PD-1-Associated Gene Expression Signature of Neoadjuvant Trastuzumab-Treated Tumors Correlates with Patient Survival in HER2-Positive Breast Cancer

 ,
,
Abstract
1. Introduction
2. Results
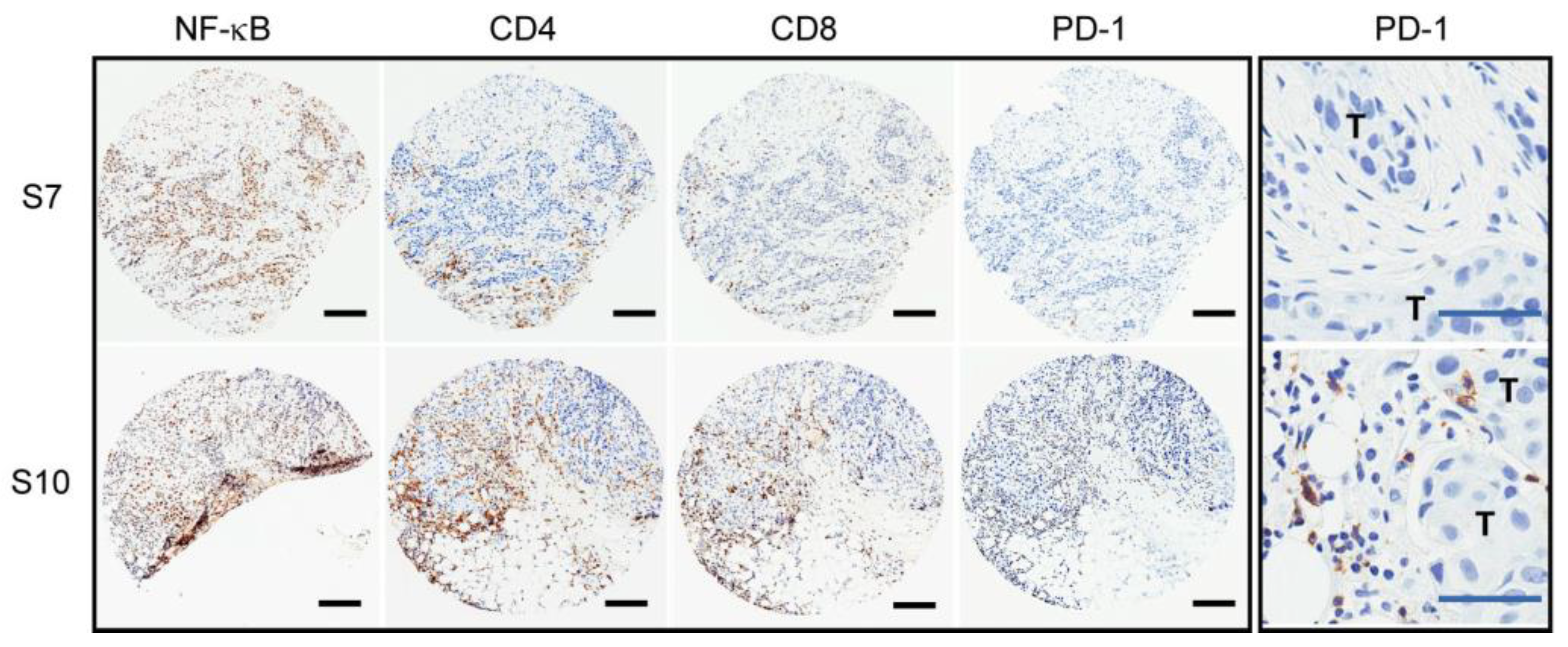
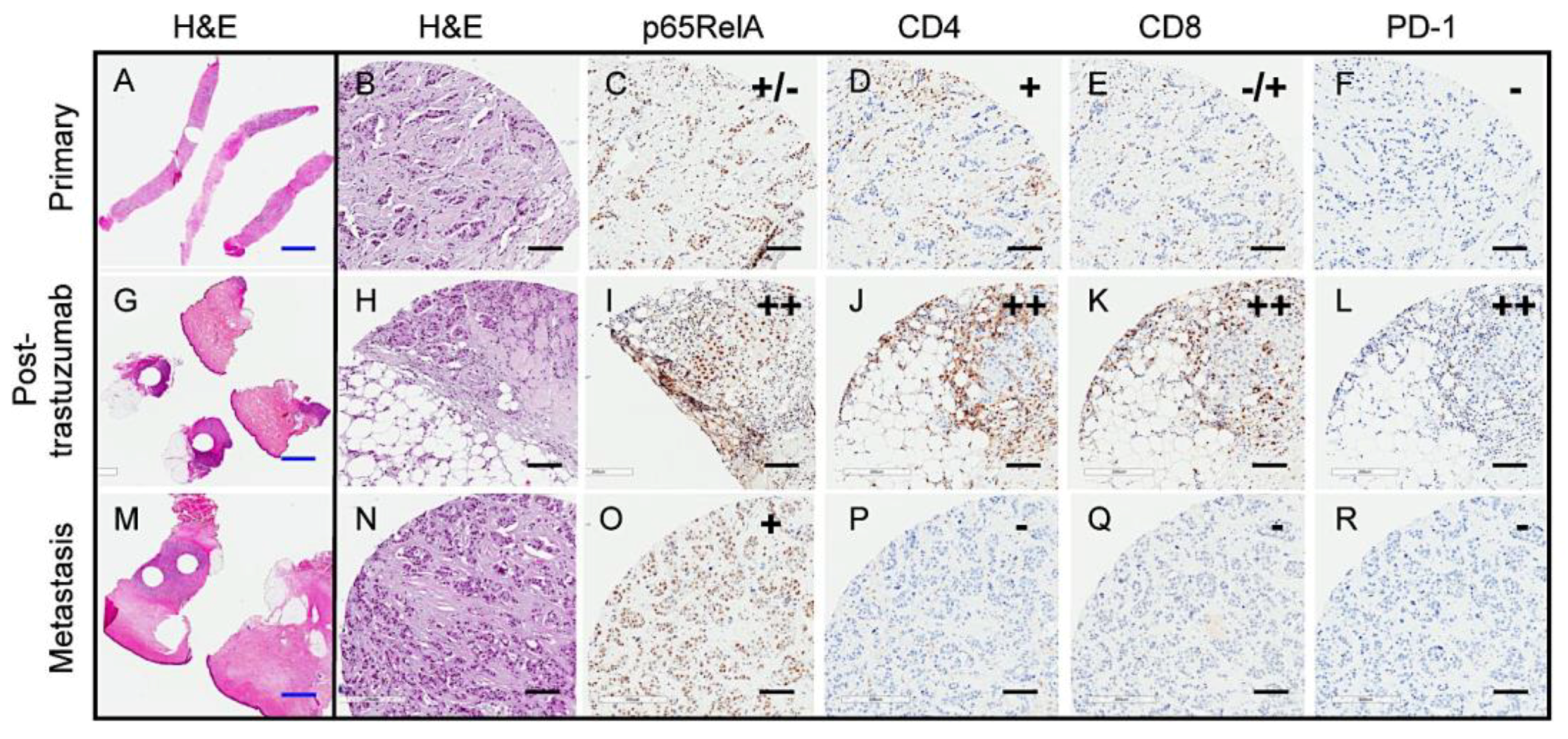
2.1. Variable PD-1 Expression in HER2-Positive Tumors from Patients Treated with Neoadjuvant Trastuzumab
2.2. No Putative Tumor Driver Mutations Associated with the PD-1 Positive Tumor Immune Environment
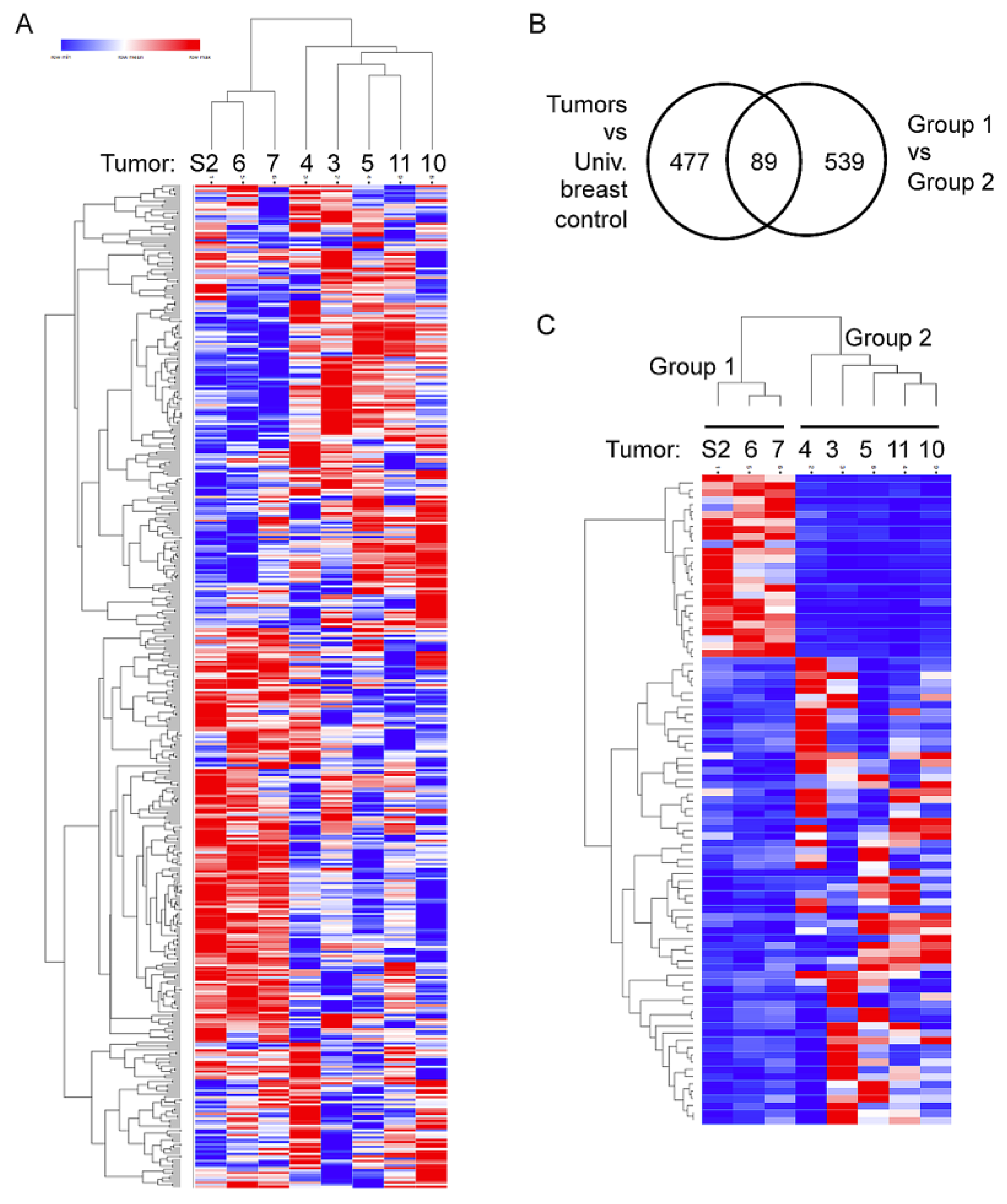
2.3. Two Distinct Tumor Groups Identified by Gene Expression Analysis
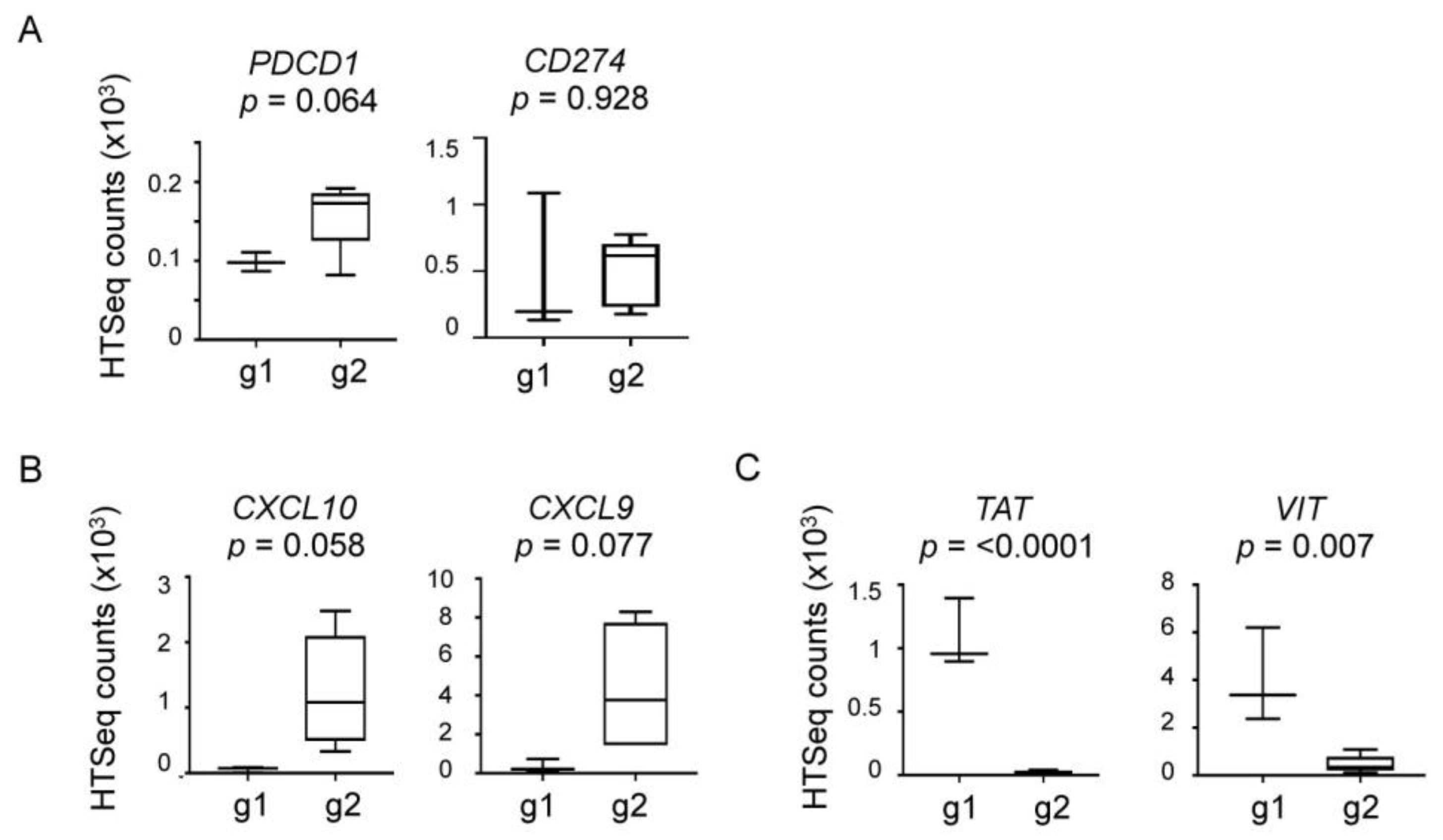
2.4. Elevated Expression of Immune and Inflammatory Genes in Group 2
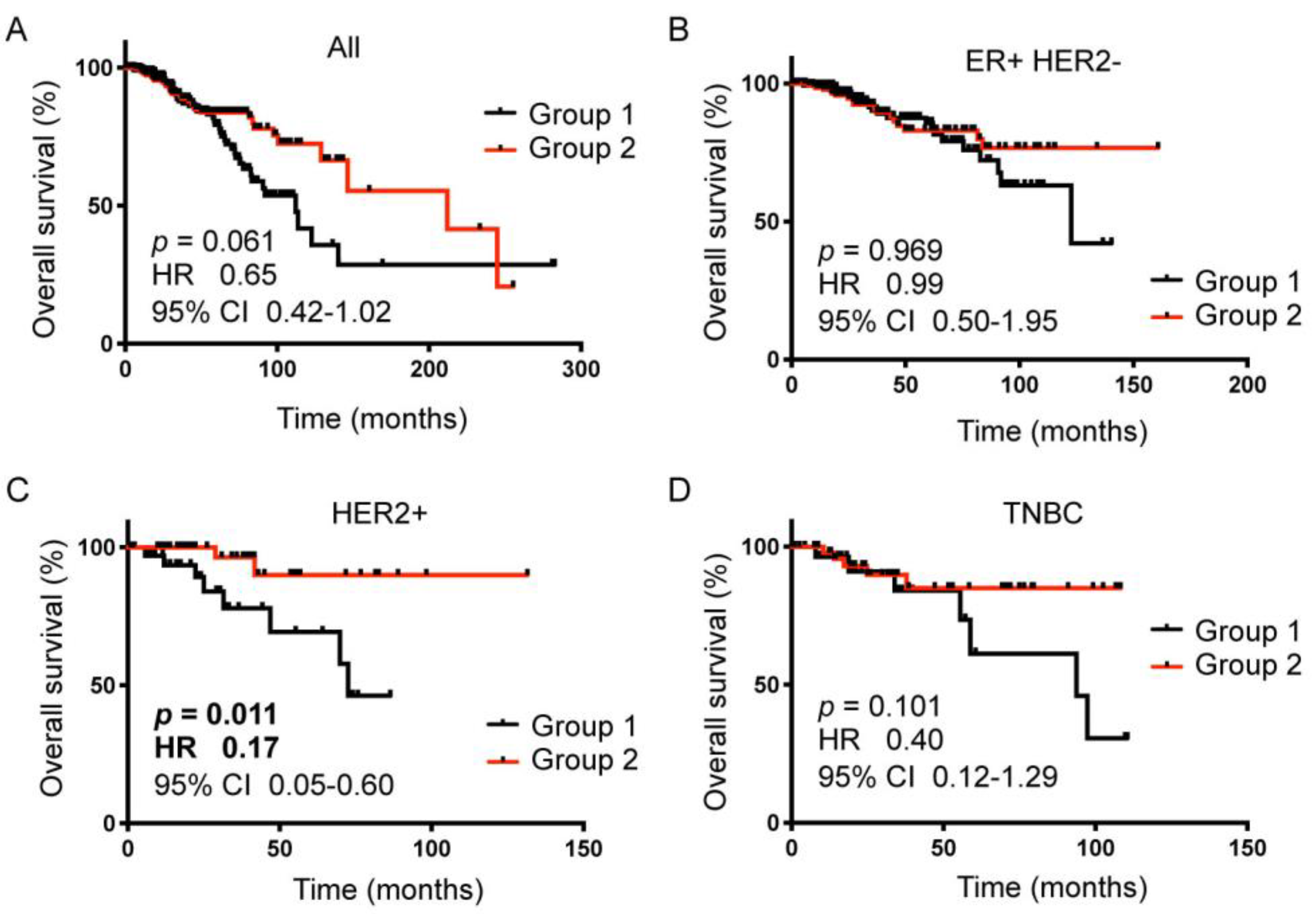
2.5. The 89-gene Signature Correlated with HER2+ Breast Cancer Patient Survival
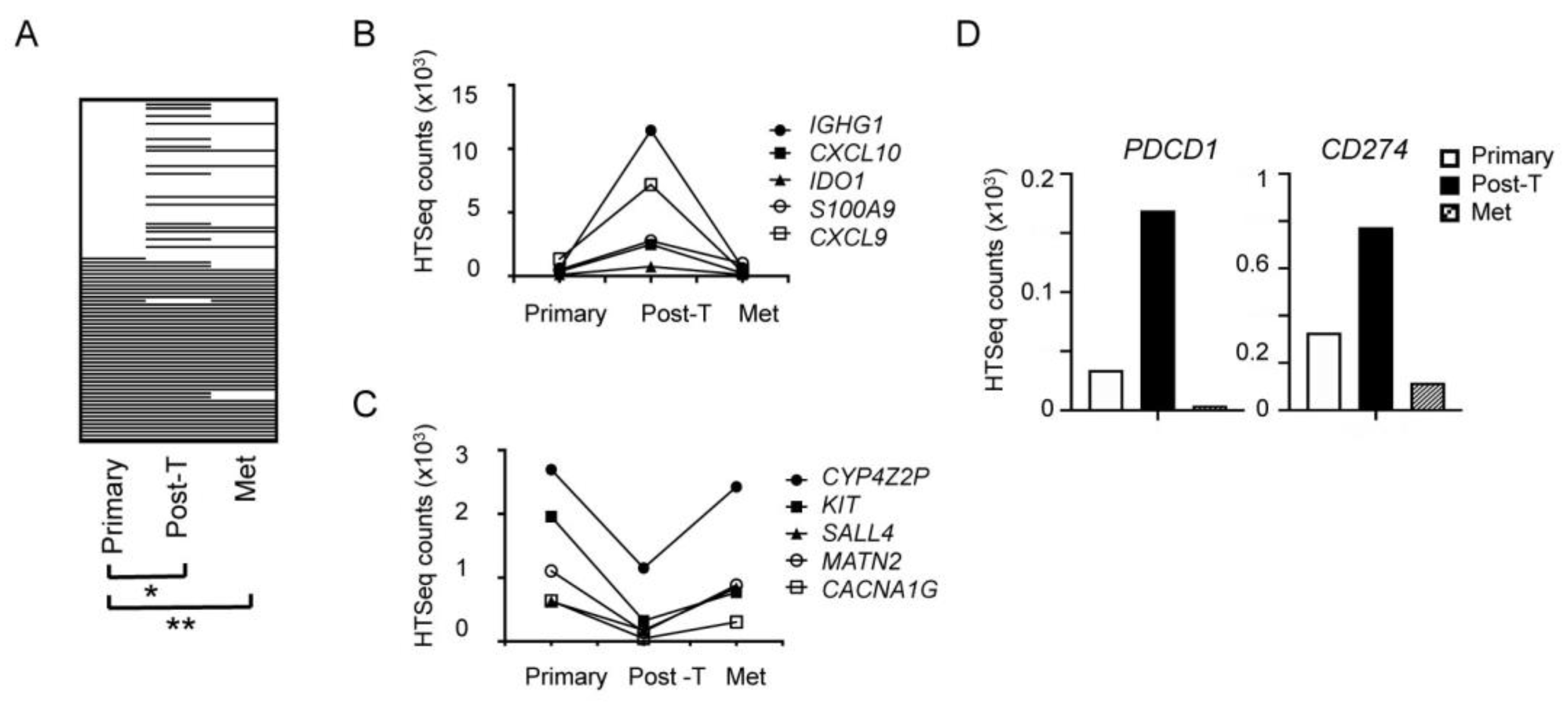
2.6. Transient Trastuzumab-Elicited Immune Activation Displayed in Longitudinal Samples from a Single Patient
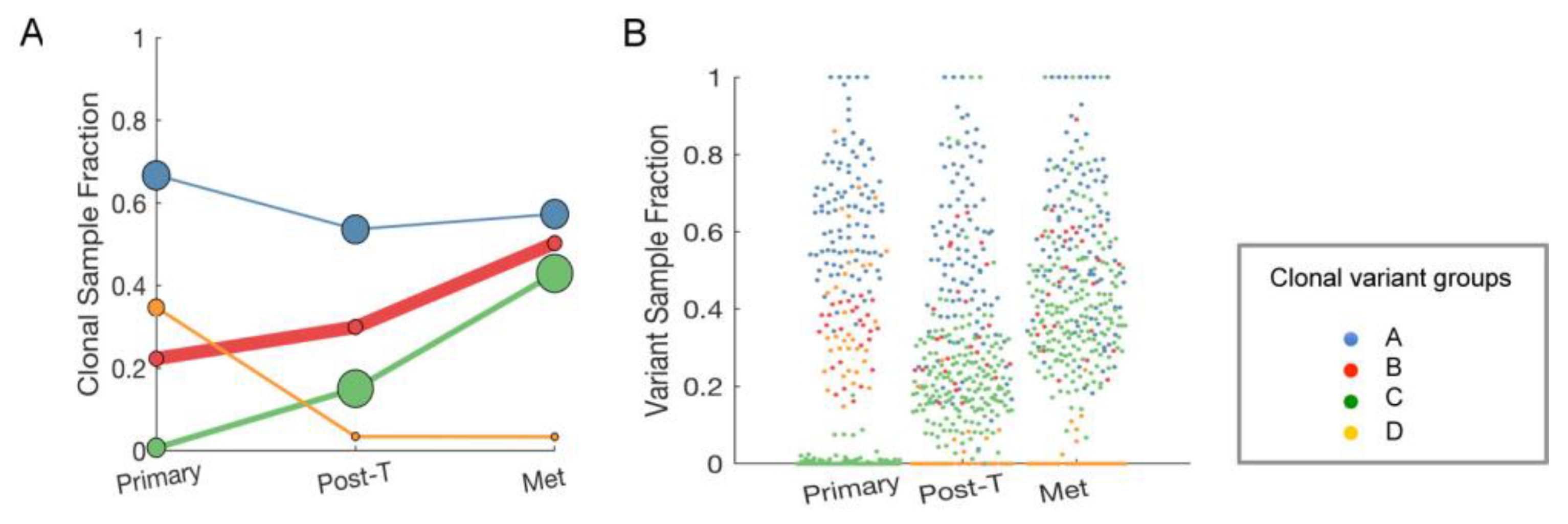
2.7. Clonal Expansion of Single Nucleotide Variants (SNVs) in the Longitudinal Samples from a Single Patient
3. Discussion
4. Materials and Methods
4.1. Patient Samples and Tumor Tissue Microarray (TMA)
4.2. Immunohistochemistry (IHC)
4.3. DNA and RNA Sequencing of Patient Samples
4.4. Sequencing Data Analysis
