Molecular Insights into Potential Contributions of Natural Polyphenols to Lung Cancer Treatment


Abstract
1. Introduction
2. Mutations and Dysregulated Signaling Pathways in Lung Cancer
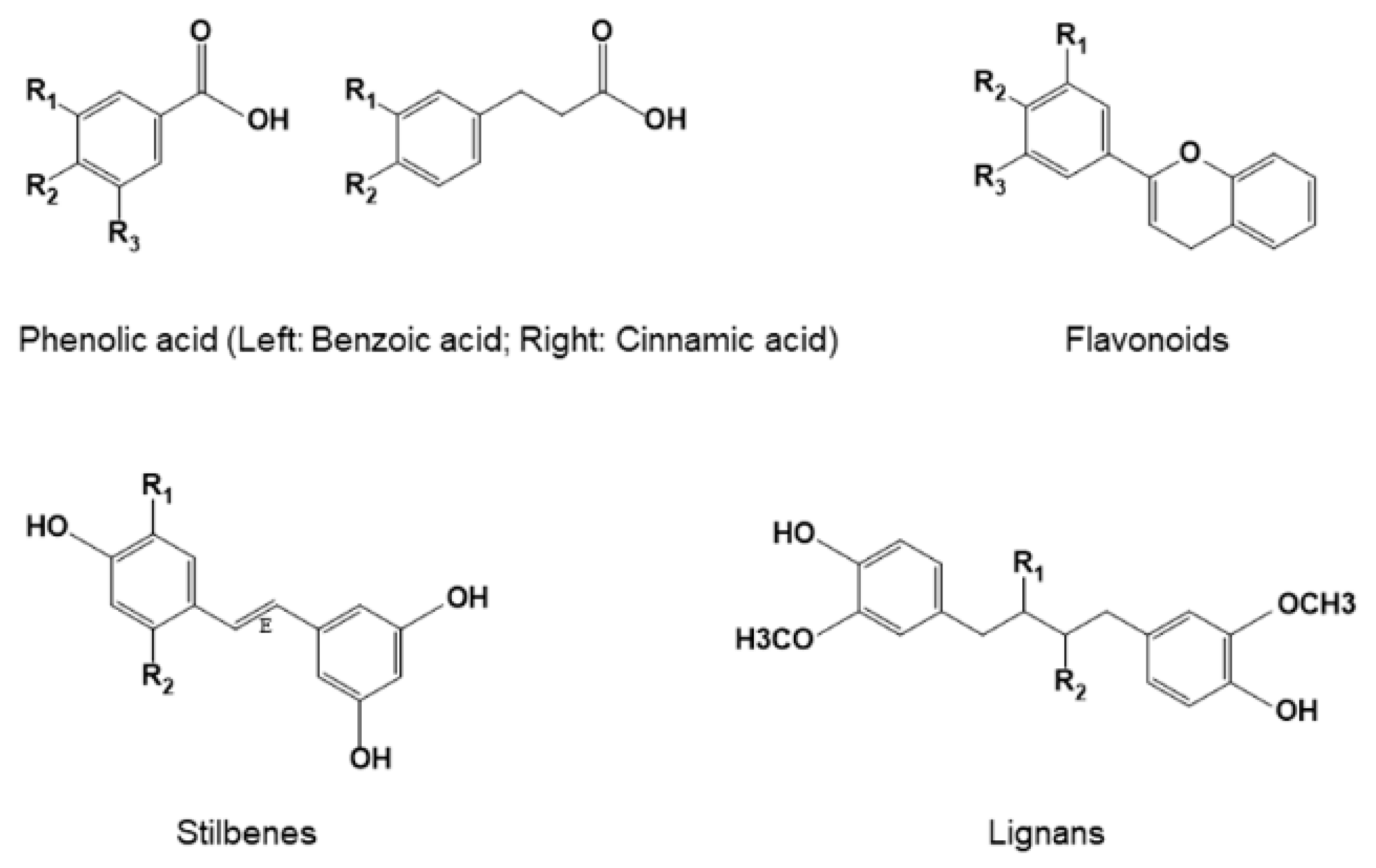
3. Classification and Structures of Natural Polyphenols
4. Molecular Underpinnings of Polyphenols in Lung Cancer Treatment
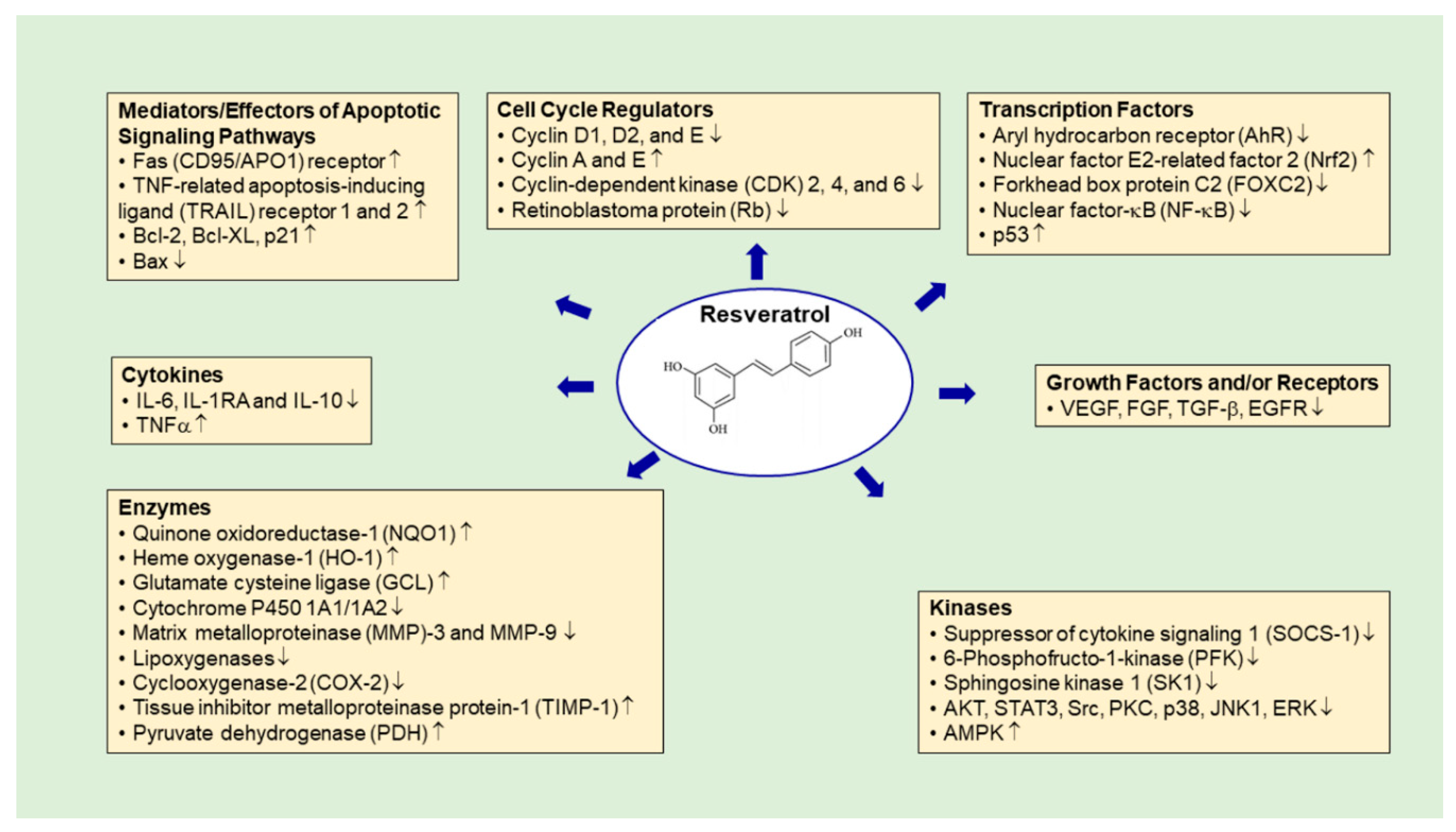
4.1. Resveratrol
4.2. Tea Catechins
4.3. Curcumin
4.4. Quercetin
4.5. Other Naturally Occurring Polyphenols
5. Conclusions and Future Perspectives
Funding
Conflicts of Interest
References
- Jemal, A.; Siegel, R.; Ward, E.; Hao, Y.; Xu, J.; Murray, T.; Thun, M.J. Cancer statistics, 2008. CA A Cancer J. Clin. 2008, 58, 71–96. [Google Scholar] [CrossRef] [PubMed]
- Herbst, R.S.; Heymach, J.V.; Lippman, S.M. Lung cancer. N. Engl. J. Med. 2008, 359, 1367–1380. [Google Scholar] [CrossRef] [PubMed]
- Pesch, B.; Kendzia, B.; Gustavsson, P.; Jockel, K.H.; Johnen, G.; Pohlabeln, H.; Olsson, A.; Ahrens, W.; Gross, I.M.; Bruske, I.; et al. Cigarette smoking and lung cancer—Relative risk estimates for the major histological types from a pooled analysis of case-control studies. Int. J. Cancer 2012, 131, 1210–1219. [Google Scholar] [CrossRef]
- Taylor, R.; Najafi, F.; Dobson, A. Meta-analysis of studies of passive smoking and lung cancer: Effects of study type and continent. Int. J. Epidemiol. 2007, 36, 1048–1059. [Google Scholar] [CrossRef]
- Field, R.W.; Withers, B.L. Occupational and environmental causes of lung cancer. Clin. Chest Med. 2012, 33, 681–703. [Google Scholar] [CrossRef]
- Hosgood, H.D.; Boffetta, P.; Greenland, S.; Lee, Y.C.; McLaughlin, J.; Seow, A.; Duell, E.J.; Andrew, A.S.; Zaridze, D.; Szeszenia-Dabrowska, N.; et al. In-home coal and wood use and lung cancer risk: A pooled analysis of the International Lung Cancer Consortium. Environ. Health Perspect. 2010, 118, 1743–1747. [Google Scholar] [CrossRef]
- Chen, C.L.; Chiou, H.Y.; Hsu, L.I.; Hsueh, Y.M.; Wu, M.M.; Chen, C.J. Ingested arsenic, characteristics of well water consumption and risk of different histological types of lung cancer in northeastern Taiwan. Environ. Res. 2010, 110, 455–462. [Google Scholar] [CrossRef]
- Brenner, D.R.; Boffetta, P.; Duell, E.J.; Bickeboller, H.; Rosenberger, A.; McCormack, V.; Muscat, J.E.; Yang, P.; Wichmann, H.E.; Brueske-Hohlfeld, I.; et al. Previous lung diseases and lung cancer risk: A pooled analysis from the International Lung Cancer Consortium. Am. J. Epidemiol. 2012, 176, 573–585. [Google Scholar] [CrossRef]
- Littman, A.J.; Thornquist, M.D.; White, E.; Jackson, L.A.; Goodman, G.E.; Vaughan, T.L. Prior lung disease and risk of lung cancer in a large prospective study. Cancer Causes Control 2004, 15, 819–827. [Google Scholar] [CrossRef]
- Feskanich, D.; Ziegler, R.G.; Michaud, D.S.; Giovannucci, E.L.; Speizer, F.E.; Willett, W.C.; Colditz, G.A. Prospective study of fruit and vegetable consumption and risk of lung cancer among men and women. J. Natl. Cancer Inst. 2000, 92, 1812–1823. [Google Scholar] [CrossRef]
- Middleton, E.; Kandaswami, C.; Theoharides, T.C. The effects of plant flavonoids on mammalian cells: Implications for inflammation, heart disease, and cancer. Pharmacol. Rev. 2000, 52, 673–751. [Google Scholar] [PubMed]
- Smith-Warner, S.A.; Spiegelman, D.; Yaun, S.S.; Albanes, D.; Beeson, W.L.; van den Brandt, P.A.; Feskanich, D.; Folsom, A.R.; Fraser, G.E.; Freudenheim, J.L.; et al. Fruits, vegetables and lung cancer: A pooled analysis of cohort studies. Int. J. Cancer 2003, 107, 1001–1011. [Google Scholar] [CrossRef] [PubMed]
- Wright, M.E.; Park, Y.; Subar, A.F.; Freedman, N.D.; Albanes, D.; Hollenbeck, A.; Leitzmann, M.F.; Schatzkin, A. Intakes of fruit, vegetables, and specific botanical groups in relation to lung cancer risk in the NIH-AARP Diet and Health Study. Am. J. Epidemiol. 2008, 168, 1024–1034. [Google Scholar] [CrossRef] [PubMed]
- Amararathna, M.; Johnston, M.R.; Rupasinghe, H.P. Plant Polyphenols as Chemopreventive Agents for Lung Cancer. Int. J. Mol. Sci. 2016, 17, 1352. [Google Scholar] [CrossRef]
- Barron, C.C.; Moore, J.; Tsakiridis, T.; Pickering, G.; Tsiani, E. Inhibition of human lung cancer cell proliferation and survival by wine. Cancer Cell Int. 2014, 14, 6. [Google Scholar] [CrossRef]
- Lim, S.L.; Goh, Y.M.; Noordin, M.M.; Rahman, H.S.; Othman, H.H.; Abu Bakar, N.A.; Mohamed, S. Morinda citrifolia edible leaf extract enhanced immune response against lung cancer. Food Funct. 2016, 7, 741–751. [Google Scholar] [CrossRef]
- Christensen, K.Y.; Naidu, A.; Parent, M.E.; Pintos, J.; Abrahamowicz, M.; Siemiatycki, J.; Koushik, A. The risk of lung cancer related to dietary intake of flavonoids. Nutr. Cancer 2012, 64, 964–974. [Google Scholar] [CrossRef]
- Grosso, G.; Godos, J.; Lamuela-Raventos, R.; Ray, S.; Micek, A.; Pajak, A.; Sciacca, S.; D’Orazio, N.; Del Rio, D.; Galvano, F. A comprehensive meta-analysis on dietary flavonoid and lignan intake and cancer risk: Level of evidence and limitations. Mol. Nutr. Food Res. 2017, 61, 1600930. [Google Scholar] [CrossRef]
- Wessner, B.; Strasser, E.M.; Koitz, N.; Schmuckenschlager, C.; Unger-Manhart, N.; Roth, E. Green tea polyphenol administration partly ameliorates chemotherapy-induced side effects in the small intestine of mice. J. Nutr. 2007, 137, 634–640. [Google Scholar] [CrossRef]
- Yao, Q.; Ye, X.; Wang, L.; Gu, J.; Fu, T.; Wang, Y.; Lai, Y.; Wang, Y.; Wang, X.; Jin, H.; et al. Protective effect of curcumin on chemotherapy-induced intestinal dysfunction. Int. J. Clin. Exp. Pathol. 2013, 6, 2342–2349. [Google Scholar]
- Khan, R.; Khan, A.Q.; Qamar, W.; Lateef, A.; Tahir, M.; Rehman, M.U.; Ali, F.; Sultana, S. Chrysin protects against cisplatin-induced colon. toxicity via amelioration of oxidative stress and apoptosis: Probable role of p38MAPK and p53. Toxicol. Appl. Pharmacol. 2012, 258, 315–329. [Google Scholar] [CrossRef] [PubMed]
- Lissoni, P.; Tancini, G.; Barni, S.; Paolorossi, F.; Ardizzoia, A.; Conti, A.; Maestroni, G. Treatment of cancer chemotherapy-induced toxicity with the pineal hormone melatonin. Support Care Cancer 1997, 5, 126–129. [Google Scholar] [CrossRef]
- Ghielmini, M.; Pagani, O.; de Jong, J.; Pampallona, S.; Conti, A.; Maestroni, G.; Sessa, C.; Cavalli, F. Double-blind randomized study on the myeloprotective effect of melatonin in combination with carboplatin and etoposide in advanced lung cancer. Br. J. Cancer 1999, 80, 1058–1061. [Google Scholar] [CrossRef] [PubMed]
- Jaakkola, K.; Lahteenmaki, P.; Laakso, J.; Harju, E.; Tykka, H.; Mahlberg, K. Treatment with antioxidant and other nutrients in combination with chemotherapy and irradiation in patients with small-cell lung cancer. Anticancer Res. 1992, 12, 599–606. [Google Scholar] [PubMed]
- Pace, A.; Savarese, A.; Picardo, M.; Maresca, V.; Pacetti, U.; Del Monte, G.; Biroccio, A.; Leonetti, C.; Jandolo, B.; Cognetti, F.; et al. Neuroprotective effect of vitamin E supplementation in patients treated with cisplatin chemotherapy. J. Clin. Oncol. 2003, 21, 927–931. [Google Scholar] [CrossRef] [PubMed]
- Schmidinger, M.; Budinsky, A.C.; Wenzel, C.; Piribauer, M.; Brix, R.; Kautzky, M.; Oder, W.; Locker, G.J.; Zielinski, C.C.; Steger, G.G. Glutathione in the prevention of cisplatin induced toxicities. A prospectively randomized pilot trial in patients with head and neck cancer and non small cell lung cancer. Wien. Klin. Wochenschr. 2000, 112, 617–623. [Google Scholar]
- Ferry, D.R.; Smith, A.; Malkhandi, J.; Fyfe, D.W.; deTakats, P.G.; Anderson, D.; Baker, J.; Kerr, D.J. Phase I clinical trial of the flavonoid quercetin: Pharmacokinetics and evidence for in vivo tyrosine kinase inhibition. Clin. Cancer Res. 1996, 2, 659–668. [Google Scholar]
- Albanes, D.; Heinonen, O.P.; Taylor, P.R.; Virtamo, J.; Edwards, B.K.; Rautalahti, M.; Hartman, A.M.; Palmgren, J.; Freedman, L.S.; Haapakoski, J.; et al. Alpha-Tocopherol and beta-carotene supplements and lung cancer incidence in the alpha-tocopherol, beta-carotene cancer prevention study: Effects of base-line characteristics and study compliance. J. Natl. Cancer Inst. 1996, 88, 1560–1570. [Google Scholar] [CrossRef]
- Pisters, K.M.; Newman, R.A.; Coldman, B.; Shin, D.M.; Khuri, F.R.; Hong, W.K.; Glisson, B.S.; Lee, J.S. Phase I trial of oral green tea extract in adult patients with solid tumors. J. Clin. Oncol. 2001, 19, 1830–1838. [Google Scholar] [CrossRef]
- Hakim, I.A.; Harris, R.B.; Brown, S.; Chow, H.H.; Wiseman, S.; Agarwal, S.; Talbot, W. Effect of increased tea consumption on oxidative DNA damage among smokers: A randomized controlled study. J. Nutr. 2003, 133, 3303–3309. [Google Scholar] [CrossRef]
- Goodman, G.E.; Thornquist, M.D.; Balmes, J.; Cullen, M.R.; Meyskens, F.L.; Omenn, G.S.; Valanis, B.; Williams, J.H. The Beta-Carotene and Retinol Efficacy Trial: Incidence of lung cancer and cardiovascular disease mortality during 6-year follow-up after stopping beta-carotene and retinol supplements. J. Natl. Cancer Inst. 2004, 96, 1743–1750. [Google Scholar] [CrossRef] [PubMed]
- Laurie, S.A.; Miller, V.A.; Grant, S.C.; Kris, M.G.; Ng, K.K. Phase I study of green tea extract in patients with advanced lung cancer. Cancer Chemother. Pharmacol. 2005, 55, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Pathak, A.K.; Bhutani, M.; Guleria, R.; Bal, S.; Mohan, A.; Mohanti, B.K.; Sharma, A.; Pathak, R.; Bhardwaj, N.K.; Prasad, K.N.; et al. Chemotherapy alone vs. chemotherapy plus high dose multiple antioxidants in patients with advanced non small cell lung cancer. J. Am. Coll. Nutr. 2005, 24, 16–21. [Google Scholar] [CrossRef]
- Osada, H.; Takahashi, T. Genetic alterations of multiple tumor suppressors and oncogenes in the carcinogenesis and progression of lung cancer. Oncogene 2002, 21, 7421–7434. [Google Scholar] [CrossRef]
- Blanco, D.; Vicent, S.; Fraga, M.F.; Fernandez-Garcia, I.; Freire, J.; Lujambio, A.; Esteller, M.; Ortiz-de-Solorzano, C.; Pio, R.; Lecanda, F.; et al. Molecular analysis of a multistep lung cancer model induced by chronic inflammation reveals epigenetic regulation of p16 and activation of the DNA damage response pathway. Neoplasia 2007, 9, 840–852. [Google Scholar] [CrossRef][Green Version]
