HSF1 Regulates Mevalonate and Cholesterol Biosynthesis Pathways

Abstract
1. Introduction
2. Results
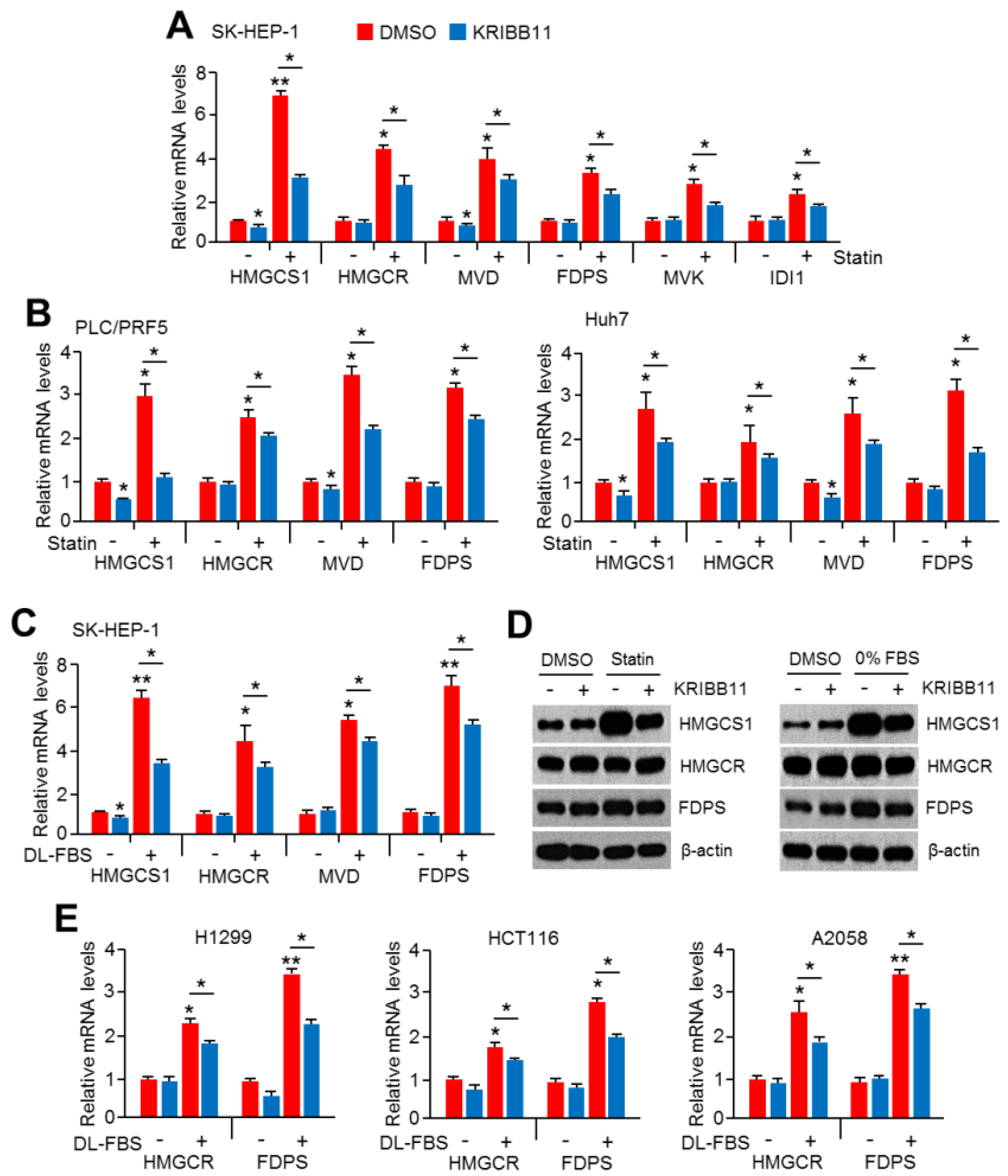
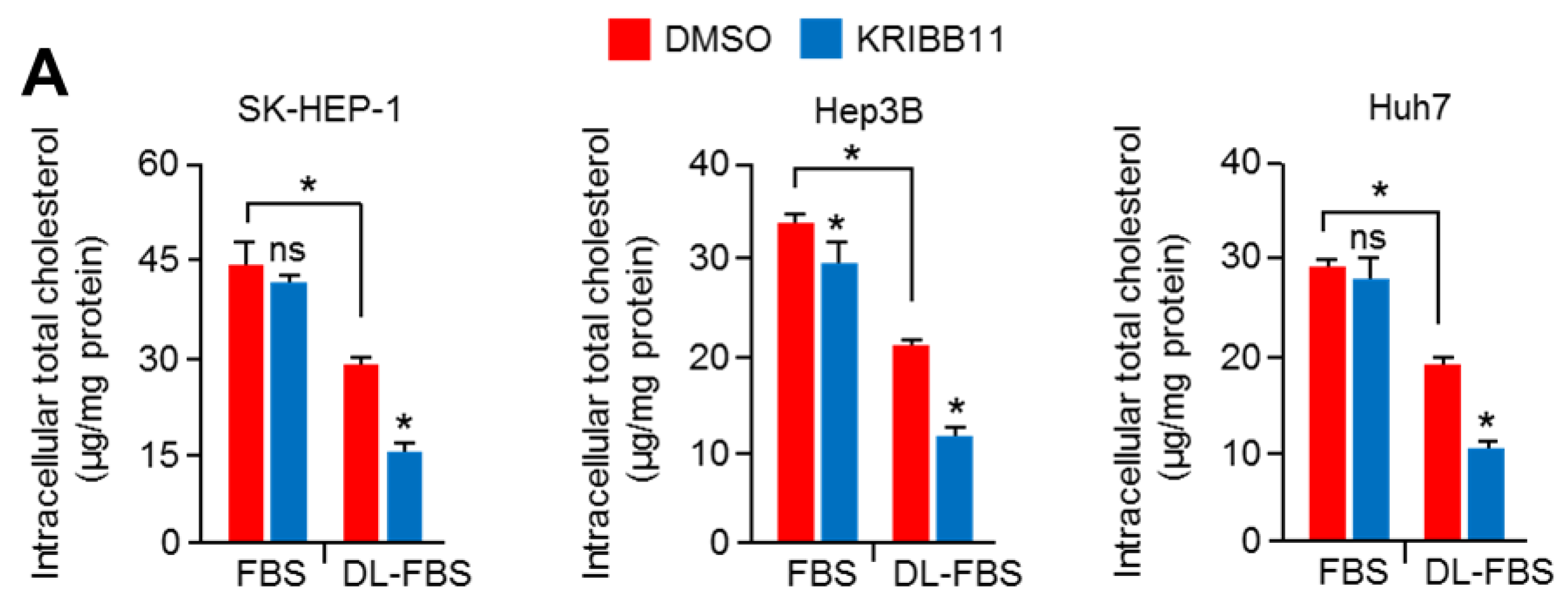
2.1. An HSF1 Inhibitor, KRIBB11, Suppressed Mevalonate and Cholesterol Biosynthesis-Related Gene Expression under Lower Cholesterol Conditions in HCC Cells
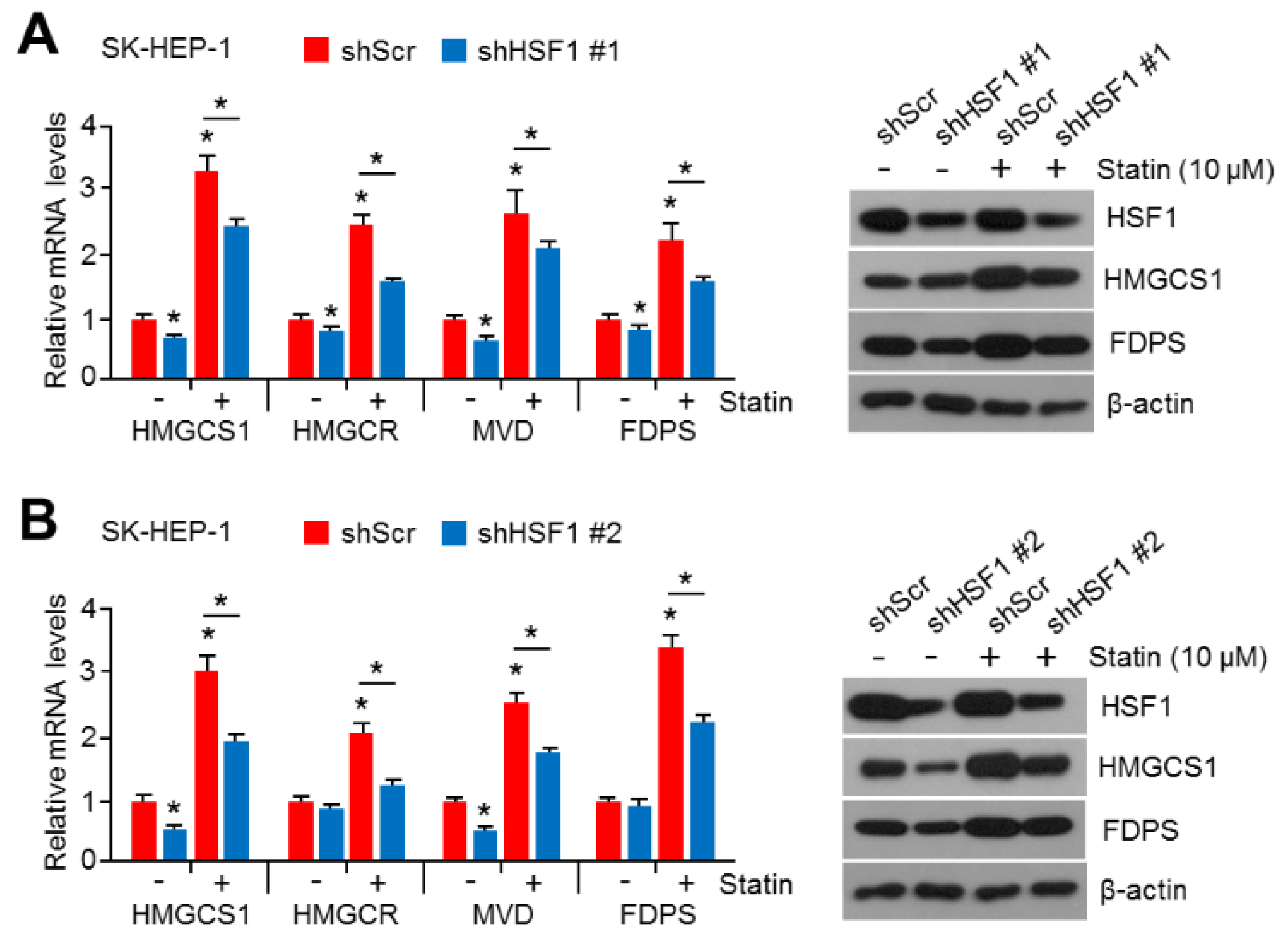
2.2. HSF1 Knock-Down Was Sufficient to Reverse Simvastatin-Induced Gene and Enzyme Expression in Cholesterol Biosynthesis
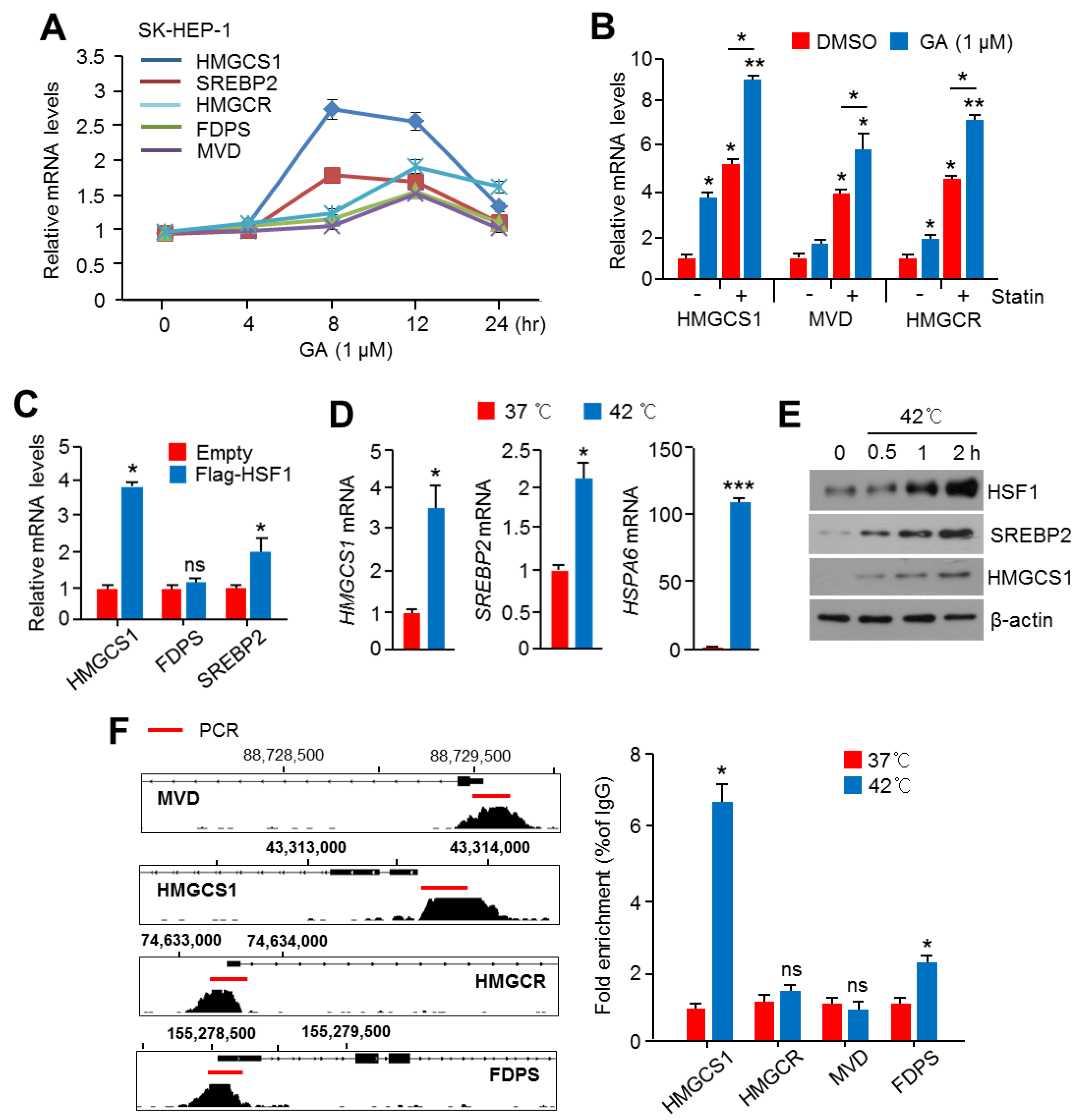
2.3. Activation of HSF1 Increased the Expression of Cholesterol Biosynthesis-Related Genes and Enzymes
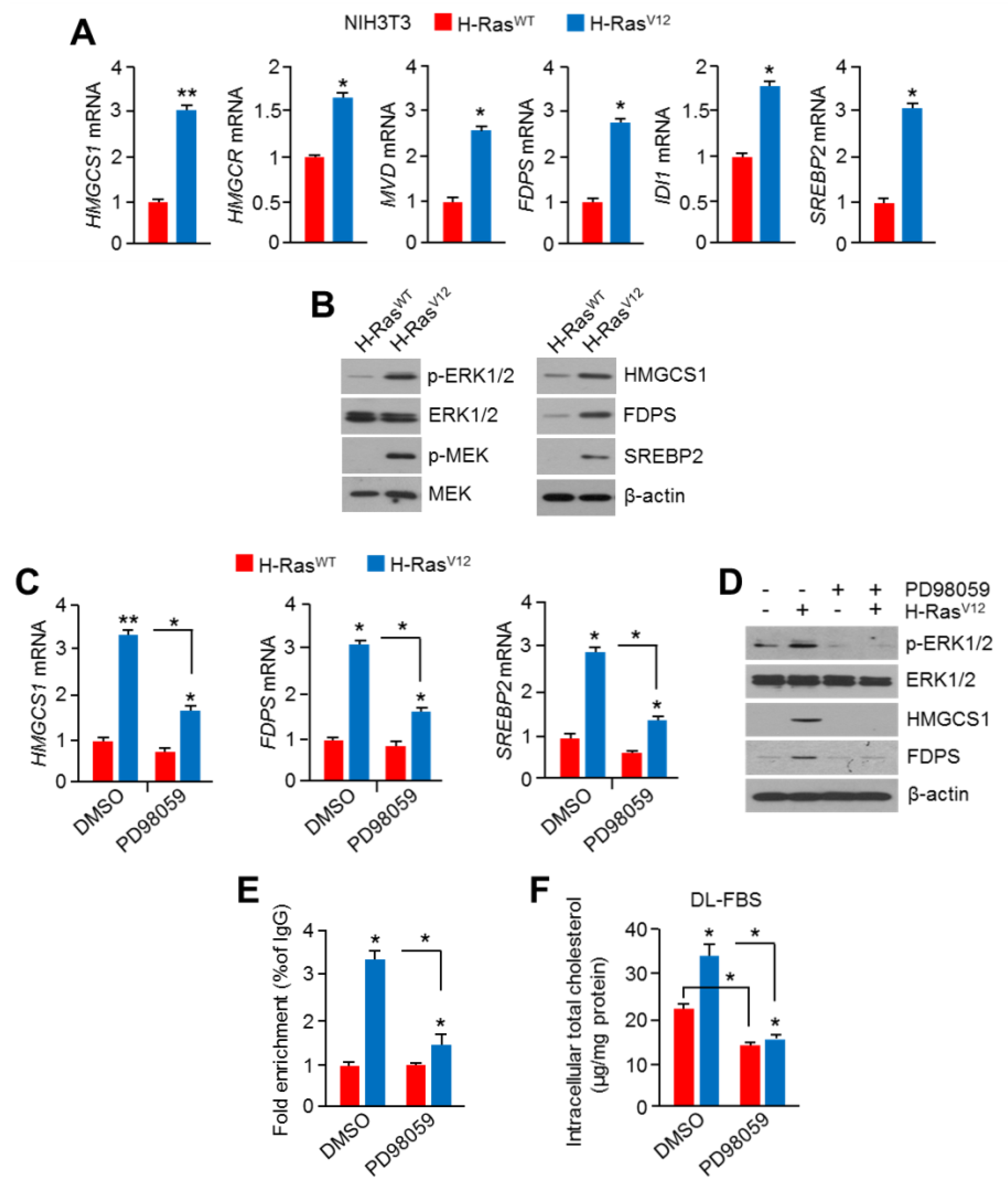
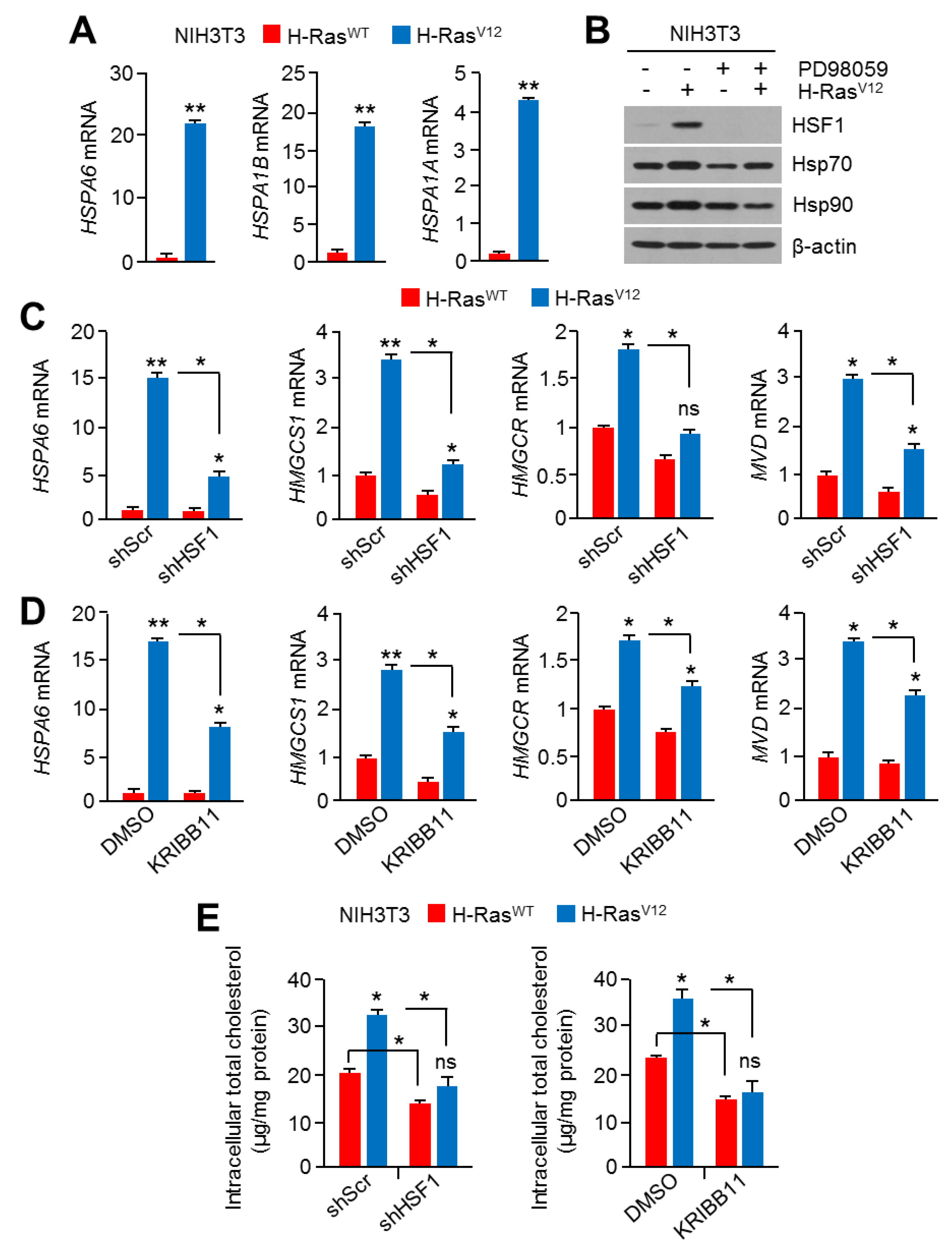
2.4. Hyperactivation of RAS-MAPK Signaling Increased Cholesterol Biosynthesis and Intracellular Cholesterol Levels
2.5. HSF1 Involved Oncogenic RAS-MAPK Signaling-Induced Cholesterol Biosynthesis
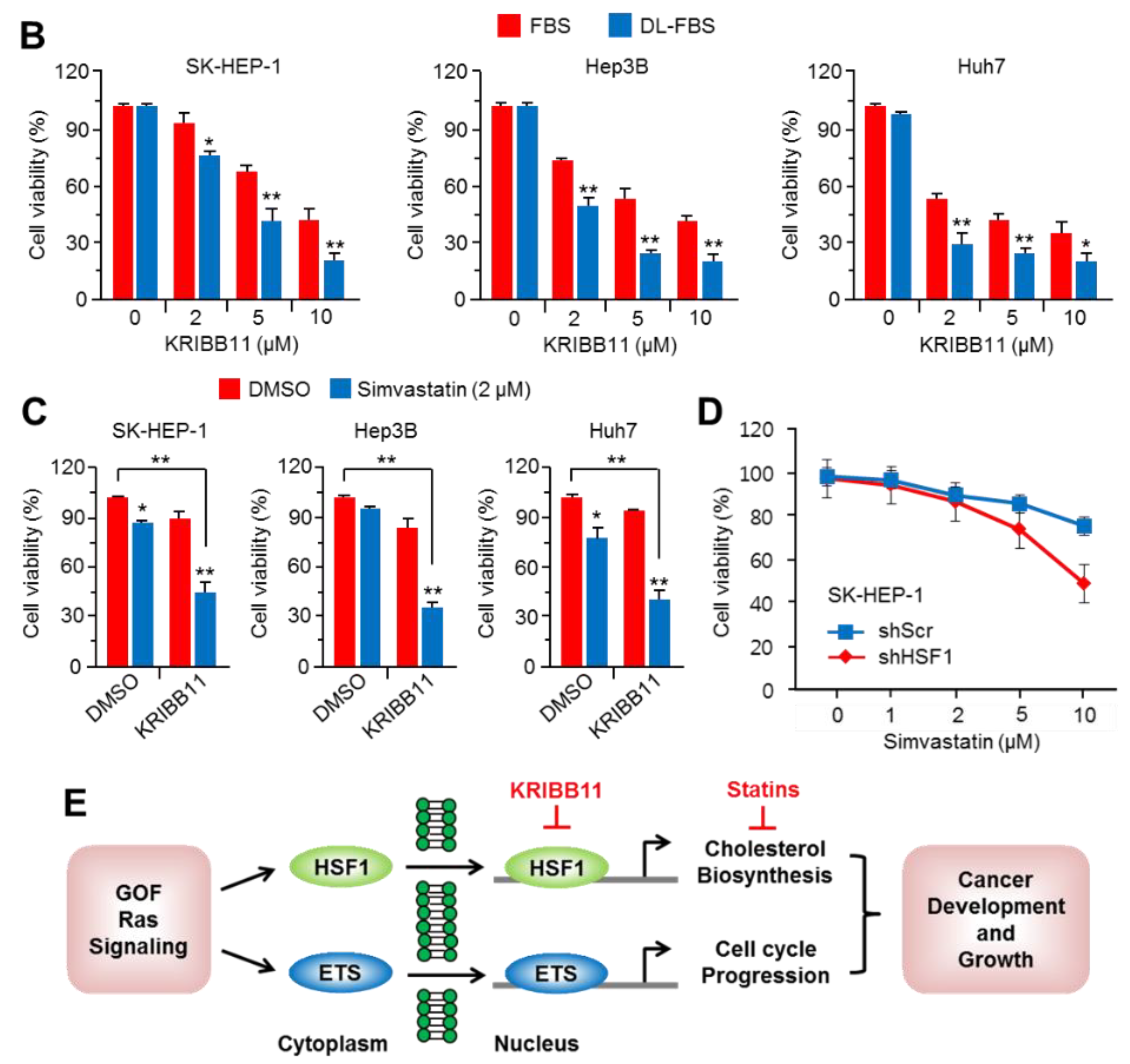
2.6. Suppression of HSF1 Sensitively Attenuated Cell Growth under Cholesterol Depletion in HCC Cells
3. Discussion
4. Materials and Methods
4.1. Reagents and Antibodies
4.2. Western Blotting
4.3. Cell Culture, Plasmids, and Generation of Stable Cell Lines
4.4. Measurement of mRNA Expression
4.5. Chromatin Immunoprecipitation (ChIP) Assay
4.6. Measurement of Intracellular Total Cholesterol
4.7. Cell Viability
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Forner, A.; Llovet, J.M.; Bruix, J. Hepatocellular carcinoma. Lancet 2012, 379, 1245–1255. [Google Scholar] [CrossRef]
- Greten, T.F.; Korangy, F.; Manns, M.P.; Malek, N.P. Molecular therapy for the treatment of hepatocellular carcinoma. Br. J. Cancer 2009, 100, 19–23. [Google Scholar] [CrossRef] [PubMed]
- Desvergne, B.; Michalik, L.; Wahli, W. Transcriptional regulation of metabolism. Physiol. Rev. 2006, 86, 465–514. [Google Scholar] [CrossRef] [PubMed]
- Tabas, I. Cholesterol in health and disease. J. Clin. Investig. 2002, 110, 583–590. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.; Zhang, W.; Li, S.; Yang, H. The role of cholesterol metabolism in cancer. Am. J. Cancer Res. 2019, 9, 219–227. [Google Scholar] [PubMed]
- Murai, T. Cholesterol lowering: role in cancer prevention and treatment. Biol. Chem. 2015, 396, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Kuzu, O.F.; Noory, M.A.; Robertson, G.P. The Role of Cholesterol in Cancer. Cancer Res. 2016, 76, 2063–2070. [Google Scholar] [CrossRef] [PubMed]