4.5. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Slamon, D.J.; Clark, G.M.; Wong, S.G.; Levin, W.J.; Ullrich, A.; McGuire, W.L. Human breast cancer: Correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science 1987, 235, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Slamon, D.J.; Leyland-Jones, B.; Shak, S.; Fuchs, H.; Paton, V.; Bajamonde, A.; Fleming, T.; Eiermann, W.; Wolter, J.; Pegram, M.; et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N. Engl. J. Med. 2001, 344, 783–792. [Google Scholar] [CrossRef] [PubMed]
- Piccart-Gebhart, M.J.; Procter, M.; Leyland-Jones, B.; Goldhirsch, A.; Untch, M.; Smith, I.; Gianni, L.; Baselga, J.; Bell, R.; Jackisch, C.; et al. Trastuzumab after adjuvant chemotherapy in HER2-Positive breast cancer. N. Engl. J. Med. 2005, 353, 1659–1672. [Google Scholar] [CrossRef] [PubMed]
- Perez, E.A.; Romond, E.H.; Suman, V.J.; Jeong, J.H.; Sledge, G.; Geyer, C.E., Jr.; Martino, S.; Rastogi, P.; Gralow, J.; Swain, S.M.; et al. Trastuzumab plus adjuvant chemotherapy for human epidermal growth factor receptor 2-Positive breast cancer: Planned joint analysis of overall survival from NSABP B-31 and NCCTG N9831. J. Clin. Oncol. 2014, 32, 3744–3752. [Google Scholar] [CrossRef] [PubMed]