- Bettio, D.; Venci, A.; Achille, V.; Alloisio, M.; Santoro, A. Lung cancer in which the hypothesis of multi-step progression is confirmed by array-CGH results: A case report. Exp. Ther. Med. 2016, 11, 98–100. [Google Scholar] [CrossRef][Green Version]
- Gazdar, A.F.; Minna, J.D. Angiogenesis and the multistage development of lung cancers. Clin. Cancer Res. 2000, 6, 1611–1612. [Google Scholar]
- McClelland, M.R.; Carskadon, S.L.; Zhao, L.; White, E.S.; Beer, D.G.; Orringer, M.B.; Pickens, A.; Chang, A.C.; Arenberg, D.A. Diversity of the angiogenic phenotype in non-small cell lung cancer. Am. J. Respir. Cell Mol. Biol. 2007, 36, 343–350. [Google Scholar] [CrossRef]
- Hilbe, W.; Manegold, C.; Pircher, A. Targeting angiogenesis in lung cancer—Pitfalls in drug development. Transl. Lung Cancer Res. 2012, 1, 122–128. [Google Scholar] [CrossRef]
- Lambert, A.W.; Wong, C.K.; Ozturk, S.; Papageorgis, P.; Raghunathan, R.; Alekseyev, Y.; Gower, A.C.; Reinhard, B.M.; Abdolmaleky, H.M.; Thiagalingam, S. Tumor Cell-Derived Periostin Regulates Cytokines That Maintain Breast Cancer Stem Cells. Mol. Cancer Res. 2016, 14, 103–113. [Google Scholar] [CrossRef]
- Popper, H.H. Progression and metastasis of lung cancer. Cancer Metastasis Rev. 2016, 35, 75–91. [Google Scholar] [CrossRef] [PubMed]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A.; Kinzler, K.W. Cancer genome landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef] [PubMed]
- Pao, W.; Girard, N. New driver mutations in non-small-cell lung cancer. Lancet Oncol. 2011, 12, 175–180. [Google Scholar] [CrossRef]
- Chan, B.A.; Hughes, B.G. Targeted therapy for non-small cell lung cancer: Current standards and the promise of the future. Transl. Lung Cancer Res. 2015, 4, 36–54. [Google Scholar] [CrossRef] [PubMed]
- Vecchiarelli, S.; Bennati, C. Oncogene addicted non-small-cell lung cancer: Current standard and hot topics. Future Oncol. 2018, 14, 3–17. [Google Scholar] [CrossRef] [PubMed]
- Facchinetti, F.; Rossi, G.; Bria, E.; Soria, J.C.; Besse, B.; Minari, R.; Friboulet, L.; Tiseo, M. Oncogene addiction in non-small cell lung cancer: Focus on ROS1 inhibition. Cancer Treat. Rev. 2017, 55, 83–95. [Google Scholar] [CrossRef] [PubMed]
- Tsakonas, G.; Ekman, S. Oncogene-addicted non-small cell lung cancer and immunotherapy. J. Thorac. Dis. 2018, 10, 1547–1555. [Google Scholar] [CrossRef] [PubMed]
- Peifer, M.; Fernandez-Cuesta, L.; Sos, M.L.; George, J.; Seidel, D.; Kasper, L.H.; Plenker, D.; Leenders, F.; Sun, R.; Zander, T.; et al. Integrative genome analyses identify key somatic driver mutations of small-cell lung cancer. Nat. Genet. 2012, 44, 1104–1110. [Google Scholar] [CrossRef] [PubMed]
- George, J.; Lim, J.S.; Jang, S.J.; Cun, Y.; Ozretic, L.; Kong, G.; Leenders, F.; Lu, X.; Fernandez-Cuesta, L.; Bosco, G.; et al. Comprehensive genomic profiles of small cell lung cancer. Nature 2015, 524, 47–53. [Google Scholar] [CrossRef]
- Brambilla, E.; Gazdar, A. Pathogenesis of lung cancer signalling pathways: Roadmap for therapies. Eur. Respir. J. 2009, 33, 1485–1497. [Google Scholar] [CrossRef]
- Ciuffreda, L.; Incani, U.C.; Steelman, L.S.; Abrams, S.L.; Falcone, I.; Curatolo, A.D.; Chappell, W.H.; Franklin, R.A.; Vari, S.; Cognetti, F.; et al. Signaling intermediates (MAPK and PI3K) as therapeutic targets in NSCLC. Curr. Pharm. Des. 2014, 20, 3944–3957. [Google Scholar] [CrossRef] [PubMed]
- Giaccia, A.J.; Kastan, M.B. The complexity of p53 modulation: Emerging patterns from divergent signals. Genes Dev. 1998, 12, 2973–2983. [Google Scholar] [CrossRef] [PubMed]
- Lohrum, M.A.; Vousden, K.H. Regulation and activation of p53 and its family members. Cell Death Differ. 1999, 6, 1162–1168. [Google Scholar] [CrossRef]
- Lee, J.S.; Yoon, A.; Kalapurakal, S.K.; Ro, J.Y.; Lee, J.J.; Tu, N.; Hittelman, W.N.; Hong, W.K. Expression of p53 oncoprotein in non-small-cell lung cancer: A favorable prognostic factor. J. Clin. Oncol. 1995, 13, 1893–1903. [Google Scholar] [CrossRef]
- Canale, M.; Petracci, E.; Delmonte, A.; Chiadini, E.; Dazzi, C.; Papi, M.; Capelli, L.; Casanova, C.; De Luigi, N.; Mariotti, M.; et al. Impact of TP53 Mutations on Outcome in EGFR-Mutated Patients Treated with First-Line Tyrosine Kinase Inhibitors. Clin. Cancer Res. 2017, 23, 2195–2202. [Google Scholar] [CrossRef]
- Shivapurkar, N.; Reddy, J.; Chaudhary, P.M.; Gazdar, A.F. Apoptosis and lung cancer: A review. J. Cell. Biochem. 2003, 88, 885–898. [Google Scholar] [CrossRef]
- Liu, G.; Pei, F.; Yang, F.; Li, L.; Amin, A.D.; Liu, S.; Buchan, J.R.; Cho, W.C. Role of Autophagy and Apoptosis in Non-Small-Cell Lung Cancer. Int. J. Mol. Sci. 2017, 18, 367. [Google Scholar] [CrossRef]
- Ouyang, L.; Shi, Z.; Zhao, S.; Wang, F.T.; Zhou, T.T.; Liu, B.; Bao, J.K. Programmed cell death pathways in cancer: A review of apoptosis, autophagy and programmed necrosis. Cell Prolif. 2012, 45, 487–498. [Google Scholar] [CrossRef]
- Li, H.; Zhu, H.; Xu, C.J.; Yuan, J. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell 1998, 94, 491–501. [Google Scholar] [CrossRef]
- Zou, H.; Li, Y.; Liu, X.; Wang, X. An APAF-1.cytochrome c multimeric complex is a functional apoptosome that activates procaspase-9. J. Biol. Chem. 1999, 274, 11549–11556. [Google Scholar] [CrossRef]
- Hata, A.N.; Engelman, J.A.; Faber, A.C. The BCL2 Family: Key Mediators of the Apoptotic Response to Targeted Anticancer Therapeutics. Cancer Discov. 2015, 5, 475–487. [Google Scholar] [CrossRef] [PubMed]
- Luthi, A.U.; Martin, S.J. The CASBAH: A searchable database of caspase substrates. Cell Death Differ. 2007, 14, 641–650. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Fennell, D.A. Caspase regulation in non-small cell lung cancer and its potential for therapeutic exploitation. Clin. Cancer Res. 2005, 11, 2097–2105. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.S.; Cho, M.C.; Lee, H.G.; Yoon, D.Y. Indole-3-carbinol induces apoptosis through p53 and activation of caspase-8 pathway in lung cancer A549 cells. Food Chem. Toxicol. Int. J. Publ. Br. Ind. Biol. Res. Assoc. 2010, 48, 883–890. [Google Scholar] [CrossRef]
- Chirumbolo, S. Dietary assumption of plant polyphenols and prevention of allergy. Curr. Pharm. Des. 2014, 20, 811–839. [Google Scholar] [CrossRef]
- Pandey, K.B.; Rizvi, S.I. Plant polyphenols as dietary antioxidants in human health and disease. Oxid. Med. Cell Longev. 2009, 2, 270–278. [Google Scholar] [CrossRef]
- Tsao, R. Chemistry and biochemistry of dietary polyphenols. Nutrients 2010, 2, 1231–1246. [Google Scholar] [CrossRef]
- Manach, C.; Scalbert, A.; Morand, C.; Remesy, C.; Jimenez, L. Polyphenols: Food sources and bioavailability. Am. J. Clin. Nutr. 2004, 79, 727–747. [Google Scholar] [CrossRef]
- Panche, A.N.; Diwan, A.D.; Chandra, S.R. Flavonoids: An overview. J. Nutr. Sci. 2016, 5, e47. [Google Scholar] [CrossRef]
- Shen, T.; Wang, X.N.; Lou, H.X. Natural stilbenes: An overview. Nat. Prod. Rep. 2009, 26, 916–935. [Google Scholar] [CrossRef]
- Teponno, R.B.; Kusari, S.; Spiteller, M. Recent advances in research on lignans and neolignans. Nat. Prod. Rep. 2016, 33, 1044–1092. [Google Scholar] [CrossRef] [PubMed]
- Haslam, E. Polyphenol-protein interactions. Biochem. J. 1974, 139, 285–288. [Google Scholar] [CrossRef] [PubMed]
- Hansch, C.; Gao, H. Comparative QSAR: Radical Reactions of Benzene Derivatives in Chemistry and Biology. Chem. Rev. 1997, 97, 2995–3060. [Google Scholar] [CrossRef]
- Nandi, S.; Vracko, M.; Bagchi, M.C. Anticancer activity of selected phenolic compounds: QSAR studies using ridge regression and neural networks. Chem. Biol. Drug Des. 2007, 70, 424–436. [Google Scholar] [CrossRef]
- Scotti, L.; Bezerra, M.F.J.; Magalhaes, M.D.R.; da Silva, M.S.; Pitta, I.R.; Scotti, M.T. SAR, QSAR and docking of anticancer flavonoids and variants: A review. Curr. Top. Med. Chem. 2012, 12, 2785–2809. [Google Scholar] [CrossRef]
- Selassie, C.; Shusterman, A.; Kapur, S.P.; Verma, R.; Zhang, L.; Hansch, C. On the toxicity of phenols to fast growing cells. A QSAR model for a radical-based toxicity. J. Chem. Soc. Perkin Trans. 1999, 2, 2729–2733. [Google Scholar] [CrossRef]
- Verma, R.P.; Kapur, S.; Barberena, O.; Shusterman, A.; Hansch, C.H.; Selassie, C.D. Synthesis, cytotoxicity, and QSAR analysis of X-thiophenols in rapidly dividing cells. Chem. Res. Toxicol. 2003, 16, 276–284. [Google Scholar] [CrossRef]
- Oliver, C.L.; Miranda, M.B.; Shangary, S.; Land, S.; Wang, S.; Johnson, D.E. (-)-Gossypol acts directly on the mitochondria to overcome Bcl-2- and Bcl-X(L)-mediated apoptosis resistance. Mol. Cancer Ther. 2005, 4, 23–31. [Google Scholar]
- Bruncko, M.; Oost, T.K.; Belli, B.A.; Ding, H.; Joseph, M.K.; Kunzer, A.; Martineau, D.; McClellan, W.J.; Mitten, M.; Ng, S.C.; et al. Studies leading to potent, dual inhibitors of Bcl-2 and Bcl-xL. J. Med. Chem. 2007, 50, 641–662. [Google Scholar] [CrossRef]
- Kitada, S.; Leone, M.; Sareth, S.; Zhai, D.; Reed, J.C.; Pellecchia, M. Discovery, characterization, and structure-activity relationships studies of proapoptotic polyphenols targeting B-cell lymphocyte/leukemia-2 proteins. J. Med. Chem. 2003, 46, 4259–4264. [Google Scholar] [CrossRef] [PubMed]
- Kitada, S.; Kress, C.L.; Krajewska, M.; Jia, L.; Pellecchia, M.; Reed, J.C. Bcl-2 antagonist apogossypol (NSC736630) displays single-agent activity in Bcl-2-transgenic mice and has superior efficacy with less toxicity compared with gossypol (NSC19048). Blood 2008, 111, 3211–3219. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Nikolovska-Coleska, Z.; Yang, C.Y.; Wang, R.; Tang, G.; Guo, J.; Shangary, S.; Qiu, S.; Gao, W.; Yang, D.; et al. Structure-based design of potent small-molecule inhibitors of anti-apoptotic Bcl-2 proteins. J. Med. Chem. 2006, 49, 6139–6142. [Google Scholar] [CrossRef] [PubMed]