- Qi, X.F.; Zheng, L.; Lee, K.J.; Kim, D.H.; Kim, C.S.; Cai, D.Q.; Wu, Z.; Qin, J.W.; Yu, Y.H.; Kim, S.K. HMG-CoA reductase inhibitors induce apoptosis of lymphoma cells by promoting ROS generation and regulating Akt, Erk and p38 signals via suppression of mevalonate pathway. Cell Death. Dis. 2013, 28, e518. [Google Scholar] [CrossRef]
- Bai, F.; Yu, Z.; Gao, X.; Gong, J.; Fan, L.; Liu, F. Simvastatin induces breast cancer cell death through oxidative stress up-regulating miR-140-5p. Aging 2019, 11, 3198–3219. [Google Scholar] [CrossRef]
- Kim, Y.S.; Lee, Y.M.; Oh, T.I.; Shin, D.H.; Kim, G.H.; Kan, S.Y.; Kang, H.; Kim, J.H.; Kim, B.M.; Yim, W.J.; et al. Emodin Sensitizes Hepatocellular Carcinoma Cells to the Anti-Cancer Effect of Sorafenib through Suppression of Cholesterol Metabolism. Int. J. Mol. Sci. 2018, 19, 3127. [Google Scholar] [CrossRef]
- Kotamraju, S.; Williams, C.L.; Kalyanaraman, B. Statin-induced breast cancer cell death: role of inducible nitric oxide and arginase-dependent pathways. Cancer Res. 2007, 67, 7386–7394. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.; Wang, X.; Song, D.; Liu, X.; Gu, Y.; Xu, Z.; Wang, X.; Zhang, X.; Ye, Q.; Tong, Z.; et al. Cholesterol Induces Epithelial-to-Mesenchymal Transition of Prostate Cancer Cells by Suppressing Degradation of EGFR through APMAP. Cancer Res. 2019, 79, 3063–3075. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Rong, X.; Palladino, E.N.D.; Wang, J.; Fogelman, A.M.; Martín, M.G.; Alrefai, W.A.; Ford, D.A.; Tontonoz, P. Phospholipid Remodeling and Cholesterol Availability Regulate Intestinal Stemness and Tumorigenesis. Cell Stem Cell 2018, 22, 206–220. [Google Scholar] [CrossRef] [PubMed]
- Ehmsen, S.; Pedersen, M.H.; Wang, G.; Terp, M.G.; Arslanagic, A.; Hood, B.L.; Conrads, T.P.; Leth-Larsen, R.; Ditzel, H.J. Increased cholesterol biosynthesis is a key characteristic of breast cancer stem cells influencing patient outcome. Cell Rep. 2019, 27, 3927–3938. [Google Scholar] [CrossRef] [PubMed]
- Manthravadi, S.; Shrestha, A.; Madhusudhana, S. Impact of statin use on cancer recurrence and mortality in breast cancer: A systematic review and meta-analysis. Int. J. Cancer 2016, 139, 1281–1288. [Google Scholar] [CrossRef] [PubMed]