- Prat, A.; Carey, L.A.; Adamo, B.; Vidal, M.; Tabernero, J.; Cortes, J.; Parker, J.S.; Perou, C.M.; Baselga, J. Molecular features and survival outcomes of the intrinsic subtypes within HER2-positive breast cancer. J. Natl. Cancer Inst. 2014, 106, dju152. [Google Scholar] [CrossRef] [PubMed]
- Carey, L.A.; Berry, D.A.; Cirrincione, C.T.; Barry, W.T.; Pitcher, B.N.; Harris, L.N.; Ollila, D.W.; Krop, I.E.; Henry, N.L.; Weckstein, D.J.; et al. Molecular Heterogeneity and Response to Neoadjuvant Human Epidermal Growth Factor Receptor 2 Targeting in CALGB 40601, a Randomized Phase III Trial of Paclitaxel Plus Trastuzumab With or Without Lapatinib. J. Clin. Oncol. 2016, 34, 542–549. [Google Scholar] [CrossRef]
- Vegran, F.; Boidot, R.; Coudert, B.; Fumoleau, P.; Arnould, L.; Garnier, J.; Causeret, S.; Fraise, J.; Dembele, D.; Lizard-Nacol, S. Gene expression profile and response to Trastuzumab-Docetaxel-Based treatment in breast carcinoma. Br. J. Cancer 2009, 101, 1357–1364. [Google Scholar] [CrossRef]
- Guarneri, V.; Dieci, M.V.; Frassoldati, A.; Maiorana, A.; Ficarra, G.; Bettelli, S.; Tagliafico, E.; Bicciato, S.; Generali, D.G.; Cagossi, K.; et al. Prospective Biomarker Analysis of the Randomized CHER-LOB Study Evaluating the Dual Anti-HER2 Treatment With Trastuzumab and Lapatinib Plus Chemotherapy as Neoadjuvant Therapy for HER2-Positive Breast Cancer. Oncologist 2015, 20, 1001–1010. [Google Scholar] [CrossRef]