- Tang, G.; Yang, C.Y.; Nikolovska-Coleska, Z.; Guo, J.; Qiu, S.; Wang, R.; Gao, W.; Wang, G.; Stuckey, J.; Krajewski, K.; et al. Pyrogallol-based molecules as potent inhibitors of the antiapoptotic Bcl-2 proteins. J. Med. Chem. 2007, 50, 1723–1726. [Google Scholar] [CrossRef]
- Tang, G.; Nikolovska-Coleska, Z.; Qiu, S.; Yang, C.Y.; Guo, J.; Wang, S. Acylpyrogallols as inhibitors of antiapoptotic Bcl-2 proteins. J. Med. Chem. 2008, 51, 717–720. [Google Scholar] [CrossRef]
- Larsen, C.A.; Bisson, W.H.; Dashwood, R.H. Tea catechins inhibit hepatocyte growth factor receptor (MET kinase) activity in human colon cancer cells: Kinetic and molecular docking studies. J. Med. Chem. 2009, 52, 6543–6545. [Google Scholar] [CrossRef]
- Ciechanover, A.; Orian, A.; Schwartz, A.L. Ubiquitin-mediated proteolysis: Biological regulation via destruction. Bioessays 2000, 22, 442–451. [Google Scholar] [CrossRef]
- Nam, S.; Smith, D.M.; Dou, Q.P. Ester bond-containing tea polyphenols potently inhibit proteasome activity in vitro and in vivo. J. Biol. Chem. 2001, 276, 13322–13330. [Google Scholar] [CrossRef]
- Kazi, A.; Wang, Z.; Kumar, N.; Falsetti, S.C.; Chan, T.H.; Dou, Q.P. Structure-activity relationships of synthetic analogs of (-)-epigallocatechin-3-gallate as proteasome inhibitors. Anticancer Res. 2004, 24, 943–954. [Google Scholar]
- Whyte, L.; Huang, Y.Y.; Torres, K.; Mehta, R.G. Molecular mechanisms of resveratrol action in lung cancer cells using dual protein and microarray analyses. Cancer Res. 2007, 67, 12007–12017. [Google Scholar] [CrossRef]
- Li, W.; Shi, Y.; Wang, R.; Pan, L.; Ma, L.; Jin, F. Resveratrol promotes the sensitivity of small-cell lung cancer H446 cells to cisplatin by regulating intrinsic apoptosis. Int. J. Oncol. 2018, 53, 2123–2130. [Google Scholar] [CrossRef] [PubMed]
- Rasheduzzaman, M.; Jeong, J.K.; Park, S.Y. Resveratrol sensitizes lung cancer cell to TRAIL by p53 independent and suppression of Akt/NF-kappaB signaling. Life Sci. 2018, 208, 208–220. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Chen, B.; Jiang, R.; Li, J.; Wang, B. Resveratrol inhibits lung cancer growth by suppressing M2-like polarization of tumor associated macrophages. Cell Immunol. 2017, 311, 86–93. [Google Scholar] [CrossRef]
- Ma, L.; Li, W.; Wang, R.; Nan, Y.; Wang, Q.; Liu, W.; Jin, F. Resveratrol enhanced anticancer effects of cisplatin on non-small cell lung cancer cell lines by inducing mitochondrial dysfunction and cell apoptosis. Int. J. Oncol. 2015, 47, 1460–1468. [Google Scholar] [CrossRef]
- Ulasli, S.S.; Celik, S.; Gunay, E.; Ozdemir, M.; Hazman, O.; Ozyurek, A.; Koyuncu, T.; Unlu, M. Anticancer effects of thymoquinone, caffeic acid phenethyl ester and resveratrol on A549 non-small cell lung cancer cells exposed to benzo (a) pyrene. Asian Pac. J. Cancer Prev. 2013, 14, 6159–6164. [Google Scholar] [CrossRef]
- Wright, C.; Iyer, A.K.V.; Yakisich, J.S.; Azad, N. Anti-Tumorigenic Effects of Resveratrol in Lung Cancer Cells Through Modulation of c-FLIP. Curr. Cancer Drug Targets 2017, 17, 669–680. [Google Scholar] [CrossRef]
- Luo, H.; Yang, A.; Schulte, B.A.; Wargovich, M.J.; Wang, G.Y. Resveratrol induces premature senescence in lung cancer cells via ROS-mediated DNA damage. PLoS ONE 2013, 8, e60065. [Google Scholar] [CrossRef]
- Luo, H.; Wang, L.; Schulte, B.A.; Yang, A.; Tang, S.; Wang, G.Y. Resveratrol enhances ionizing radiation-induced premature senescence in lung cancer cells. Int. J. Oncol. 2013, 43, 1999–2006. [Google Scholar] [CrossRef]
- Zhu, Y.; He, W.; Gao, X.; Li, B.; Mei, C.; Xu, R.; Chen, H. Resveratrol overcomes gefitinib resistance by increasing the intracellular gefitinib concentration and triggering apoptosis, autophagy and senescence in PC9/G NSCLC cells. Sci. Rep. 2015, 5, 17730. [Google Scholar] [CrossRef]
- Nie, P.; Hu, W.; Zhang, T.; Yang, Y.; Hou, B.; Zou, Z. Synergistic Induction of Erlotinib-Mediated Apoptosis by Resveratrol in Human Non-Small-Cell Lung Cancer Cells by Down-Regulating Survivin and Up-Regulating PUMA. Cell Physiol. Biochem. 2015, 35, 2255–2271. [Google Scholar] [CrossRef]
- Ko, J.C.; Syu, J.J.; Chen, J.C.; Wang, T.J.; Chang, P.Y.; Chen, C.Y.; Jian, Y.T.; Jian, Y.J.; Lin, Y.W. Resveratrol Enhances Etoposide-Induced Cytotoxicity through Down-Regulating ERK1/2 and AKT-Mediated X-ray Repair Cross-Complement Group 1 (XRCC1) Protein Expression in Human Non-Small-Cell Lung Cancer Cells. Basic Clin. Pharm. Toxicol. 2015, 117, 383–391. [Google Scholar] [CrossRef] [PubMed]
- Bae, S.; Lee, E.M.; Cha, H.J.; Kim, K.; Yoon, Y.; Lee, H.; Kim, J.; Kim, Y.J.; Lee, H.G.; Jeung, H.K.; et al. Resveratrol alters microRNA expression profiles in A549 human non-small cell lung cancer cells. Mol. Cells 2011, 32, 243–249. [Google Scholar] [CrossRef] [PubMed]
- Han, Z.; Yang, Q.; Liu, B.; Wu, J.; Li, Y.; Yang, C.; Jiang, Y. MicroRNA-622 functions as a tumor suppressor by targeting K-Ras and enhancing the anticarcinogenic effect of resveratrol. Carcinogenesis 2012, 33, 131–139. [Google Scholar] [CrossRef]
- Yu, Y.H.; Chen, H.A.; Chen, P.S.; Cheng, Y.J.; Hsu, W.H.; Chang, Y.W.; Chen, Y.H.; Jan, Y.; Hsiao, M.; Chang, T.Y.; et al. MiR-520h-mediated FOXC2 regulation is critical for inhibition of lung cancer progression by resveratrol. Oncogene 2013, 32, 431–443. [Google Scholar] [CrossRef]
- Lee, E.J.; Min, H.Y.; Joo Park, H.; Chung, H.J.; Kim, S.; Nam Han, Y.; Lee, S.K. G2/M cell cycle arrest and induction of apoptosis by a stilbenoid, 3,4,5-trimethoxy-4’-bromo-cis-stilbene, in human lung cancer cells. Life Sci. 2004, 75, 2829–2839. [Google Scholar] [CrossRef]
- Wang, X.; Wang, D.; Zhao, Y. Effect and Mechanism of Resveratrol on the Apoptosis of Lung Adenocarcinoma Cell Line A549. Cell Biochem. Biophys. 2015, 73, 527–531. [Google Scholar] [CrossRef]
- He, L.; Fan, F.; Hou, X.; Gao, C.; Meng, L.; Meng, S.; Huang, S.; Wu, H. Resveratrol suppresses pulmonary tumor metastasis by inhibiting platelet-mediated angiogenic responses. J. Surg. Res. 2017, 217, 113–122. [Google Scholar] [CrossRef]
- Li, X.; Wang, D.; Zhao, Q.C.; Shi, T.; Chen, J. Resveratrol Inhibited Non-small Cell Lung Cancer Through Inhibiting STAT-3 Signaling. Am. J. Med. Sci. 2016, 352, 524–530. [Google Scholar] [CrossRef]
- Li, W.; Ma, X.; Li, N.; Liu, H.; Dong, Q.; Zhang, J.; Yang, C.; Liu, Y.; Liang, Q.; Zhang, S.; et al. Resveratrol inhibits Hexokinases II mediated glycolysis in non-small cell lung cancer via targeting Akt signaling pathway. Exp. Cell Res. 2016, 349, 320–327. [Google Scholar] [CrossRef]
- Lee, Y.S.; Doonan, B.B.; Wu, J.M.; Hsieh, T.C. Combined metformin and resveratrol confers protection against UVC-induced DNA damage in A549 lung cancer cells via modulation of cell cycle checkpoints and DNA repair. Oncol. Rep. 2016, 35, 3735–3741. [Google Scholar] [CrossRef][Green Version]
- Sahin, E.; Baycu, C.; Koparal, A.T.; Burukoglu Donmez, D.; Bektur, E. Resveratrol reduces IL-6 and VEGF secretion from co-cultured A549 lung cancer cells and adipose-derived mesenchymal stem cells. Tumour Biol. J. Int. Soc. Oncodev. Biol. Med. 2016, 37, 7573–7582. [Google Scholar] [CrossRef] [PubMed]
- Yuan, L.; Zhang, Y.; Xia, J.; Liu, B.; Zhang, Q.; Liu, J.; Luo, L.; Peng, Z.; Song, Z.; Zhu, R. Resveratrol induces cell cycle arrest via a p53-independent pathway in A549 cells. Mol. Med. Rep. 2015, 11, 2459–2464. [Google Scholar] [CrossRef] [PubMed]
- Bai, T.; Dong, D.S.; Pei, L. Synergistic antitumor activity of resveratrol and miR-200c in human lung cancer. Oncol. Rep. 2014, 31, 2293–2297. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhang, H.; Tang, L.; Chen, H.; Wu, C.; Zhao, M.; Yang, Y.; Chen, X.; Liu, G. Resveratrol inhibits TGF-beta1-induced epithelial-to-mesenchymal transition and suppresses lung cancer invasion and metastasis. Toxicology 2013, 303, 139–146. [Google Scholar] [CrossRef]
- Liao, H.F.; Kuo, C.D.; Yang, Y.C.; Lin, C.P.; Tai, H.C.; Chen, Y.Y.; Chen, Y.J. Resveratrol enhances radiosensitivity of human non-small cell lung cancer NCI-H838 cells accompanied by inhibition of nuclear factor-kappa B activation. J. Radiat. Res. 2005, 46, 387–393. [Google Scholar] [CrossRef]
- Liu, P.L.; Tsai, J.R.; Charles, A.L.; Hwang, J.J.; Chou, S.H.; Ping, Y.H.; Lin, F.Y.; Chen, Y.L.; Hung, C.Y.; Chen, W.C.; et al. Resveratrol inhibits human lung adenocarcinoma cell metastasis by suppressing heme oxygenase 1-mediated nuclear factor-kappaB pathway and subsequently downregulating expression of matrix met alloproteinases. Mol. Nutr. Food Res. 2010, 54, 196–204. [Google Scholar] [CrossRef]
- Zhao, W.; Bao, P.; Qi, H.; You, H. Resveratrol down-regulates survivin and induces apoptosis in human multidrug-resistant SPC-A-1/CDDP cells. Oncol. Rep. 2010, 23, 279–286. [Google Scholar] [CrossRef]
- Kim, Y.A.; Lee, W.H.; Choi, T.H.; Rhee, S.H.; Park, K.Y.; Choi, Y.H. Involvement of p21WAF1/CIP1, pRB, Bax and NF-kappaB in induction of growth arrest and apoptosis by resveratrol in human lung carcinoma A549 cells. Int. J. Oncol. 2003, 23, 1143–1149. [Google Scholar]
- Karthikeyan, S.; Hoti, S.L.; Prasad, N.R. Resveratrol loaded gelatin nanoparticles synergistically inhibits cell cycle progression and constitutive NF-kappaB activation, and induces apoptosis in non-small cell lung cancer cells. Biomed. Pharmacother. 2015, 70, 274–282. [Google Scholar] [CrossRef]
- Thomas, E.; Gopalakrishnan, V.; Hegde, M.; Kumar, S.; Karki, S.S.; Raghavan, S.C.; Choudhary, B. A Novel Resveratrol Based Tubulin Inhibitor Induces Mitotic Arrest and Activates Apoptosis in Cancer Cells. Sci. Rep. 2016, 6, 34653. [Google Scholar] [CrossRef]
- Savio, M.; Ferraro, D.; Maccario, C.; Vaccarone, R.; Jensen, L.D.; Corana, F.; Mannucci, B.; Bianchi, L.; Cao, Y.; Stivala, L.A. Resveratrol analogue 4,4’-dihydroxy-trans-stilbene potently inhibits cancer invasion and metastasis. Sci. Rep. 2016, 6, 19973. [Google Scholar] [CrossRef] [PubMed]
- Malhotra, A.; Nair, P.; Dhawan, D.K. Premature mitochondrial senescence and related ultrastructural changes during lung carcinogenesis modulation by curcumin and resveratrol. Ultrastruct. Pathol. 2012, 36, 179–184. [Google Scholar] [CrossRef] [PubMed]