- McGlynn, K.A.; Divine, G.W.; Sahasrabuddhe, V.V.; Engel, L.S.; VanSlooten, A.; Wells, K.; Yood, M.U.; Alford, S.H. Statin use and risk of hepatocellular carcinoma in a U.S. population. Cancer Epidemiol. 2014, 38, 523–527. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, S.F.; Nordestgaard, B.G.; Bojesen, S.E. Statin use and reduced cancer-related mortality. N. Engl. J. Med. 2012, 367, 1792–1802. [Google Scholar] [CrossRef]
- Jiang, S.Y.; Li, H.; Tang, J.J.; Wang, J.; Luo, J.; Liu, B.; Wang, J.K.; Shi, X.J.; Cui, H.W.; Tang, J.; et al. Discovery of a potent HMG-CoA reductase degrader that eliminates statin-induced reductase accumulation and lowers cholesterol. Nat. Commun. 2018, 9, e5138. [Google Scholar] [CrossRef]
- Pineda, A.; Cubeddu, L.X. Statin rebound or withdrawal syndrome: does it exist? Curr. Atheroscler. Rep. 2011, 13, 23–30. [Google Scholar] [CrossRef]
- Daskalopoulou, S.S. When statin therapy stops: implications for the patient. Curr. Opin. Cardiol. 2009, 24, 454–460. [Google Scholar] [CrossRef]
- Dai, C.; Sampson, S.B. HSF1: Guardian of Proteostasis in Cancer. Trends Cell Biol. 2016, 26, 17–28. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Pastor, R.; Burchfiel, E.T.; Thiele, D.J. Regulation of heat shock transcription factors and their roles in physiology and disease. Nat. Rev. Mol. Cell Biol. 2018, 19, 4–19. [Google Scholar] [CrossRef] [PubMed]
- Hahn, J.S.; Hu, Z.; Thiele, D.J.; Iyer, V.R. Genome-wide analysis of the biology of stress responses through heat shock transcription factor. Mol. Cell Biol. 2004, 24, 5249–5256. [Google Scholar] [CrossRef] [PubMed]
- Dai, S.; Tang, Z.; Cao, J.; Zhou, W.; Li, H.; Sampson, S.; Dai, C. Suppression of the HSF1-mediated proteotoxic stress response by the metabolic stress sensor AMPK. EMBO J. 2015, 34, 275–293. [Google Scholar] [CrossRef] [PubMed]
- Qiao, A.; Jin, X.; Pang, J.; Moskophidis, D.; Mivechi, N.F. The transcriptional regulator of the chaperone response HSF1 controls hepatic bioenergetics and protein homeostasis. J. Cell Biol. 2017, 216, 723–741. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Xu, L.; Alberobello, A.T.; Gavrilova, O.; Bagattin, A.; Skarulis, M.; Liu, J.; Finkel, T.; Mueller, E. Celastrol Protects against Obesity and Metabolic Dysfunction through Activation of a HSF1-PGC1α Transcriptional Axis. Cell Metab. 2015, 22, 695–708. [Google Scholar] [CrossRef] [PubMed]
- Jin, X.; Moskophidis, D.; Mivechi, N.F. Heat shock transcription factor 1 is a key determinant of HCC development by regulating hepatic steatosis and metabolic syndrome. Cell Metab. 2011, 14, 91–103. [Google Scholar] [CrossRef]