- Lesurf, R.; Griffith, O.L.; Griffith, M.; Hundal, J.; Trani, L.; Watson, M.A.; Aft, R.; Ellis, M.J.; Ota, D.; Suman, V.J.; et al. Genomic characterization of HER2-Positive breast cancer and response to neoadjuvant trastuzumab and chemotherapy-results from the ACOSOG Z1041 (Alliance) trial. Ann. Oncol. 2017, 28, 1070–1077. [Google Scholar] [CrossRef]
- Tanioka, M.; Fan, C.; Parker, J.S.; Hoadley, K.A.; Hu, Z.; Li, Y.; Hyslop, T.M.; Pitcher, B.N.; Soloway, M.G.; Spears, P.A.; et al. Integrated Analysis of RNA and DNA from the Phase III Trial CALGB 40601 Identifies Predictors of Response to Trastuzumab-Based Neoadjuvant Chemotherapy in HER2-Positive Breast Cancer. Clin. Cancer Res. 2018, 24, 5292–5304. [Google Scholar] [CrossRef]
- Caswell-Jin, J.L.; McNamara, K.; Reiter, J.G.; Sun, R.; Hu, Z.; Ma, Z.; Ding, J.; Suarez, C.J.; Tilk, S.; Raghavendra, A.; et al. Clonal replacement and heterogeneity in breast tumors treated with neoadjuvant HER2-targeted therapy. Nat. Commun. 2019, 10, 657. [Google Scholar] [CrossRef] [PubMed]
- Bianchini, G.; Gianni, L. The immune system and response to HER2-Targeted treatment in breast cancer. Lancet Oncol. 2014, 15, e58–e68. [Google Scholar] [CrossRef]