- Weng, C.J.; Yang, Y.T.; Ho, C.T.; Yen, G.C. Mechanisms of apoptotic effects induced by resveratrol, dibenzoylmethane, and their analogues on human lung carcinoma cells. J. Agric. Food Chem. 2009, 57, 5235–5243. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.O.; Lee, H.J.; Hwang, H.S.; Ahn, K.S.; Chae, C.; Kang, K.S.; Lu, J.; Kim, S.H. Potent inhibition of Lewis lung cancer growth by heyneanol A from the roots of Vitis amurensis through apoptotic and anti-angiogenic activities. Carcinogenesis 2006, 27, 2059–2069. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, R.; Sasaki, K.; Yoshida, K. Identification of epigallocatechin-3-gallate in green tea polyphenols as a potent inducer of p53-dependent apoptosis in the human lung cancer cell line A549. Toxicol. In Vitro 2009, 23, 834–839. [Google Scholar] [CrossRef]
- Okabe, S.; Suganuma, M.; Hayashi, M.; Sueoka, E.; Komori, A.; Fujiki, H. Mechanisms of growth inhibition of human lung cancer cell line, PC-9, by tea polyphenols. Jpn. J. Cancer Res. 1997, 88, 639–643. [Google Scholar] [CrossRef]
- Honda, Y.; Takigawa, N.; Ichihara, E.; Ninomiya, T.; Kubo, T.; Ochi, N.; Yasugi, M.; Murakami, T.; Yamane, H.; Tanimoto, M.; et al. Effects of (-)-epigallocatechin-3-gallate on EGFR- or Fusion Gene-driven Lung Cancer Cells. Acta Med. Okayama 2017, 71, 505–512. [Google Scholar] [CrossRef]
- Okabe, S.; Fujimoto, N.; Sueoka, N.; Suganuma, M.; Fujiki, H. Modulation of gene expression by (-)-epigallocatechin gallate in PC-9 cells using a cDNA expression array. Biol. Pharm. Bull. 2001, 24, 883–886. [Google Scholar] [CrossRef]
- Zhou, H.; Chen, J.X.; Yang, C.S.; Yang, M.Q.; Deng, Y.; Wang, H. Gene regulation mediated by microRNAs in response to green tea polyphenol EGCG in mouse lung cancer. BMC Genom. 2014, 15, S3. [Google Scholar] [CrossRef]
- Suganuma, M.; Kurusu, M.; Suzuki, K.; Tasaki, E.; Fujiki, H. Green tea polyphenol stimulates cancer preventive effects of celecoxib in human lung cancer cells by upregulation of GADD153 gene. Int. J. Cancer 2006, 119, 33–40. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, X.; Han, L.; Zhou, Y.; Sun, S. Green tea polyphenol EGCG reverse cisplatin resistance of A549/DDP cell line through candidate genes demethylation. Biomed. Pharmacother. 2015, 69, 285–290. [Google Scholar] [CrossRef] [PubMed]
- Amin, A.R.; Wang, D.; Zhang, H.; Peng, S.; Shin, H.J.; Brandes, J.C.; Tighiouart, M.; Khuri, F.R.; Chen, Z.G.; Shin, D.M. Enhanced anti-tumor activity by the combination of the natural compounds (-)-epigallocatechin-3-gallate and luteolin: Potential role of p53. J. Biol. Chem. 2010, 285, 34557–34565. [Google Scholar] [CrossRef] [PubMed]
- Ganguly, C.; Saha, P.; Panda, C.K.; Das, S. Inhibition of growth, induction of apoptosis and alteration of gene expression by tea polyphenols in the highly metastatic human lung cancer cell line NCI-H460. Asian Pac. J. Cancer Prev. 2005, 6, 326–331. [Google Scholar] [PubMed]
- Banerjee, S.; Manna, S.; Mukherjee, S.; Pal, D.; Panda, C.K.; Das, S. Black tea polyphenols restrict benzopyrene-induced mouse lung cancer progression through inhibition of Cox-2 and induction of caspase-3 expression. Asian Pac. J. Cancer Prev. 2006, 7, 661–666. [Google Scholar] [PubMed]
- Banerjee, S.; Manna, S.; Saha, P.; Panda, C.K.; Das, S. Black tea polyphenols suppress cell proliferation and induce apoptosis during benzo(a)pyrene-induced lung carcinogenesis. Eur. J. Cancer Prev. 2005, 14, 215–221. [Google Scholar] [CrossRef]
- Gu, Q.; Hu, C.; Chen, Q.; Xia, Y.; Feng, J.; Yang, H. Development of a rat model by 3,4-benzopyrene intra-pulmonary injection and evaluation of the effect of green tea drinking on p53 and bcl-2 expression in lung carcinoma. Cancer Detect. Prev. 2009, 32, 444–451. [Google Scholar] [CrossRef]
- Gu, Q.; Hu, C.; Chen, Q.; Xia, Y. Tea polyphenols prevent lung from preneoplastic lesions and effect p53 and bcl-2 gene expression in rat lung tissues. Int. J. Clin. Exp. Pathol. 2013, 6, 1523–1531. [Google Scholar]
- Roy, P.; Nigam, N.; Singh, M.; George, J.; Srivastava, S.; Naqvi, H.; Shukla, Y. Tea polyphenols inhibit cyclooxygenase-2 expression and block activation of nuclear factor-kappa B and Akt in diethylnitrosoamine induced lung tumors in Swiss mice. Investig. New Drugs 2010, 28, 466–471. [Google Scholar] [CrossRef]
- Pan, J.; Zhang, Q.; Xiong, D.; Vedell, P.; Yan, Y.; Jiang, H.; Cui, P.; Ding, F.; Tichelaar, J.W.; Wang, Y.; et al. Transcriptomic analysis by RNA-seq reveals AP-1 pathway as key regulator that green tea may rely on to inhibit lung tumorigenesis. Mol. Carcinog. 2014, 53, 19–29. [Google Scholar] [CrossRef]
- Lu, Q.Y.; Yang, Y.; Jin, Y.S.; Zhang, Z.F.; Heber, D.; Li, F.P.; Dubinett, S.M.; Sondej, M.A.; Loo, J.A.; Rao, J.Y. Effects of green tea extract on lung cancer A549 cells: Proteomic identification of proteins associated with cell migration. Proteomics 2009, 9, 757–767. [Google Scholar] [CrossRef]
- Lu, G.; Xiao, H.; You, H.; Lin, Y.; Jin, H.; Snagaski, B.; Yang, C.S. Synergistic inhibition of lung tumorigenesis by a combination of green tea polyphenols and atorvastatin. Clin. Cancer Res. 2008, 14, 4981–4988. [Google Scholar] [CrossRef] [PubMed]
- Izdebska, M.; Klimaszewska-Wisniewska, A.; Halas, M.; Gagat, M.; Grzanka, A. Green tea extract induces protective autophagy in A549 non-small lung cancer cell line. Postepy. Hig. Med. Dosw. 2015, 69, 1478–1484. [Google Scholar]
- Radhakrishna, P.G.; Srivastava, A.S.; Hassanein, T.I.; Chauhan, D.P.; Carrier, E. Induction of apoptosis in human lung cancer cells by curcumin. Cancer Lett. 2004, 208, 163–170. [Google Scholar] [CrossRef] [PubMed]
- Saha, A.; Kuzuhara, T.; Echigo, N.; Fujii, A.; Suganuma, M.; Fujiki, H. Apoptosis of human lung cancer cells by curcumin mediated through up-regulation of “growth arrest and DNA damage inducible genes 45 and 153”. Biol. Pharm. Bull. 2010, 33, 1291–1299. [Google Scholar] [CrossRef]
- Wu, S.H.; Hang, L.W.; Yang, J.S.; Chen, H.Y.; Lin, H.Y.; Chiang, J.H.; Lu, C.C.; Yang, J.L.; Lai, T.Y.; Ko, Y.C.; et al. Curcumin induces apoptosis in human non-small cell lung cancer NCI-H460 cells through ER stress and caspase cascade- and mitochondria-dependent pathways. Anticancer Res. 2010, 30, 2125–2133. [Google Scholar]
- Li, Y.; Zhang, S.; Geng, J.X.; Hu, X.Y. Curcumin inhibits human non-small cell lung cancer A549 cell proliferation through regulation of Bcl-2/Bax and cytochrome C. Asian Pac. J. Cancer Prev. 2013, 14, 4599–4602. [Google Scholar] [CrossRef]
- Yang, C.L.; Ma, Y.G.; Xue, Y.X.; Liu, Y.Y.; Xie, H.; Qiu, G.R. Curcumin induces small cell lung cancer NCI-H446 cell apoptosis via the reactive oxygen species-mediated mitochondrial pathway and not the cell death receptor pathway. DNA Cell Biol. 2012, 31, 139–150. [Google Scholar] [CrossRef]
- Jin, H.; Qiao, F.; Wang, Y.; Xu, Y.; Shang, Y. Curcumin inhibits cell proliferation and induces apoptosis of human non-small cell lung cancer cells through the upregulation of miR-192-5p and suppression of PI3K/Akt signaling pathway. Oncol. Rep. 2015, 34, 2782–2789. [Google Scholar] [CrossRef]
- Wang, A.; Wang, J.; Zhang, S.; Zhang, H.; Xu, Z.; Li, X. Curcumin inhibits the development of non-small cell lung cancer by inhibiting autophagy and apoptosis. Exp. Ther. Med. 2017, 14, 5075–5080. [Google Scholar] [CrossRef]
- Liu, F.; Gao, S.; Yang, Y.; Zhao, X.; Fan, Y.; Ma, W.; Yang, D.; Yang, A.; Yu, Y. Curcumin induced autophagy anticancer effects on human lung adenocarcinoma cell line A549. Oncol. Lett. 2017, 14, 2775–2782. [Google Scholar] [CrossRef]
- Liao, H.; Wang, Z.; Deng, Z.; Ren, H.; Li, X. Curcumin inhibits lung cancer invasion and metastasis by attenuating GLUT1/MT1-MMP/MMP2 pathway. Int. J. Clin. Exp. Med. 2015, 8, 8948–8957. [Google Scholar] [PubMed]
- Tsai, J.R.; Liu, P.L.; Chen, Y.H.; Chou, S.H.; Cheng, Y.J.; Hwang, J.J.; Chong, I.W. Curcumin Inhibits Non-Small Cell Lung Cancer Cells Metastasis through the Adiponectin/NF-kappab/MMPs Signaling Pathway. PLoS ONE 2015, 10, e0144462. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.Q.; Wei, X.Y.; Li, W.L.; Kanchana, K.; Xu, C.C.; Chen, D.H.; Chou, P.H.; Jin, R.; Wu, J.Z.; Liang, G. Curcumin analogue A501 induces G2/M arrest and apoptosis in non-small cell lung cancer cells. Asian Pac. J. Cancer Prev. 2014, 15, 6893–6898. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Lee, Y.M.; Chang, G.C.; Yu, S.L.; Hsieh, W.Y.; Chen, J.J.; Chen, H.W.; Yang, P.C. Curcumin induces EGFR degradation in lung adenocarcinoma and modulates p38 activation in intestine: The versatile adjuvant for gefitinib therapy. PLoS ONE 2011, 6, e23756. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.H.; Kang, H.S.; Kim, I.K.; Lee, H.Y.; Ha, J.H.; Yeo, C.D.; Kang, H.H.; Moon, H.S.; Lee, S.H. Curcumin sensitizes human lung cancer cells to apoptosis and metastasis synergistically combined with carboplatin. Exp. Biol. Med. 2015, 240, 1416–1425. [Google Scholar] [CrossRef] [PubMed]
- Kuo, P.C.; Liu, H.F.; Chao, J.I. Survivin and p53 modulate quercetin-induced cell growth inhibition and apoptosis in human lung carcinoma cells. J. Biol. Chem. 2004, 279, 55875–55885. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.T.; Tran, E.; Nguyen, T.H.; Do, P.T.; Huynh, T.H.; Huynh, H. The role of activated MEK-ERK pathway in quercetin-induced growth inhibition and apoptosis in A549 lung cancer cells. Carcinogenesis 2004, 25, 647–659. [Google Scholar] [CrossRef] [PubMed]
- Youn, H.; Jeong, J.C.; Jeong, Y.S.; Kim, E.J.; Um, S.J. Quercetin potentiates apoptosis by inhibiting nuclear factor-kappaB signaling in H460 lung cancer cells. Biol. Pharm. Bull. 2013, 36, 944–951. [Google Scholar] [CrossRef]
- Izumi, H.; Takahashi, M.; Uramoto, H.; Nakayama, Y.; Oyama, T.; Wang, K.Y.; Sasaguri, Y.; Nishizawa, S.; Kohno, K. Monocarboxylate transporters 1 and 4 are involved in the invasion activity of human lung cancer cells. Cancer Sci. 2011, 102, 1007–1013. [Google Scholar] [CrossRef]
- Zhu, X.; Ma, P.; Peng, D.; Wang, Y.; Wang, D.; Chen, X.; Zhang, X.; Song, Y. Quercetin suppresses lung cancer growth by targeting Aurora B kinase. Cancer Med. 2016, 5, 3156–3165. [Google Scholar] [CrossRef]