- Cigliano, A.; Wang, C.; Pilo, M.G.; Szydlowska, M.; Brozzetti, S.; Latte, G.; Pes, G.M.; Pascale, R.M.; Seddaiu, M.A.; Vidili, G.; et al. Inhibition of HSF1 suppresses the growth of hepatocarcinoma cell lines in vitro and AKT-driven hepatocarcinogenesis in mice. Oncotarget 2017, 8, 54149–54159. [Google Scholar] [CrossRef][Green Version]
- Cigliano, A.; Pilo, M.G.; Li, L.; Latte, G.; Szydlowska, M.; Simile, M.M.; Paliogiannis, P.; Che, L.; Pes, G.M.; Palmieri, G.; et al. Deregulated c-Myc requires a functional HSF1 for experimental and human hepatocarcinogenesis. Oncotarget 2017, 8, 90638–90650. [Google Scholar] [CrossRef]
- Mullen, P.J.; Yu, R.; Longo, J.; Archer, M.C.; Penn, L.Z. The interplay between cell signalling and the mevalonate pathway in cancer. Nat. Rev. Cancer 2016, 16, 718–731. [Google Scholar] [CrossRef]
- Luu, W.; Sharpe, L.J.; Stevenson, J.; Brown, A.J. Akt acutely activates the cholesterogenic transcription factor SREBP-2. Biochem. Biophys. Acta. 2012, 1823, 458–464. [Google Scholar] [CrossRef] [PubMed]
- Demoulin, J.B.; Ericsson, J.; Kallin, A.; Rorsman, C.; Rönnstrand, L.; Heldin, C.H. Platelet-derived growth factor stimulates membrane lipid synthesis through activation of phosphatidylinositol 3-kinase and sterol regulatory element-binding proteins. J. Biol. Chem. 2004, 279, 35392–35402. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.H.; Yao, M.; Lee, T.S.; Zhu, Y.; Martins-Green, M.; Shyy, J.Y. Vascular endothelial growth factor activation of sterol regulatory element binding protein: a potential role in angiogenesis. Circ. Res. 2004, 95, 471–478. [Google Scholar] [CrossRef] [PubMed]
- Fleischmann, M.; Iynedjian, P.B. Regulation of sterol regulatory-element binding protein 1 gene expression in liver: role of insulin and protein kinase B/cAkt. Biochem. J. 2000, 349, 13–17. [Google Scholar] [CrossRef] [PubMed]
- Porstmann, T.; Griffiths, B.; Chung, Y.L.; Delpuech, O.; Griffiths, J.R.; Downward, J.; Schulze, A. PKB/Akt induces transcription of enzymes involved in cholesterol and fatty acid biosynthesis via activation of SREBP. Oncogene 2005, 24, 6465–6481. [Google Scholar] [CrossRef] [PubMed]
- Sundqvist, A.; Bengoechea-Alonso, M.T.; Ye, X.; Lukiyanchuk, V.; Jin, J.; Harper, J.W.; Ericsson, J. Control of lipid metabolism by phosphorylation-dependent degradation of the SREBP family of transcription factors by SCF(Fbw7). Cell Metab. 2005, 1, 379–391. [Google Scholar] [CrossRef]