- Loi, S.; Michiels, S.; Salgado, R.; Sirtaine, N.; Jose, V.; Fumagalli, D.; Kellokumpu-Lehtinen, P.L.; Bono, P.; Kataja, V.; Desmedt, C.; et al. Tumor infiltrating lymphocytes are prognostic in triple negative breast cancer and predictive for trastuzumab benefit in early breast cancer: Results from the FinHER trial. Ann. Oncol. 2014, 25, 1544–1550. [Google Scholar] [CrossRef]
- Kochi, M.; Iwamoto, T.; Niikura, N.; Bianchini, G.; Masuda, S.; Mizoo, T.; Nogami, T.; Shien, T.; Motoki, T.; Taira, N.; et al. Tumour-Infiltrating lymphocytes (TILs)-Related genomic signature predicts chemotherapy response in breast cancer. Breast Cancer Res. Treat. 2018, 167, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Ceeraz, S.; Nowak, E.C.; Noelle, R.J. B7 family checkpoint regulators in immune regulation and disease. Trends Immunol. 2013, 34, 556–563. [Google Scholar] [CrossRef]
- Francisco, L.M.; Sage, P.T.; Sharpe, A.H. The PD-1 pathway in tolerance and autoimmunity. Immunol. Rev. 2010, 236, 219–242. [Google Scholar] [CrossRef]
- Thommen, D.S.; Schumacher, T.N. T Cell Dysfunction in Cancer. Cancer Cell 2018, 33, 547–562. [Google Scholar] [CrossRef]
- Sharma, P.; Allison, J.P. The future of immune checkpoint therapy. Science 2015, 348, 56–61. [Google Scholar] [CrossRef]
- Muenst, S.; Soysal, S.D.; Gao, F.; Obermann, E.C.; Oertli, D.; Gillanders, W.E. The presence of programmed death 1 (PD-1)-Positive Tumor-Infiltrating lymphocytes is associated with poor prognosis in human breast cancer. Breast Cancer Res. Treat. 2013, 139, 667–676. [Google Scholar] [CrossRef]