- Klimaszewska-Wisniewska, A.; Halas-Wisniewska, M.; Izdebska, M.; Gagat, M.; Grzanka, A.; Grzanka, D. Antiproliferative and antimetastatic action of quercetin on A549 non-small cell lung cancer cells through its effect on the cytoskeleton. Acta Histochem. 2017, 119, 99–112. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.H.; Lai, S.L.; Chen, W.S.; Hung, W.Y.; Chow, J.M.; Hsiao, M.; Lee, W.J.; Chien, M.H. Quercetin suppresses the metastatic ability of lung cancer through inhibiting Snail-dependent Akt activation and Snail-independent ADAM9 expression pathways. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 1746–1758. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.C.; Chan, S.T.; Chang, C.N.; Yu, P.S.; Chuang, C.H.; Yeh, S.L. Quercetin and chrysin inhibit nickel-induced invasion and migration by downregulation of TLR4/NF-kappaB signaling in A549cells. Chem. Biol. Interact. 2018, 292, 101–109. [Google Scholar] [CrossRef]
- Chan, S.T.; Yang, N.C.; Huang, C.S.; Liao, J.W.; Yeh, S.L. Quercetin enhances the antitumor activity of trichostatin A through upregulation of p53 protein expression in vitro and in vivo. PLoS ONE 2013, 8, e54255. [Google Scholar] [CrossRef]
- Lee, S.H.; Lee, E.J.; Min, K.H.; Hur, G.Y.; Lee, S.H.; Lee, S.Y.; Kim, J.H.; Shin, C.; Shim, J.J.; In, K.H.; et al. Quercetin Enhances Chemosensitivity to Gemcitabine in Lung Cancer Cells by Inhibiting Heat Shock Protein 70 Expression. Clin. Lung Cancer 2015, 16, 235–243. [Google Scholar] [CrossRef]
- Lee, H.; Kim, Y.; Jeong, J.H.; Ryu, J.H.; Kim, W.Y. ATM/CHK/p53 Pathway Dependent Chemopreventive and Therapeutic Activity on Lung Cancer by Pterostilbene. PLoS ONE 2016, 11, e0162335. [Google Scholar] [CrossRef]
- Chen, Z.; Jin, K.; Gao, L.; Lou, G.; Jin, Y.; Yu, Y.; Lou, Y. Anti-tumor effects of bakuchiol, an analogue of resveratrol, on human lung adenocarcinoma A549 cell line. Eur. J. Pharmacol. 2010, 643, 170–179. [Google Scholar] [CrossRef]
- Park, J.J.; Hwang, S.J.; Park, J.H.; Lee, H.J. Chlorogenic acid inhibits hypoxia-induced angiogenesis via down-regulation of the HIF-1alpha/AKT pathway. Cell. Oncol. 2015, 38, 111–118. [Google Scholar] [CrossRef]
- Khan, N.; Afaq, F.; Khusro, F.H.; Mustafa Adhami, V.; Suh, Y.; Mukhtar, H. Dual inhibition of phosphatidylinositol 3-kinase/Akt and mammalian target of rapamycin signaling in human nonsmall cell lung cancer cells by a dietary flavonoid fisetin. Int. J. Cancer 2012, 130, 1695–1705. [Google Scholar] [CrossRef]
- Zhuo, W.; Zhang, L.; Zhu, Y.; Zhu, B.; Chen, Z. Fisetin, a dietary bioflavonoid, reverses acquired Cisplatin-resistance of lung adenocarcinoma cells through MAPK/Survivin/Caspase pathway. Am. J. Transl. Res. 2015, 7, 2045–2052. [Google Scholar]
- Klimaszewska-Wisniewska, A.; Halas-Wisniewska, M.; Tadrowski, T.; Gagat, M.; Grzanka, D.; Grzanka, A. Paclitaxel and the dietary flavonoid fisetin: A synergistic combination that induces mitotic catastrophe and autophagic cell death in A549 non-small cell lung cancer cells. Cancer Cell Int. 2016, 16, 10. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Ho, W.S. Combination of liquiritin, isoliquiritin and isoliquirigenin induce apoptotic cell death through upregulating p53 and p21 in the A549 non-small cell lung cancer cells. Oncol. Rep. 2014, 31, 298–304. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, A.; DaCunha, D.C.; Barros, L.; Caires, H.R.; Xavier, C.P.R.; Ferreira, I.; Vasconcelos, M.H. Eucalyptus globulus Labill. decoction extract inhibits the growth of NCI-H460 cells by increasing the p53 levels and altering the cell cycle profile. Food Funct. 2019, 6. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.G.; Yao, W.N.; Zhang, B.; Hua, J.; Liang, D.; Wang, H.S. Lung cancer and matrix met alloproteinases inhibitors of polyphenols from Selaginella tamariscina with suppression activity of migration. Bioorg. Med. Chem. Lett. 2018, 28, 2413–2417. [Google Scholar] [CrossRef] [PubMed]
- Moore, J.; Megaly, M.; MacNeil, A.J.; Klentrou, P.; Tsiani, E. Rosemary extract reduces Akt/mTOR/p70S6K activation and inhibits proliferation and survival of A549 human lung cancer cells. Biomed. Pharmacother. 2016, 83, 725–732. [Google Scholar] [CrossRef]
- Tang, X.L.; Yan, L.; Zhu, L.; Jiao, D.M.; Chen, J.; Chen, Q.Y. Salvianolic acid A reverses cisplatin resistance in lung cancer A549 cells by targeting c-met and attenuating Akt/mTOR pathway. J. Pharm. Sci. 2017, 135, 1–7. [Google Scholar] [CrossRef]
- Liu, Y.; Tong, Y.; Yang, X.; Li, F.; Zheng, L.; Liu, W.; Wu, J.; Ou, R.; Zhang, G.; Hu, M.; et al. Novel histone deacetylase inhibitors derived from Magnolia officinalis significantly enhance TRAIL-induced apoptosis in non-small cell lung cancer. Pharmacol. Res. 2016, 111, 113–125. [Google Scholar] [CrossRef]
- Husari, A.; Hashem, Y.; Zaatari, G.; El Sabban, M. Pomegranate Juice Prevents the Formation of Lung Nodules Secondary to Chronic Cigarette Smoke Exposure in an Animal Model. Oxid. Med. Cell Longev. 2017, 2017, 6063201. [Google Scholar] [CrossRef]
- Narayan, C.; Kumar, A. Antineoplastic and immunomodulatory effect of polyphenolic components of Achyranthes aspera (PCA) extract on urethane induced lung cancer in vivo. Mol. Biol. Rep. 2014, 41, 179–191. [Google Scholar] [CrossRef]
- Aichinger, G.; Pahlke, G.; Nagel, L.J.; Berger, W.; Marko, D. Bilberry extract, its major polyphenolic compounds, and the soy isoflavone genistein antagonize the cytostatic drug erlotinib in human epithelial cells. Food Funct. 2016, 7, 3628–3636. [Google Scholar] [CrossRef]
- Aggarwal, B.B.; Bhardwaj, A.; Aggarwal, R.S.; Seeram, N.P.; Shishodia, S.; Takada, Y. Role of resveratrol in prevention and therapy of cancer: Preclinical and clinical studies. Anticancer Res. 2004, 24, 2783–2840. [Google Scholar] [PubMed]
- Salehi, B.; Mishra, A.P.; Nigam, M.; Sener, B.; Kilic, M.; Sharifi-Rad, M.; Fokou, P.V.T.; Martins, N.; Sharifi-Rad, J. Resveratrol: A Double-Edged Sword in Health Benefits. Biomedicines 2018, 6, 91. [Google Scholar] [CrossRef]
- Poulose, S.M.; Thangthaeng, N.; Miller, M.G.; Shukitt-Hale, B. Effects of pterostilbene and resveratrol on brain and behavior. Neurochem. Int. 2015, 89, 227–233. [Google Scholar] [CrossRef] [PubMed]
- Ko, J.H.; Sethi, G.; Um, J.Y.; Shanmugam, M.K.; Arfuso, F.; Kumar, A.P.; Bishayee, A.; Ahn, K.S. The Role of Resveratrol in Cancer Therapy. Int. J. Mol. Sci. 2017, 18, 2589. [Google Scholar] [CrossRef]
- Whitlock, N.C.; Baek, S.J. The anticancer effects of resveratrol: Modulation of transcription factors. Nutr. Cancer 2012, 64, 493–502. [Google Scholar] [CrossRef]
- Bergman, M.; Levin, G.S.; Bessler, H.; Djaldetti, M.; Salman, H. Resveratrol affects the cross talk between immune and colon cancer cells. Biomed. Pharmacother. 2013, 67, 43–47. [Google Scholar] [CrossRef]
- Su, J.L.; Chen, P.B.; Chen, Y.H.; Chen, S.C.; Chang, Y.W.; Jan, Y.H.; Cheng, X.; Hsiao, M.; Hung, M.C. Downregulation of microRNA miR-520h by E1A contributes to anticancer activity. Cancer Res. 2010, 70, 5096–5108. [Google Scholar] [CrossRef]
- Graham, H.N. Green tea composition, consumption, and polyphenol chemistry. Prev. Med. 1992, 21, 334–350. [Google Scholar] [CrossRef]
- Balentine, D.A.; Wiseman, S.A.; Bouwens, L.C. The chemistry of tea flavonoids. Crit. Rev. Food Sci. Nutr. 1997, 37, 693–704. [Google Scholar] [CrossRef]
- Colomer, R.; Sarrats, A.; Lupu, R.; Puig, T. Natural Polyphenols and their Synthetic Analogs as Emerging Anticancer Agents. Curr. Drug Targets 2017, 18, 147–159. [Google Scholar] [CrossRef]
- Sekher Pannala, A.; Chan, T.S.; O’Brien, P.J.; Rice-Evans, C.A. Flavonoid B-ring chemistry and antioxidant activity: Fast reaction kinetics. Biochem. Biophys. Res. Commun. 2001, 282, 1161–1168. [Google Scholar] [CrossRef] [PubMed]
- Burda, S.; Oleszek, W. Antioxidant and antiradical activities of flavonoids. J. Agric. Food Chem. 2001, 49, 2774–2779. [Google Scholar] [CrossRef] [PubMed]
- Sang, S.; Lambert, J.D.; Ho, C.T.; Yang, C.S. The chemistry and biotransformation of tea constituents. Pharmacol. Res. 2011, 64, 87–99. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.S.; Wang, X.; Lu, G.; Picinich, S.C. Cancer prevention by tea: Animal studies, molecular mechanisms and human relevance. Nat. Rev. Cancer 2009, 9, 429–439. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.S.; Wang, H.; Li, G.X.; Yang, Z.; Guan, F.; Jin, H. Cancer prevention by tea: Evidence from laboratory studies. Pharmacol. Res. 2011, 64, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.S.; Wang, H.; Chen, J.X.; Zhang, J. Effects of Tea Catechins on Cancer Signaling Pathways. Enzymes 2014, 36, 195–221. [Google Scholar] [CrossRef] [PubMed]
- Miyata, Y.; Matsuo, T.; Araki, K.; Nakamura, Y.; Sagara, Y.; Ohba, K.; Sakai, H. Anticancer Effects of Green Tea and the Underlying Molecular Mechanisms in Bladder Cancer. Medicines 2018, 5, 87. [Google Scholar] [CrossRef]
- Wu, X.; Yu, H.; Amos, C.I.; Hong, W.K.; Spitz, M.R. Joint effect of insulin-like growth factors and mutagen sensitivity in lung cancer risk. J. Natl. Cancer Inst. 2000, 92, 737–743. [Google Scholar] [CrossRef][Green Version]
- Wang, H.; Bian, S.; Yang, C.S. Green tea polyphenol EGCG suppresses lung cancer cell growth through upregulating miR-210 expression caused by stabilizing HIF-1alpha. Carcinogenesis 2011, 32, 1881–1889. [Google Scholar] [CrossRef]
- Liu, F.; Cao, X.; Liu, Z.; Guo, H.; Ren, K.; Quan, M.; Zhou, Y.; Xiang, H.; Cao, J. Casticin suppresses self-renewal and invasion of lung cancer stem-like cells from A549 cells through down-regulation of pAkt. Acta Biochim. Biophys. Sin. 2014, 46, 15–21. [Google Scholar] [CrossRef]
- Singh, B.N.; Shankar, S.; Srivastava, R.K. Green tea catechin, epigallocatechin-3-gallate (EGCG): Mechanisms, perspectives and clinical applications. Biochem. Pharmacol. 2011, 82, 1807–1821. [Google Scholar] [CrossRef] [PubMed]
- Vallianou, N.G.; Evangelopoulos, A.; Schizas, N.; Kazazis, C. Potential anticancer properties and mechanisms of action of curcumin. Anticancer Res. 2015, 35, 645–651. [Google Scholar] [PubMed]
