2009, 106, 13820–13825. [Google Scholar] [CrossRef]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.J.; Nieto, M.A. Epithelial-Mesenchymal Transitions in Development and Disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef]
- Olalla, L.; Gutierrez, A.; Jimenez, A.J.; Lopez-Tellez, J.F.; Khan, Z.U.; Perez, J.; Alonso, F.J.; de la Rosa, V.; Campos-Sandoval, J.A.; Segura, J.A.; et al. Expression of the scaffolding PDZ protein glutaminase-interacting protein in mammalian brain. J. Neurosci. Res. 2008, 86, 281–292. [Google Scholar] [CrossRef]
- Hiscox, S.; Jiang, W.G.; Obermeier, K.; Taylor, K.; Morgan, L.; Burmi, R.; Barrow, D.; Nicholson, R.I. Tamoxifen resistance in MCF7 cells promotes EMT-like behaviour and involves modulation of β-catenin phosphorylation. Int. J. Cancer 2006, 118, 290–301. [Google Scholar] [CrossRef]
- Huber, M.A.; Kraut, N.; Beug, H. Molecular requirements for epithelial-mesenchymal transition during tumor progression. Curr. Opin. Cell Biol. 2005, 17, 548–558. [Google Scholar] [CrossRef]
- Brabletz, T.; Jung, A.; Spaderna, S.; Hlubek, F.; Kirchner, T. Opinion: Migrating cancer stem cells—an integrated concept of malignant tumour progression. Nat. Rev. Cancer 2005, 5, 744–749. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.; Cai, Y.; Liu, J.; Wang, Z.; Wu, Q.; Zhang, Z.; Yang, C.J.; Yuan, L.; Ouyang, G. Twist2 contributes to breast cancer progression by promoting an epithelial-mesenchymal transition and cancer stem-like cell self-renewal. Oncogene 2011, 30, 4707–4720. [Google Scholar] [CrossRef] [PubMed]
- Chiou, S.H.; Wang, M.L.; Chou, Y.T.; Chen, C.J.; Hong, C.F.; Hsieh, W.J.; Chang, H.T.; Chen, Y.S.; Lin, T.W.; Hsu, H.S.; et al. Coexpression of Oct4 and Nanog enhances malignancy in lung adenocarcinoma by inducing cancer stem cell-like properties and epithelial-mesenchymal transdifferentiation. Cancer Res. 2010, 70, 10433–10444. [Google Scholar] [CrossRef] [PubMed]
- Wicha, M.S.; Liu, S.; Dontu, G. Cancer stem cells: An old idea--a paradigm shift. Cancer Res. 2006, 66, 1883–1890. [Google Scholar] [CrossRef]
- Mani, S.A.; Guo, W.; Liao, M.J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef]
- Taube, J.H.; Herschkowitz, J.I.; Komurov, K.; Zhou, A.Y.; Gupta, S.; Yang, J.; Hartwell, K.; Onder, T.T.; Gupta, P.B.; Evans, K.W.; et al. Core epithelial-to-mesenchymal transition interactome gene-expression signature is associated with claudin-low and metaplastic breast cancer subtypes. Proc. Natl. Acad. Sci. USA 2010, 107, 15449–15454. [Google Scholar] [CrossRef]
- Paranjape, A.N.; Soundararajan, R.; Werden, S.J.; Joseph, R.; Taube, J.H.; Liu, H.; Rodriguez-Canales, J.; Sphyris, N.; Wistuba, I.; Miura, N.; et al. Inhibition of FOXC2 restores epithelial phenotype and drug sensitivity in prostate cancer cells with stem-cell properties. Oncogene 2016. [Google Scholar] [CrossRef]
- Werden, S.J.; Sphyris, N.; Sarkar, T.R.; Paranjape, A.N.; LaBaff, A.M.; Taube, J.H.; Hollier, B.G.; Ramirez-Pena, E.Q.; Soundararajan, R.; den Hollander, P.; et al. Phosphorylation of serine 367 of FOXC2 by p38 regulates ZEB1 and breast cancer metastasis, without impacting primary tumor growth. Oncogene 2016, 35, 5977–5988. [Google Scholar] [CrossRef]