- Sun, S.; Fei, X.; Mao, Y.; Wang, X.; Garfield, D.H.; Huang, O.; Wang, J.; Yuan, F.; Sun, L.; Yu, Q.; et al. PD-1(+) immune cell infiltration inversely correlates with survival of operable breast cancer patients. Cancer Immunol. Immunother. 2014, 63, 395–406. [Google Scholar] [CrossRef]
- Sabatier, R.; Finetti, P.; Mamessier, E.; Adelaide, J.; Chaffanet, M.; Ali, H.R.; Viens, P.; Caldas, C.; Birnbaum, D.; Bertucci, F. Prognostic and predictive value of PDL1 expression in breast cancer. Oncotarget 2015, 6, 5449–5464. [Google Scholar] [CrossRef] [PubMed]
- Bae, S.B.; Cho, H.D.; Oh, M.H.; Lee, J.H.; Jang, S.H.; Hong, S.A.; Cho, J.; Kim, S.Y.; Han, S.W.; Lee, J.E.; et al. Expression of Programmed Death Receptor Ligand 1 with High Tumor-Infiltrating Lymphocytes Is Associated with Better Prognosis in Breast Cancer. J. Breast Cancer 2016, 19, 242–251. [Google Scholar] [CrossRef] [PubMed]
- Varadan, V.; Gilmore, H.; Miskimen, K.L.; Tuck, D.; Parsai, S.; Awadallah, A.; Krop, I.E.; Winer, E.P.; Bossuyt, V.; Somlo, G.; et al. Immune Signatures Following Single Dose Trastuzumab Predict Pathologic Response to PreoperativeTrastuzumab and Chemotherapy in HER2-Positive Early Breast Cancer. Clin. Cancer Res. 2016, 22, 3249–3259. [Google Scholar] [CrossRef] [PubMed]