- Tomeh, M.A.; Hadianamrei, R.; Zhao, X. A Review of Curcumin and Its Derivatives as Anticancer Agents. Int. J. Mol. Sci. 2019, 20, 1033. [Google Scholar] [CrossRef]
- Adekola, K.; Rosen, S.T.; Shanmugam, M. Glucose transporters in cancer metabolism. Curr. Opin. Oncol. 2012, 24, 650–654. [Google Scholar] [CrossRef]
- Liu, Y.; Cao, Y.; Zhang, W.; Bergmeier, S.; Qian, Y.; Akbar, H.; Colvin, R.; Ding, J.; Tong, L.; Wu, S.; et al. A small-molecule inhibitor of glucose transporter 1 downregulates glycolysis, induces cell-cycle arrest, and inhibits cancer cell growth in vitro and in vivo. Mol. Cancer Ther. 2012, 11, 1672–1682. [Google Scholar] [CrossRef]
- Meng, Y.; Xu, X.; Luan, H.; Li, L.; Dai, W.; Li, Z.; Bian, J. The progress and development of GLUT1 inhibitors targeting cancer energy metabolism. Future Med. Chem. 2019, 11, 2333–2352. [Google Scholar] [CrossRef]
- Rauf, A.; Imran, M.; Khan, I.A.; Ur-Rehman, M.; Gilani, S.A.; Mehmood, Z.; Mubarak, M.S. Anticancer potential of quercetin: A comprehensive review. Phytother. Res. 2018, 32, 2109–2130. [Google Scholar] [CrossRef]
- Garrido, C.; Schmitt, E.; Cande, C.; Vahsen, N.; Parcellier, A.; Kroemer, G. HSP27 and HSP70: Potentially oncogenic apoptosis inhibitors. Cell Cycle 2003, 2, 579–584. [Google Scholar] [CrossRef]
- Meng, S.; Cao, J.; Feng, Q.; Peng, J.; Hu, Y. Roles of chlorogenic Acid on regulating glucose and lipids metabolism: A review. Evid. Based Complementary Altern. Med. 2013, 2013, 801457. [Google Scholar] [CrossRef]
- Santana-Galvez, J.; Cisneros-Zevallos, L.; Jacobo-Velazquez, D.A. Chlorogenic Acid: Recent Advances on Its Dual Role as a Food Additive and a Nutraceutical against Metabolic Syndrome. Molecules 2017, 22, 358. [Google Scholar] [CrossRef]
- Naveed, M.; Hejazi, V.; Abbas, M.; Kamboh, A.A.; Khan, G.J.; Shumzaid, M.; Ahmad, F.; Babazadeh, D.; FangFang, X.; Modarresi-Ghazani, F.; et al. Chlorogenic acid (CGA): A pharmacological review and call for further research. Biomed. Pharmacother. 2018, 97, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.C.; Zhang, Y.; Dai, B.L.; Ma, Y.J.; Zhang, Q.; Wang, Y.; Yang, H. Chlorogenic acid prevents inflammatory responses in IL1betastimulated human SW1353 chondrocytes, a model for osteoarthritis. Mol. Med. Rep. 2017, 16, 1369–1375. [Google Scholar] [CrossRef] [PubMed]
- Karunanidhi, A.; Thomas, R.; van Belkum, A.; Neela, V. In vitro antibacterial and antibiofilm activities of chlorogenic acid against clinical isolates of Stenotrophomonas maltophilia including the trimethoprim/sulfamethoxazole resistant strain. Biomed. Res. Int. 2013, 2013, 392058. [Google Scholar] [CrossRef] [PubMed]
- Sung, W.S.; Lee, D.G. Antifungal action of chlorogenic acid against pathogenic fungi, mediated by membrane disruption. Pure Appl. Chem. 2010, 82, 219–226. [Google Scholar] [CrossRef]
- Shimizu, M.; Yoshimi, N.; Yamada, Y.; Matsunaga, K.; Kawabata, K.; Hara, A.; Moriwaki, H.; Mori, H. Suppressive effects of chlorogenic acid on N-methyl-N-nitrosourea-induced glandular stomach carcinogenesis in male F344 rats. J. Toxicol. Sci. 1999, 24, 433–439. [Google Scholar] [CrossRef]
- Matsunaga, K.; Katayama, M.; Sakata, K.; Kuno, T.; Yoshida, K.; Yamada, Y.; Hirose, Y.; Yoshimi, N.; Mori, H. Inhibitory Effects of Chlorogenic Acid on Azoxymethane-induced Colon Carcinogenesis in Male F344 Rats. Asian Pac. J. Cancer Prev. 2002, 3, 163–166. [Google Scholar]
- Kasai, H.; Fukada, S.; Yamaizumi, Z.; Sugie, S.; Mori, H. Action of chlorogenic acid in vegetables and fruits as an inhibitor of 8-hydroxydeoxyguanosine formation in vitro and in a rat carcinogenesis model. Food Chem. Toxicol. Int. J. Publ. Br. Ind. Biol. Res. Assoc. 2000, 38, 467–471. [Google Scholar] [CrossRef]
- Ignatowicz, E.; Balana, B.; Vulimiri, S.V.; Szaefer, H.; Baer-Dubowska, W. The effect of plant phenolics on the formation of 7,12-dimethylbenz[a]anthracene-DNA adducts and TPA-stimulated polymorphonuclear neutrophils chemiluminescence in vitro. Toxicology 2003, 189, 199–209. [Google Scholar] [CrossRef]
- Kang, T.Y.; Yang, H.R.; Zhang, J.; Li, D.; Lin, J.; Wang, L.; Xu, X. The studies of chlorogenic Acid antitumor mechanism by gene chip detection: The immune pathway gene expression. J. Anal. Methods Chem. 2013, 2013, 617243. [Google Scholar] [CrossRef]
- Khan, N.; Afaq, F.; Syed, D.N.; Mukhtar, H. Fisetin, a novel dietary flavonoid, causes apoptosis and cell cycle arrest in human prostate cancer LNCaP cells. Carcinogenesis 2008, 29, 1049–1056. [Google Scholar] [CrossRef]
- Syed, D.N.; Adhami, V.M.; Khan, M.I.; Mukhtar, H. Inhibition of Akt/mTOR signaling by the dietary flavonoid fisetin. Anticancer Agents Med. Chem. 2013, 13, 995–1001. [Google Scholar] [CrossRef] [PubMed]
- Yang, P.M.; Tseng, H.H.; Peng, C.W.; Chen, W.S.; Chiu, S.J. Dietary flavonoid fisetin targets caspase-3-deficient human breast cancer MCF-7 cells by induction of caspase-7-associated apoptosis and inhibition of autophagy. Int. J. Oncol. 2012, 40, 469–478. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kashyap, D.; Garg, V.K.; Tuli, H.S.; Yerer, M.B.; Sak, K.; Sharma, A.K.; Kumar, M.; Aggarwal, V.; Sandhu, S.S. Fisetin and Quercetin: Promising Flavonoids with Chemopreventive Potential. Biomolecules 2019, 9, 174. [Google Scholar] [CrossRef]
- D’Archivio, M.; Filesi, C.; Vari, R.; Scazzocchio, B.; Masella, R. Bioavailability of the polyphenols: Status and controversies. Int. J. Mol. Sci. 2010, 11, 1321–1342. [Google Scholar] [CrossRef]
- Teng, H.; Chen, L. Polyphenols and bioavailability: An update. Crit. Rev. Food Sci. Nutr. 2019, 59, 2040–2051. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Polyphenol Compounds or Extracts | Mechanisms | In Vitro and/or In Vivo Models | References |
|---|---|---|---|
| Resveratrol | Induction of apoptosis by up-regulation of p53 and p21, activation of the caspases and disruption of the mitochondrial membrane complex. Cell cycle arrest at the G1 phase. Alterations in expressions of cyclin A, chk1, CDC27, and Eg5. Anti-tumor effect mediated by transforming growth factor-β pathway, particularly through the Smad proteins. i.e., down-regulation of the Smad activators 2 and 4 and up-regulation of the repressor Smad 7 | A549 human NSCLC cell line | [90] |
| Resveratrol | Induction of apoptosis as a result of mitochondrial depolarization, release of cytochrome c from the mitochondrial compartment to the cytoplasm, apoptosis-inducing factor translocation from the mitochondrial compartment to the nucleus, and altered protein levels of Bcl-2, Bcl-xL and Bax | H446 human SCLC cells | [91] |
| Resveratrol | Induction of TRAIL-mediated apoptosis through suppression of NF-κB and downregulation of anti-apoptotic factors Bcl-2 and Bcl-xl | A549 and HCC15 human NSCLC cells | [92] |
| Resveratrol | Suppressed M2-like polarization of tumor associated macrophages and inhibited STAT3 activity | A549 and H1299 human NSCLC cells. Lewis lung cancer (LLC) s.c. xenograft model (Intraperitoneal (i.p.) administration) a | [93] |
| Resveratrol | Resveratrol enhancing the effects of cisplatin on inhibition of cancer cell proliferation, induction of cell apoptosis, depolarization of mitochondrial membrane potential, release of cytochrome c, upregulation of Bax, downregulation of Bcl-2 | H520 and H838 human NSCLC cell lines | [94] |
| Resveratrol | Upregulation of p21 and TRAIL receptor 1 and 2 expression, and downregulation of Bcl2, cyclin D, NF-κB and IKK1 expression | A549 human NSCLC cell line | [95] |
| Resveratrol | Increase in production of hydrogen peroxide (H2O2), activation of Bid, PARP and caspase 8, and downregulation of pEGFR, pAkt, c-FLIP and NF-κB protein expression | H460 human NSCLC cells | [96] |
| Resveratrol | Suppress of tumor cell growth via an apoptosis-independent mechanism involving induction of premature senescence by increasing P53 and p21 expression and ROS production and decreasing EF1A expression | A549 and H460 human NSCLC cell lines | [97] |
| Resveratrol | Enhancing ionizing radiation through increased production of ROS, and induction of DNA double-strand breaks and senescence induction | A549 and H460 human NSCLC cell lines | [98] |
| Resveratrol | Resveratrol overcoming gefitinib resistance by increasing the intracellular gefitinib concentration through inhibition of CYP1A1 and ABCG2 and by inducing cell apoptosis, autophagy, cell cycle arrest and senescence through increase in expression of cleaved caspase-3, LC3B-II, p53 and p21 | PC9/G human NSCLC cells | [99] |
| Resveratrol | Resveratrol-enhanced erlotinib-mediated apoptosis through decreasing survivin expression and induction of PUMA expression | H460, A549, PC-9 and H1975 human NSCLC cell lines | [100] |
| Resveratrol | Resveratrol-enhanced etoposide-Induced cytotoxicity through down-regulating ERK1/2 and AKT-mediated X-ray repair cross-complement group 1 (XRCC1) protein expression | H1703 and H1975 human NSCLC cell lines | [101] |
| Resveratrol | Modulation of the expression of specific miRNAs with potential target genes involved in apoptosis, cell cycle regulation, cell proliferation, and differentiation | A549 human NSCLC cell line | [102] |
| Resveratrol | Upregulation of miR-622 leading to suppression of K-Ras mRNA translation without affecting its accumulation levels | 16HBE-T human bronchial epithelial cell line and H460 human NSCLC cell line. | [103] |
| Resveratrol | Inhibition of lung cancer progression by downregulating miR-500h, which subsequently leads to downregulation of PP2A expression, inactivation of AKT/NF-kB and downregulation of FOXC2 | CL1-5, A549, H322, H520 and H1435 human NSCLC cell lines | [104] |
| Resveratrol | Induction of G2/M cell cycle arrest through downregulation of checkpoint protein cyclin B1. Induction of apoptosis by increasing p53 and p21 expression and the release of cytochrome c in the cytosol | A549 human NSCLC cell line | [105] |