- Kotzka, J.; Lehr, S.; Roth, G.; Avci, H.; Knebel, B.; Muller-Wieland, D. Insulin-activated Erk-mitogen-activated protein kinases phosphorylate sterol regulatory element-binding Protein-2 at serine residues 432 and 455 in vivo. J. Biol. Chem. 2004, 279, 22404–22411. [Google Scholar] [CrossRef] [PubMed]
- Roth, G.; Kotzka, J.; Kremer, L.; Lehr, S.; Lohaus, C.; Meyer, H.E.; Krone, W.; Müller-Wieland, D. MAP kinases Erk1/2 phosphorylate sterol regulatory element-binding protein (SREBP)-1a at serine 117 in vitro. J. Biol. Chem. 2000, 275, 33302–33307. [Google Scholar] [CrossRef]
- Yoon, Y.J.; Kim, J.A.; Shin, K.D.; Shin, D.S.; Han, Y.M.; Lee, Y.J.; Lee, J.S.; Kwon, B.M.; Han, D.C. KRIBB11 inhibits HSP70 synthesis through inhibition of heat shock factor 1 function by impairing the recruitment of positive transcription elongation factor b to the hsp70 promoter. J. Biol. Chem. 2011, 286, 1737–1747. [Google Scholar] [CrossRef]
- Downward, J. Targeting RAS signalling pathways in cancer therapy. Nat. Rev. Cancer 2003, 3, 11–22. [Google Scholar] [CrossRef]
- Dai, C.; Santagata, S.; Tang, Z.; Shi, J.; Cao, J.; Kwon, H.; Bronson, R.T.; Whitesell, L.; Lindquist, S. Loss of tumor suppressor NF1 activates HSF1 to promote carcinogenesis. J. Clin. Investig. 2012, 122, 3742–3754. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, F.; Sun, Q.; Patel, D.; Stommel, J.M. Cholesterol Metabolism: A Potential Therapeutic Target in Glioblastoma. Cancers 2019, 11, 146. [Google Scholar] [CrossRef] [PubMed]
- Kuan, Y.C.; Hashidume, T.; Shibata, T.; Uchida, K.; Shimizu, M.; Inoue, J.; Sato, R. Heat Shock Protein 90 Modulates Lipid Homeostasis by Regulating the Stability and Function of Sterol Regulatory Element-binding Protein (SREBP) and SREBP Cleavage-activating Protein. J. Biol. Chem. 2017, 292, 3016–3028. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Medarde, A.; Santos, E. Ras in cancer and developmental diseases. Genes. Cancer 2011, 2, 344–358. [Google Scholar] [CrossRef] [PubMed]
- Dai, C.; Whitesell, L.; Rogers, A.B.; Lindquist, S. Heat shock factor 1 is a powerful multifaceted modifier of carcinogenesis. Cell 2007, 130, 1005–1018. [Google Scholar] [CrossRef] [PubMed]
- Fang, F.; Chang, R.; Yang, L. Heat shock factor 1 promotes invasion and metastasis of hepatocellular carcinoma in vitro and in vivo. Cancers 2012, 118, 1782–1794. [Google Scholar] [CrossRef] [PubMed]