- Biswas, D.K.; Shi, Q.; Baily, S.; Strickland, I.; Ghosh, S.; Pardee, A.B.; Iglehart, J.D. NF-kappa B activation in human breast cancer specimens and its role in cell proliferation and apoptosis. Proc. Natl. Acad. Sci. USA 2004, 101, 10137–10142. [Google Scholar] [CrossRef] [PubMed]
- Curtis, C.; Shah, S.P.; Chin, S.F.; Turashvili, G.; Rueda, O.M.; Dunning, M.J.; Speed, D.; Lynch, A.G.; Samarajiwa, S.; Yuan, Y.; et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature 2012, 486, 346–352. [Google Scholar] [CrossRef] [PubMed]
- Kandoth, C.; McLellan, M.D.; Vandin, F.; Ye, K.; Niu, B.; Lu, C.; Xie, M.; Zhang, Q.; McMichael, J.F.; Wyczalkowski, M.A.; et al. Mutational landscape and significance across 12 major cancer types. Nature 2013, 502, 333–339. [Google Scholar] [CrossRef] [PubMed]
- da Huang, W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]
- Griffith, J.W.; Sokol, C.L.; Luster, A.D. Chemokines and chemokine receptors: Positioning cells for host defense and immunity. Annu. Rev. Immunol. 2014, 32, 659–702. [Google Scholar] [CrossRef]
- Stephens, P.J.; Tarpey, P.S.; Davies, H.; Van Loo, P.; Greenman, C.; Wedge, D.C.; Nik-Zainal, S.; Martin, S.; Varela, I.; Bignell, G.R.; et al. The landscape of cancer genes and mutational processes in breast cancer. Nature 2012, 486, 400–404. [Google Scholar] [CrossRef]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Carvajal, R.D.; Sosman, J.A.; Atkins, M.B.; et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N. Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef]