| Resveratrol | Inhibition of A549 cell proliferation through the reduction of the ratio of Bcl-2/Bax through activation of p53, thus activating the caspase-3- dependent apoptotic cascade and induces apoptosis | A549 human NSCLC cells. | [106] |
| Resveratrol | Attenuated A549 cell-induced platelet secretion and angiogenic responses in vitro and suppressed A549 lung cancer metastasis and angiogenesis in vivo through inhibition of platelets-mediated angiogenic responses induced by [106] adenosine diphosphate (ADP) through increased cGMP generation and cGMP-mediated vasodilator-stimulated phosphoprotein phosphorylation along with reduced intracellular Ca2+ mobilization | A549 human NSCLC cells, and A549 subcutaneous (s.c.) xenograft tumors in nude mice (i.p. administration) a | [107] |
| Resveratrol | Anticancer effects attributable to inhibition of STAT-3 Signaling | A549 human NSCLC cells | [108] |
| Resveratrol | Inhibition of anchorage-dependent and -independent growth of NSCLC cells by decreasing EGFR and downstream kinases Akt and ERK1/2 activation, and subsequent impairment of hexokinase II (HK2)-mediated glycolysis by inhibiting HK2 expression mediated by the Akt signaling pathway | H460, H1650 and HCC827 human NSCLC cells. H460 s.c. xenograft model (i.p. administration) a | [109] |
| Resveratrol | Synergism between Resveratrol and Metformin attributable to the suppression of DNA damage based on the downregulation of γH2AX/p53/p-chk2, inhibition of cell cycle progression via modulation of cyclin E/cdk2, Rb, p21 cyclin B1/cdk1 and plk1/cdc25c and enhancement of DNA repair indicated by the upregulation of p53R2 | A549 human NSCLC cells | [110] |
| Resveratrol | Inhibition of the release of IL-6 and VEGF for co-cultured A549 lung cancer cells and adipose-derived mesenchymal stem cells | Co-cultured A549 human lung cancer cells and adipose-derived mesenchymal stem cells | [111] |
| Resveratrol | Induction of cell cycle arrest in the G0/G1 phase by downregulating the expression levels of cyclin D1, cyclin-dependent kinase (CDK)4 and CDK6, and upregulating the expression levels of the CDK inhibitors, p21 and p27 | A549 human NSCLC cell line | [112] |
| Resveratrol | miR-200c sensitized tumor cell response to resveratrol by targeting reversion-inducing cysteine-rich protein with Kazal motifs (RECK), followed by activation of the JNK signaling pathway and ER stress | H460 human NSCLC cell line | [113] |
| Resveratrol | Suppression of invasion and metastasis through reversal of TGF-β1-induced EMT through increasing E-cadherin expression and repressing Fibronectin, Vimentin, Snail1 and Slug expression | A549 human NSCLC cell line | [114] |
| Resveratrol | Enhancing the radiosensitivity through NF-κB inhibition and S-phase arrest | NCI-H838 human NSCLC cell line | [115] |
| Resveratrol | Anti-metastasis effect attributable to the inhibition of expression of MMP-9/MMP-2 by suppression of HO-1, which in part results from the suppression of NF-κB-dependent signaling pathway | A549 human NSCLC cell line | [116] |
| Resveratrol | Inhibition of the proliferation of SPC-A-1/CDDP cells, induction of apoptosis and cell cycle arrest at phase between G0-G1 and S phase or at the G2/M phase by downregulating survivin | Human multidrug-resistant SPC-A-1/CDDP cells | [117] |
| Resveratrol | Anti-proliferative effect associated with inhibition of the phosphorylation of the retinoblastoma protein (pRB) and induction of cyclin-dependent kinase (Cdk) inhibitor p21WAF1/CIP. Induction of apoptosis associated with activation of caspase-3, shift in Bax/Bcl-xL ratio and inhibition of transcriptional activity of NF-κB | A549 human NSCLC cell line | [118] |
| Resveratrol loaded gelatin nanoparticles | Induction of cell death through inhibition of cell cycle progression and constitutive NF-κB activation by altering the expression of p53, p21, caspase-3, Bax, Bcl-2 and NF-κB | H460 human NSCLC cell line | [119] |
| SS28 (a synthetic Resveratrol analog) | Inhibition of Tubulin polymerization during cell division to cause cell cycle arrest at G2/M phase of the cell cycle | A549 human NSCLC cell line | [120] |
| 4,4’-Dihydroxy-trans-stilbene (DHS) (a resveratrol analog) | Inhibition on anchorage-dependent or -independent cell growth, leading to impairment of the cell cycle progression with reduction of cell numbers arresting at the G1 phase | Murine Lewis lung carcinoma (LLC) cell line | [121] |
| Curcumin and resveratrol alone or in combination | Improvement of lung histoarchitecture and ultrahistoarchitecture during benzopyrene-induced lung carcinogenesis in mice | 3,4-Benzopyrene-induced mouse lung carcinoma model (Oral (p.o.) administration) a | [122] |
| Resveratrol and dibenzoylmethane | Induction of apoptosis through activation of caspase-9 and caspase-3 and subsequent cleavage of PARP | A549 and CH27 human NSCLC cell lines | [123] |
| Heyneanol A (HA) (A tetramer of resveratrol) | Induction of caspase-mediated cancer cell apoptosis by inducing cleavage of caspase-9 and caspase-3 and suppression of basic fibroblast growth factor (bFGF)-induced tumor angiogenesis. | In vivo Lewis lung tumor model (i.p. administration) a | [124] |
| EGCG, ECG, EGC and EC | Induction of apoptosis through a p53-dependent pathway. | A549 human NSCLC cell line | [125] |
| EGCG | Induction of G2-M arrest. Incorporation into cytosol and nuclei | PC-9 human NSCLC cell line | [126] |
| EGCG | EGCG inhibited cell growth through decreasing the phosphorylation of Akt and ERK irrespective of EGFR-, ALK- or ROS1-dependency. The antiangiogenic effect of EGCG might be attributable to the inhibition of HIF-1α | PC-9, RPC-9, H1975, H2228 and HCC78 human NSCLC cell lines (EGFR- or fusion gene-driven tumor cells) and xenograft models (p.o. administration) a | [127] |
| EGCG | Downregulation of gene expression of NF-κB inducing kinase (NIK), death-associated protein kinase 1 (DAPK 1), RhoB and tyrosine-protein kinase (SKY), and upregulation of the retinoic acid receptor alpha1 gene expression | PC-9 human NSCLC cell line | [128] |
| EGCG | Induction of miRNA profile changes, which modulate several regulatory networks associated to AKT, NF-κB, MAP kinases, and cell cycle | 4-(Methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) induced mouse lung cancer (p.o. administration) a | [129] |
| EGCG | Co-treatment with celecoxib synergistically inducing apoptosis by upregulation of growth arrest and DNA damage-inducible 153 (GADD153) through the ERK signaling pathway | A549, ChaGo K-1 and PC-9 human NSCLC cell lines | [130] |
| EGCG | Co-treatment of EGCG with cisplatin resulting in proliferation inhibition, cell cycle arrest in G1 phase, increase in apoptosis along with inhibition of DNA methyltransferase (DNMT) activity and histone deacetylase (HDAC) activity, reversal of hypermethylated status and downregulated expression of GAS1, TIMP4, ICAM1 and WISP2 genes | Cisplatin-resistant A549 (A549/DDP) human NSCLC cell line and A549/DDP xenograft tumor model (i.p. administration) | [131] |
| EGCG and Luteolin | Enhanced antitumor effect attributable to ATM (ataxia telangiectasia mutated) kinase-dependent Ser15 phosphorylation of p53 as a consequence of DNA double strand break | H292, A549 and H460 human NSCLC cell lines expressing wild-type p53; A549 xenograft tumor model (p.o. administration) a | [132] |
| Tea polyphenols | Upregulation of p53 expression and downregulation of Bcl-2 expression with no influence on H-Ras and c-Myc expressions | NCI-H460 human NSCLC cell line | [133] |
| Black tea polyphenols | Inhibition of Cox-1 and induction of caspase-3 and caspase-7 expression | 3,4-Benzopyrene induced mouse lung tumor model | [134] |
| Black tea polyphenols | Suppressing cell proliferation and inducing apoptosis | 3,4-Benzopyrene induced mouse lung tumor model | [135] |
| Green tea polyphenols | Preventive effect against lung cancer by upregulating p53 and downregulating Bcl-2 | 3,4-Benzopyrene induced rat lung tumor model | [136] |
| Tea polyphenols | Increase in p53 expression and decrease in Bcl-2 expression | 3,4-Benzopyrene-induced rat lung carcinoma model | [137] |
| Tea polyphenols | Inhibition of Akt and cyclooxygenase-2 expression, and inactivation of nuclear factor-kappa B via blocking phosphorylation and subsequent degradation of IkappaB alpha | Diethylnitrosoamine- induced mouse lung tumor model (p.o. administration) a | [138] |
| Green tea polyphenols | TAM67-mediated changes in gene expression involving the downregulation of activator protein-1 (AP-1) | H1299 human NSCLC cell line and SPON 10 mouse lung tumor cell line | [139] |
| Green tea extracts | Modulation of the expression of 14 proteins involved in calcium-binding, cytoskeleton and motility, metabolism, detoxification, or gene regulation | A549 human NSCLC cell line | [140] |
| Green tea polyphenols | Synergistic antitumor effect with atorvastatin attributable to increased apoptosis, reduced Mcl-1 level and increased cleaved caspase-3 and cleaved poly(ADP)-ribose polymerase (PARP) | H1299 and H460 human NSCLC cell lines. 4-(Methylnitrosaminao)-1-(3-pyridyl)-1-butanone induced mouse lung tumor model (p.o. administration) a | [141] |
| Green tea extract | Induction of protective autophagy | A549 human NSCLC cell line | [142] |
| Thymoquinone (TQ) | Upregulation of Bax and downregulation of Bcl-2 expression and increase in the Bax/Bcl-2 ratio. Decrease in the expression of cyclin D, NF-κB and IKK1 and increase in the expression of p21 and TRAIL receptor 1 and 2 expression | A549 human NSCLC cell line | [95] |
| Curcumin | Induction of apoptosis through p53-independent pathway by downregulation of Bcl-2 and Bcl-xL expression | A549 and H1299 human NSCLC cell lines | [143] |
| Curcumin | Induction of cell cycle arrest at the G1/S phase and apoptosis through up-regulation of GADD45 and GADD153 | PC-9 human NSCLC cell line | [144] |
| Curcumin | Induction of cell cycle arrest at the G2/M phase and apoptosis through upregulation of Bax and Bad expression, downregulation of Bcl-2, Bcl-xL and XIAP expression, increase in ROS, intracellular Ca2+ and endoplasmic reticulum stress, activation of GRP78 and GADD153 proteins and FAS/caspase-8 pathway | H460 human NSCLC cell line | [145] |