- Freed-Pastor, W.A.; Mizuno, H.; Zhao, X.; Langerød, A.; Moon, S.H.; Rodriguez-Barrueco, R.; Barsotti, A.; Chicas, A.; Li, W.; Polotskaia, A.; et al. Mutant p53 disrupts mammary tissue architecture via the mevalonate pathway. Cell 2012, 148, 244–258. [Google Scholar] [CrossRef]
- Parrales, A.; Thoenen, E.; Iwakuma, T. The interplay between mutant p53 and the mevalonate pathway. Cell Death Differ. 2018, 25, 460–470. [Google Scholar] [CrossRef]
- Hussain, S.P.; Schwank, J.; Staib, F.; Wang, X.W.; Harris, C.C. TP53 mutations and hepatocellular carcinoma: insights into the etiology and pathogenesis of liver cancer. Oncogene 2007, 26, 2166–2176. [Google Scholar] [CrossRef]
- Ponchel, F.; Puisieux, A.; Tabone, E.; Michot, J.P.; Fröschl, G.; Morel, A.P.; Frébourg, T.; Fontanière, B.; Oberhammer, F.; Ozturk, M. Hepatocarcinoma-specific mutant p53-249ser induces mitotic activity but has no effect on transforming growth factor beta 1-mediated apoptosis. Cancer Res. 1994, 54, 2064–2068. [Google Scholar]
- Li, D.; Yallowitz, A.; Ozog, L.; Marchenko, N. A gain-of-function mutant p53-HSF1 feed forward circuit governs adaptation of cancer cells to proteotoxic stress. Cell Death Dis. 2014, 5, e1194. [Google Scholar] [CrossRef] [PubMed]
- Che, L.; Chi, W.; Qiao, Y.; Zhang, J.; Song, X.; Liu, Y.; Li, L.; Jia, J.; Pilo, M.G.; Wang, J.; et al. Cholesterol biosynthesis supports the growth of hepatocarcinoma lesions depleted of fatty acid synthase in mice and humans. Gut 2019, 0, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Mansourian, P.G.; Yoneda, M.; Krishna Rao, M.; Martinez, F.J.; Thomas, E.; Schiff, E.R. Effects of statins on the risk of hepatocellular carcinoma. Gastroenterol. Hepatol. 2014, 10, 417–426. [Google Scholar]
- Nayan, M.; Punjani, N.; Juurlink, D.N.; Finelli, A.; Austin, P.C.; Kulkarni, G.S.; Uleryk, E.; Hamilton, R.J. Statin use and kidney cancer survival outcomes: A systematic review and meta-analysis. Cancer Treat Rev. 2017, 52, 105–116. [Google Scholar] [CrossRef] [PubMed]
- Cardwell, C.R.; Mc Menamin, Ú.; Hughes, C.M.; Murray, L.J. Statin use and survival from lung cancer: A population-based cohort study. Cancer Epidemiol. Biomarkers Prev. 2015, 24, 833–841. [Google Scholar] [CrossRef] [PubMed]