- Lidell, M.E.; Seifert, E.L.; Westergren, R.; Heglind, M.; Gowing, A.; Sukonina, V.; Arani, Z.; Itkonen, P.; Wallin, S.; Westberg, F.; et al. The adipocyte-expressed forkhead transcription factor Foxc2 regulates metabolism through altered mitochondrial function. Diabetes 2011, 60, 427–435. [Google Scholar] [CrossRef]
- Gan, L.; Liu, Z.; Feng, F.; Wu, T.; Luo, D.; Hu, C.; Sun, C. Foxc2 coordinates inflammation and browning of white adipose by leptin-STAT3-PRDM16 signal in mice. Int. J. Obes. (Lond.) 2018, 42, 252–259. [Google Scholar] [CrossRef]
- Luo, Y.; Burrington, C.M.; Graff, E.C.; Zhang, J.; Judd, R.L.; Suksaranjit, P.; Kaewpoowat, Q.; Davenport, S.K.; O’Neill, A.M.; Greene, M.W. Metabolic phenotype and adipose and liver features in a high-fat Western diet-induced mouse model of obesity-linked NAFLD. Am. J. Physiol. Endocrinol. Metab. 2016, 310, E418–E439. [Google Scholar] [CrossRef] [PubMed]
- Gan, L.; Liu, Z.; Chen, Y.; Dan, L.; Feng, F.; Liu, G.; Sun, C. alpha-MSH and Foxc2 promote fatty acid oxidation through C/EBPbeta negative transcription in mice adipose tissue. Sci. Rep. 2016, 6, 36661. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Suraokar, M.; Darnay, B.G.; Hollier, B.G.; Shaiken, T.E.; Asano, T.; Chen, C.H.; Chang, B.H.; Lu, Y.; Mills, G.B.; et al. BSTA promotes mTORC2-mediated phosphorylation of Akt1 to suppress expression of FoxC2 and stimulate adipocyte differentiation. Sci. Signal. 2013, 6, ra2. [Google Scholar] [CrossRef] [PubMed]
- Hakansson, J.; Eliasson, B.; Smith, U.; Enerback, S. Adipocyte mitochondrial genes and the forkhead factor FOXC2 are decreased in type 2 diabetes patients and normalized in response to rosiglitazone. Diabetol. Metab. Syndr. 2011, 3, 32. [Google Scholar] [CrossRef]
- Hao, Q.; Hansen, J.B.; Petersen, R.K.; Hallenborg, P.; Jorgensen, C.; Cinti, S.; Larsen, P.J.; Steffensen, K.R.; Wang, H.; Collins, S.; et al. ADD1/SREBP1c activates the PGC1-alpha promoter in brown adipocytes. Biochim. Biophys. Acta 2010, 1801, 421–429. [Google Scholar] [CrossRef]
- Xue, Y.; Cao, R.; Nilsson, D.; Chen, S.; Westergren, R.; Hedlund, E.M.; Martijn, C.; Rondahl, L.; Krauli, P.; Walum, E.; et al. FOXC2 controls Ang-2 expression and modulates angiogenesis, vascular patterning, remodeling, and functions in adipose tissue. Proc. Natl. Acad. Sci. USA 2008, 105, 10167–10172. [Google Scholar] [CrossRef]
- Gronning, L.M.; Baillie, G.S.; Cederberg, A.; Lynch, M.J.; Houslay, M.D.; Enerback, S.; Tasken, K. Reduced PDE4 expression and activity contributes to enhanced catecholamine-induced cAMP accumulation in adipocytes from FOXC2 transgenic mice. FEBS Lett. 2006, 580, 4126–4130. [Google Scholar] [CrossRef]
- Kim, J.K.; Kim, H.J.; Park, S.Y.; Cederberg, A.; Westergren, R.; Nilsson, D.; Higashimori, T.; Cho, Y.R.; Liu, Z.X.; Dong, J.; et al. Adipocyte-specific overexpression of FOXC2 prevents diet-induced increases in intramuscular fatty acyl CoA and insulin resistance. Diabetes 2005, 54, 1657–1663. [Google Scholar] [CrossRef]
- Foulkes, W.D.; Smith, I.E.; Reis-Filho, J.S. Triple-negative breast cancer. N. Engl. J. Med. 2010, 363, 1938–1948. [Google Scholar] [CrossRef]