| Curcumin | Induction of apoptosis through a mitochondria-dependent mechanism as manifested by the decrease in the mitochondrial membrane potential, releasing cytochrome c from mitochondria to cytoplasm | A549 human NSCLC cell line | [146] |
| Curcumin | Induction of apoptosis via the ROS-mediated mitochondrial pathway accompanied by increased Bax expression and decreased expression of Bcl-2 and Bcl-xL | H446 human SCLC cell line | [147] |
| Curcumin | Inhibition of tumor cell proliferation and induction of apoptosis through upregulation of miR-192-5p and suppression of the PI3K/Akt signaling pathway | A549 human NSCLC cell line | [148] |
| Curcumin | Enhancing autophagy and apoptosis through inaction of PI3K/mTOR signaling pathway | A549 and H1299 human NSCLC cell lines | [149] |
| Curcumin | Induction of autophagy leading to suppression of tumor cell proliferation. | A549 human NSCLC cell line | [150] |
| Curcumin | Inhibition of tumor cell invasion and metastasis through attenuating GLUT1/MT1-MMP/MMP2 pathway | A549 human NSCLC cell line and xenograft tumor model (i.p. administration) a | [151] |
| Curcumin | Inhibition of tumor cell metastasis through inhibition of the adiponectin/NF-κB/MMPs signaling pathway | A549 human NSCLC cell line and xenograft tumor model (i.p. administration) a | [152] |
| A501 (Curcumin analogue) | Induction of cell cycle arrest at the G2/M phase and apoptosis through decreasing the expression of cyclinB1, cdc-2, Bcl-2, while increasing the expression of p53, cleaved caspase-3 and Bax | A549 and H460 human NSCLC cell lines | [153] |
| Curcumin and gefitinib | Potentiating the antitumor effect of gefitinib in gefitinib-resistant tumor cells through induction of endogenous EGFR protein degradation and downregulation of EGFR and AKT protein expression. Reduction of the gefitinib-induced villi damage and apoptosis in mouse intestine through attenuating gefitinib-induced p38 activation | CL1-5, A549 and H1975 human NSCLC cell lines and xenograft models (p.o. administration) a | [154] |
| Curcumin and carboplatin | Synergistic antitumor activity mediated by multiple mechanisms involving suppression of NF-kB via inhibition of the Akt/IKKα pathway, enhancement of ERK1/2 activity and downregulation of MMP-2 and MMP-9 expression | A549 human NSCLC cell line | [155] |
| Quercetin | Induction of cell cycle arrest at the G2/M phase and apoptosis through increased expression of cyclin B1 and phosph-cdc2 (T161), survivin, total p53, phosphor-p53 (S15) and p21 proteins | A549 and H1299 human NSCLC cell lines | [156] |
| Quercetin | Induction of apoptosis through activation of MEK-ERK pathway, inactivation of Akt and alteration in the expression of Bcl-2 family | A549 human NSCLC cell line | [157] |
| Quercetin | Proapoptosis activity through multiple mechanisms including upregulated the expression of genes associated with the death pathway, the JNK pathway, the IL1 receptor pathway, the caspase cascade, the NF-κB pathway and cell cycle arrest, and downregulated the expression of genes related to cell proliferation | H460 human NSCLC cell line | [158] |
| Quercetin | Anti-invasion activity through inhibition of monocarboxylate transporter 1 | A110L human lung cancer cell line | [159] |
| Quercetin | Targeting aurora B kinase | A549 human NSCLC cell line and xenograft model (i.p. administration) a | [160] |
| Quercetin | Trigger Bcl-2/Bax-mediated apoptosis, necrosis and mitotic catastrophe. Inhibition of cell migration through disassembly of microfilaments, microtubules and vimentin filaments and inhibition of vimentin and N-cadherin expression | A549 human NSCLC cell line | [161] |
| Quercetin | Suppression of in vitro cell migration/invasion and in vivo bone metastasis through inhibition of Snail-dependent Akt activation and Snail-independent ADAM9 expression pathways | A549 and HCC827 human NSCLC cell lines and A549 xenograft model (i.p. administration) a | [162] |
| Quercetin and chrysin | Suppressed the secretion of cytokines, IL-1β, IL-6, TNF-α and IL-10, and decreased the phosphorylation of IKKβ and IκB, the nuclear level of p65 (NF-κB) as well as the expression of MMP-9 in A549cells exposed to nickel | A549 human NSCLC cell line | [163] |
| Quercetin and trichostatin A | Enhancing the antitumor activity of trichostatin A through upregulation of p53 expression | A549 and H1299 human NSCLC cell lines and A549 xenograft model (i.p. administration) a | [164] |
| Quercetin and gemcitabine | Promoting apoptosis and sensitizing tumor response to gemcitabine via inhibition of HSP70 expression | A549 and H4650 human NSCLC cell lines | [165] |
| Caffeic acid phenethyl ester (CAPE) | Upregulation of Bax, p21 and TRAIL receptor 1 and 2 expression, and downregulation of cyclin D expression | A549 human NSCLC cell line | [95] |
| Pterostilbene | Exhibition of p53-dependent chemotherapeutic effects through the ATM/CHK/p53 tumor suppressive pathway leading to cell senescence | Precancerous human bronchial epithelial cell lines, HBECR and HBECR/p53i, with normal p53 and suppressed p53 expression, respectively | [166] |
| Bakuchiol | Increase in reactive oxygen species production, decrease in mitochondrial membrane potential (∆Ψm), cell cycle arrest at S phase, caspase 9/3 activation, p53 and Bax up-regulation, and Bcl-2 downregulation | A549 human NSCLC cell line | [167] |
| Chlorogenic acid (CGA) | Decrease in hypoxia-induced HIF-1α protein level and suppression of the transcriptional activity of HIF-1α under hypoxic conditions, leading to antiangiogenic activity through inhibition of HIF-1α/AKT pathway and decrease in VEGF expression | A549 human NSCLC cell line | [168] |
| Fisetin | Inhibition of cell growth through concomitant suppression of PI3K/Akt and mTOR signaling | A549 and H1792 human NSCLC cell lines | [169] |
| Fisetin | Enhancing cisplatin cytotoxicity in cisplatin-resistant cells by modulation of the MAPK/survivin/caspase pathway | A549 human NSCLC cell line | [170] |
| Fisetin | Synergistic interaction between paclitaxel and fisetin due to the induction of mitotic catastrophe probably through the promotion of multipolar spindle formation. Mitotic catastrophe induced protective autophagy against apoptosis, which then switched to the autophagic cell death | A549 human NSCLC cell line | [171] |
| Liquiritin, isoliquiritin and isoliquirigenin | Induction of apoptosis and cell cycle at the G2/M phase by increasing p53, p21 and BAX expression and decreasing PCNA, MDM2, p-GSK-3β, p-Akt, p-c-Raf, p-PTEN, caspase-3, pro-caspase-8, pro-caspase-9, PARP and Bcl-2 expression | A549 human NSCLC cell line | [172] |
| Eucalyptus globulus Labill | Cell cycle arrest in the G0/G1 phase. Increase in the expression of p53, p21 and cyclin D1 proteins | NCI-H460 human NSCLC cell line | [173] |
| Polyphenols isolated from | Suppressed cell migration by targeting MMP-9. Induced cell apoptosis through intrinsic apoptosis pathways, accompanied by increasing the expression of Bax and caspase-3 | A549 human NSCLC cell line | [174] |
| Rosemary extract | Reduced total and phosphorylated/activated Akt, mTOR and p70S6K levels | A549 human NSCLC cell line | [175] |
| Salvianolic acid A | Salvianolic acid A enhanced sensitivity to cisplatin through suppression of the c-met/AKT/mTOR signaling pathway | A549 human lung cancer cisplatin resistance cell line (A549/DDP) | [176] |
| Red wine | Inhibition of basal and EGF-stimulated Akt and Erk signals and enhancement of total and phosphorylated levels of p53, leading to inhibition of A549 cell proliferation and clonogenic survival | A549 human NSCLC cell line | [15] |
| Magnolol and polyphenol mixture (PM) derived from Magnolia officinalis | Induction of cell apoptosis by arresting the cell cycle in the G0/G1 phase while simultaneously activating various pro-apoptotic signals, including TRAIL-R2 (DR5), Bax, caspase 3, cleaved caspase 3, and cleaved PARP | A549 and H1299 human NSCLC cell lines | [177] |
| Pomegranate juice | Attenuated the formation of lung nodules and reduced PHH3 (marker of mitotic activity) and HIF-1α expression | Two-month-old adult male AJ mice (p.o. administration) a | [178] |
| Polyphenolic compounds of Achyranthes aspera (PCA) extract | Downregulation of the expression of pro-inflammatory cytokines IL-1β, IL-6 and TNF-α, TFs, NF-κB and Stat3, and upregulation of the expression of pro-apoptotic proteins Bax and p53. Increase in the activities and expression of antioxidant enzymes GST, GR, CAT, and SOD. Decrease in the activity and expression of LDH enzymes | Urethane-induced mouse lung cancer in vivo model (p.o. administration) a | [179] |
| Bilberry extract (BE), genistein (GEN), delphinidin-3-O-glucoside (D3G), delphinidin (DE), gallic acid (GA), and phloroglucinol aldehyde (PGA) | Antagonistic interactions of BE, D3G, DEL and GEN with erlotinib. No effect of GA on erlotinib, while synergistic interaction of PGA with erlotinib. Mechanism unknown | A431 human epithelial cell line | [180] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, Q.; Pan, H.; Li, J. Molecular Insights into Potential Contributions of Natural Polyphenols to Lung Cancer Treatment. Cancers 2019, 11, 1565. https://doi.org/10.3390/cancers11101565
Zhou Q, Pan H, Li J. Molecular Insights into Potential Contributions of Natural Polyphenols to Lung Cancer Treatment. Cancers. 2019; 11(10):1565. https://doi.org/10.3390/cancers11101565
Chicago/Turabian StyleZhou, Qingyu, Hua Pan, and Jing Li. 2019. "Molecular Insights into Potential Contributions of Natural Polyphenols to Lung Cancer Treatment" Cancers 11, no. 10: 1565. https://doi.org/10.3390/cancers11101565
APA StyleZhou, Q., Pan, H., & Li, J. (2019). Molecular Insights into Potential Contributions of Natural Polyphenols to Lung Cancer Treatment. Cancers, 11(10), 1565. https://doi.org/10.3390/cancers11101565