- Alfaqih, M.A.; Allott, E.H.; Hamilton, R.J.; Freeman, M.R.; Freedland, S.J. The current evidence on statin use and prostate cancer prevention: are we there yet? Nat. Rev. Urol. 2017, 14, 107–119. [Google Scholar] [CrossRef] [PubMed]
- Gray, R.T.; Coleman, H.G.; Hughes, C.; Murray, L.J.; Cardwell, C.R. Statin use and survival in colorectal cancer: Results from a population-based cohort study and an updated systematic review and meta-analysis. Cancer Epidemiol. 2016, 45, 71–81. [Google Scholar] [CrossRef]
- Huang, W.Y.; Li, C.H.; Lin, C.L.; Liang, J.A. Long-term statin use in patients with lung cancer and dyslipidemia reduces the risk of death. Oncotarget 2016, 7, 42208–42215. [Google Scholar] [CrossRef]
- Berndt, N.; Hamilton, A.D.; Sebti, S.M. Targeting protein prenylation for cancer therapy. Nat. Rev. Cancer 2011, 11, 775–791. [Google Scholar] [CrossRef]
- Xia, Z.; Tan, M.M.; Wong, W.W.; Dimitroulakos, J.; Minden, M.D.; Penn, L.Z. Blocking protein geranylgeranylation is essential for lovastatin-induced apoptosis of human acute myeloid leukemia cells. Leukemia 2001, 9, 1398–1407. [Google Scholar] [CrossRef]
- Yanae, M.; Tsubaki, M.; Satou, T.; Itoh, T.; Imano, M.; Yamazoe, Y.; Nishida, S. Statin-induced apoptosis via the suppression of ERK1/2 and Akt activation by inhibition of the geranylgeranyl-pyrophosphate biosynthesis in glioblastoma. J. Exp. Clin. Cancer Res. 2011, 30, e74. [Google Scholar] [CrossRef] [PubMed]
- Kamel, W.A.; Sugihara, E.; Nobusue, H.; Yamaguchi-Iwai, S.; Onishi, N.; Maki, K.; Fukuchi, Y.; Matsuo, K.; Muto, A.; Saya, H.; et al. Simvastatin-induced apoptosis in osteosarcoma cells: A key role of RhoA-AMPK/p38 MAPK signaling in antitumor activity. Mol. Cancer Ther. 2017, 1, 182–192. [Google Scholar] [CrossRef] [PubMed]
- Vazquez, F.; Lim, J.H.; Chim, H.; Bhalla, K.; Girnun, G.; Pierce, K.; Clish, C.B.; Granter, S.R.; Widlund, H.R.; Spiegelman, B.M.; et al. PGC1α expression defines a subset of human melanoma tumors with increased mitochondrial capacity and resistance to oxidative stress. Cancer Cell 2013, 23, 287–301. [Google Scholar] [CrossRef] [PubMed]
- Park, J.W.; Kim, S.; Bahk, Y.Y. A proteomic approach for dissecting H-Ras signaling networks in NIH/3T3 mouse embryonic fibroblast cells. Proteomics 2006, 6, 2433–2443. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Grammatikakis, N.; Siganou, A.; Calderwood, S.K. Regulation of molecular chaperone gene transcription involves the serine phosphorylation, 14-3-3 epsilon binding, and cytoplasmic sequestration of heat shock factor 1. Mol. Cell Biol. 2003, 23, 6013–6026. [Google Scholar] [CrossRef]