- Lee, S.Y.; Jeon, H.M.; Ju, M.K.; Jeong, E.K.; Kim, C.H.; Park, H.G.; Han, S.I.; Kang, H.S. Dlx-2 and glutaminase upregulate epithelial-mesenchymal transition and glycolytic switch. Oncotarget 2016, 7, 7925–7939. [Google Scholar] [CrossRef]
- Shaw, F.L.; Harrison, H.; Spence, K.; Ablett, M.P.; Simoes, B.M.; Farnie, G.; Clarke, R.B. A detailed mammosphere assay protocol for the quantification of breast stem cell activity. J. Mammary Gland Biol. Neoplasia 2012, 17, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Hollier, B.G.; Tinnirello, A.A.; Werden, S.J.; Evans, K.W.; Taube, J.H.; Sarkar, T.R.; Sphyris, N.; Shariati, M.; Kumar, S.V.; Battula, V.L.; et al. FOXC2 expression links epithelial-mesenchymal transition and stem cell properties in breast cancer. Cancer Res. 2013, 73, 1981–1992. [Google Scholar] [CrossRef] [PubMed]
- Bhowmik, S.K.; Ramirez-Pena, E.; Arnold, J.M.; Putluri, V.; Sphyris, N.; Michailidis, G.; Putluri, N.; Ambs, S.; Sreekumar, A.; Mani, S.A. EMT-induced metabolite signature identifies poor clinical outcome. Oncotarget 2015, 6, 42651–42660. [Google Scholar] [CrossRef] [PubMed]
- Perez-Gomez, C.; Campos-Sandoval, J.A.; Alonso, F.J.; Segura, J.A.; Manzanares, E.; Ruiz-Sanchez, P.; Gonzalez, M.E.; Marquez, J.; Mates, J.M. Co-expression of glutaminase K and L isoenzymes in human tumour cells. Biochem. J. 2005, 386, 535–542. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Zhang, C.; Wu, R.; Sun, Y.; Levine, A.; Feng, Z. Glutaminase 2, a novel p53 target gene regulating energy metabolism and antioxidant function. Proc. Natl. Acad. Sci. USA 2010, 107, 7455–7460. [Google Scholar] [CrossRef] [PubMed]
- Martin-Rufian, M.; Nascimento-Gomes, R.; Higuero, A.; Crisma, A.R.; Campos-Sandoval, J.A.; Gomez-Garcia, M.C.; Cardona, C.; Cheng, T.; Lobo, C.; Segura, J.A.; et al. Both GLS silencing and GLS2 overexpression synergize with oxidative stress against proliferation of glioma cells. J. Mol. Med. (Berl.) 2014, 92, 277–290. [Google Scholar] [CrossRef]
- Szeliga, M.; Obara-Michlewska, M.; Matyja, E.; Lazarczyk, M.; Lobo, C.; Hilgier, W.; Alonso, F.J.; Marquez, J.; Albrecht, J. Transfection with liver-type glutaminase cDNA alters gene expression and reduces survival, migration and proliferation of T98G glioma cells. Glia 2009, 57, 1014–1023. [Google Scholar] [CrossRef]
- Szeliga, M.; Bogacinska-Karas, M.; Rozycka, A.; Hilgier, W.; Marquez, J.; Albrecht, J. Silencing of GLS and overexpression of GLS2 genes cooperate in decreasing the proliferation and viability of glioblastoma cells. Tumour. Biol. 2014, 35, 1855–1862. [Google Scholar] [CrossRef]
- Obre, E.; Rossignol, R. Emerging concepts in bioenergetics and cancer research: Metabolic flexibility, coupling, symbiosis, switch, oxidative tumors, metabolic remodeling, signaling and bioenergetic therapy. Int. J. Biochem. Cell Biol. 2015, 59, 167–181. [Google Scholar] [CrossRef]
- Marquez, J.; Alonso, F.J.; Mates, J.M.; Segura, J.A.; Martin-Rufian, M.; Campos-Sandoval, J.A. Glutamine Addiction In Gliomas. Neurochem. Res. 2017, 42, 1735–1746. [Google Scholar] [CrossRef]
- Wise, D.R.; Thompson, C.B. Glutamine addiction: A new therapeutic target in cancer. Trends Biochem. Sci. 2010, 35, 427–433. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Liu, J.; Zhao, Y.; Yue, X.; Zhu, Y.; Wang, X.; Wu, H.; Blanco, F.; Li, S.; Bhanot, G.; et al. Glutaminase 2 is a novel negative regulator of small GTPase Rac1 and mediates p53 function in suppressing metastasis. Elife 2016, 5, e10727. [Google Scholar] [CrossRef] [PubMed]