- Luo, C.; Lim, J.H.; Lee, Y.; Granter, S.R.; Thomas, A.; Vazquez, F.; Widlund, H.R.; Puigserver, P. A PGC1α-mediated transcriptional axis suppresses melanoma metastasis. Nature 2016, 537, 422–426. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward Primer | Reverse Primer |
|---|---|---|
| HMGCS1 (h) | TGGCAGGGAGTCTTGGTACT | TCCCACTCCAAATGATGACA |
| HMGCR (h) | GATGGGAGGCCACAAAGAG | TTCGGTGGCCTCTAGTGAGA |
| MVD (h) | AACATCGCGGTCATCAAGTA | TTAACTGGTCCTGGTGCAGA |
| FDPS (h) | AGCCAAGGAAACAGGATG | TCCATGATGTCATCTGCCAC |
| MVK (h) | GAGGTCGCCAGCTCTCCA | GAACTTGAGCAGCCTGTTTCTGA |
| HMGCS1 (m) | TGGCAGGGAGTCTTGGTACT | TCCCACTCCAAATGATGACA |
| HMGCR (m) | TGTCCCCACTATGACTTCCC | TCGGTGGCCTCTAGTGAGAT |
| MVD (m) | ATGGCCTCAGAAAAGCCTCAG | TGGTCGTTTTTAGCTGGTCCT |
| FDPS (m) | GGAGGTCCTAGAGTACAATGCC | AAGCCTGGAGCAGTTCTACAC |
| IDI1 (m) | GGTTCAGCTTCTAGCGGAGA | TCGCCTGGGTTACTTAATGG |
| HSPA6 (m) | CCAAATGCAAGACAAGTGTCG | TTCTAGCTTTGGAGGGAAAG |
| HSPA1B (m) | CCCTACCATTGAGGAGGTG | AAACTCGTACAGAAGGTGGC |
| HSPA1A (m) | GGCAAGATCAGCGAGGCC | TCTCTGCATGTAGAAACCGC |
| Gene | Forward Primer | Reverse Primer |
|---|---|---|
| HMGCS1 | CGCTGGAGAGATGGTCAAAT | CTTTCAGGTGAAGCCTAGTTCT |
| HMGCR | GAGAACATAAGCAGGGAGGTAAA | TTCCTGTGCGAACCTTACAG |
| MVD | AAGGTGGGCGTCTCAAATAC | CCTGTACGTTTCACAGGAGAAG |
| FDPS | AGCTGCCCAGGAAGATAATG | CAATGGAAGGGCTGAAGTCTA |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kang, H.; Oh, T.; Bahk, Y.Y.; Kim, G.-H.; Kan, S.-Y.; Shin, D.H.; Kim, J.H.; Lim, J.-H. HSF1 Regulates Mevalonate and Cholesterol Biosynthesis Pathways. Cancers 2019, 11, 1363. https://doi.org/10.3390/cancers11091363
Kang H, Oh T, Bahk YY, Kim G-H, Kan S-Y, Shin DH, Kim JH, Lim J-H. HSF1 Regulates Mevalonate and Cholesterol Biosynthesis Pathways. Cancers. 2019; 11(9):1363. https://doi.org/10.3390/cancers11091363
Chicago/Turabian StyleKang, Hyeji, Taerim Oh, Young Yil Bahk, Geon-Hee Kim, Sang-Yeon Kan, Dong Hoon Shin, Ji Hyung Kim, and Ji-Hong Lim. 2019. "HSF1 Regulates Mevalonate and Cholesterol Biosynthesis Pathways" Cancers 11, no. 9: 1363. https://doi.org/10.3390/cancers11091363
APA StyleKang, H., Oh, T., Bahk, Y. Y., Kim, G.-H., Kan, S.-Y., Shin, D. H., Kim, J. H., & Lim, J.-H. (2019). HSF1 Regulates Mevalonate and Cholesterol Biosynthesis Pathways. Cancers, 11(9), 1363. https://doi.org/10.3390/cancers11091363