- Tsai, J.H.; Yang, J. Epithelial-mesenchymal plasticity in carcinoma metastasis. Genes Dev. 2013, 27, 2192–2206. [Google Scholar] [CrossRef] [PubMed]
- May, C.D.; Sphyris, N.; Evans, K.W.; Werden, S.J.; Guo, W.; Mani, S.A. Epithelial-mesenchymal transition and cancer stem cells: A dangerously dynamic duo in breast cancer progression. Breast Cancer Res. 2011, 13. [Google Scholar] [CrossRef] [PubMed]
- Wein, L.; Loi, S. Mechanisms of resistance of chemotherapy in early-stage triple negative breast cancer (TNBC). Breast 2017, 34 (Suppl. 1), S27–S30. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Prijic, S.; Urban, B.C.; Tisza, M.J.; Zuo, Y.; Li, L.; Tan, Z.; Chen, X.; Mani, S.A.; Chang, J.T. Candidate Antimetastasis Drugs Suppress the Metastatic Capacity of Breast Cancer Cells by Reducing Membrane Fluidity. Cancer Res. 2016, 76, 2037–2049. [Google Scholar] [CrossRef]
- 2016_01_28 stddata Run. Available online: http://gdac.broadinstitute.org/runs/stddata__2016_01_28/ (accessed on 13 October 2019).
- Possemato, R.; Marks, K.M.; Shaul, Y.D.; Pacold, M.E.; Kim, D.; Birsoy, K.; Sethumadhavan, S.; Woo, H.K.; Jang, H.G.; Jha, A.K.; et al. Functional genomics reveal that the serine synthesis pathway is essential in breast cancer. Nature 2011, 476, 346–350. [Google Scholar] [CrossRef]
- Davarinejad, H. Quantifications of Western Blots with ImageJ. 2017. Available online: http://www.yorku.ca/yisheng/Internal/Protocols/ImageJ.pdf (accessed on 13 October 2019).





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramirez-Peña, E.; Arnold, J.; Shivakumar, V.; Joseph, R.; Vidhya Vijay, G.; den Hollander, P.; Bhangre, N.; Allegakoen, P.; Prasad, R.; Conley, Z.; et al. The Epithelial to Mesenchymal Transition Promotes Glutamine Independence by Suppressing GLS2 Expression. Cancers 2019, 11, 1610. https://doi.org/10.3390/cancers11101610
Ramirez-Peña E, Arnold J, Shivakumar V, Joseph R, Vidhya Vijay G, den Hollander P, Bhangre N, Allegakoen P, Prasad R, Conley Z, et al. The Epithelial to Mesenchymal Transition Promotes Glutamine Independence by Suppressing GLS2 Expression. Cancers. 2019; 11(10):1610. https://doi.org/10.3390/cancers11101610
Chicago/Turabian StyleRamirez-Peña, Esmeralda, James Arnold, Vinita Shivakumar, Robiya Joseph, Geraldine Vidhya Vijay, Petra den Hollander, Neeraja Bhangre, Paul Allegakoen, Rishika Prasad, Zachary Conley, and et al. 2019. "The Epithelial to Mesenchymal Transition Promotes Glutamine Independence by Suppressing GLS2 Expression" Cancers 11, no. 10: 1610. https://doi.org/10.3390/cancers11101610
APA StyleRamirez-Peña, E., Arnold, J., Shivakumar, V., Joseph, R., Vidhya Vijay, G., den Hollander, P., Bhangre, N., Allegakoen, P., Prasad, R., Conley, Z., Matés, J. M., Márquez, J., Chang, J. T., Vasaikar, S., Soundararajan, R., Sreekumar, A., & Mani, S. A. (2019). The Epithelial to Mesenchymal Transition Promotes Glutamine Independence by Suppressing GLS2 Expression. Cancers, 11(10), 1610. https://doi.org/10.3390/cancers11101610