- Nanda, R.; Chow, L.Q.; Dees, E.C.; Berger, R.; Gupta, S.; Geva, R.; Pusztai, L.; Pathiraja, K.; Aktan, G.; Cheng, J.D.; et al. Pembrolizumab in Patients With Advanced Triple-Negative Breast Cancer: Phase Ib KEYNOTE-012 Study. J. Clin. Oncol. 2016, 34, 2460–2467. [Google Scholar] [CrossRef] [PubMed]
- Gibson, J. Anti-PD-L1 for metastatic Triple-Negative breast cancer. Lancet Oncol. 2015, 16, e264. [Google Scholar] [CrossRef]
- Loi, S.; Giobbie-Hurder, A.; Gombos, A.; Bachelot, T.; Hui, R.; Curigliano, G.; Campone, M.; Biganzoli, L.; Bonnefoi, H.; Jerusalem, G.; et al. Pembrolizumab plus trastuzumab in Trastuzumab-Resistant, advanced, HER2-Positive breast cancer (PANACEA): A Single-Arm, multicentre, phase 1b-2 trial. Lancet Oncol. 2019, 20, 371–382. [Google Scholar] [CrossRef]
- Iwai, Y.; Ishida, M.; Tanaka, Y.; Okazaki, T.; Honjo, T.; Minato, N. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc. Natl. Acad. Sci. USA 2002, 99, 12293–12297. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Gilligan, B.M.; Yuan, J.; Li, T. Current status and perspectives in translational biomarker research for PD-1/PD-L1 immune checkpoint blockade therapy. J. Hematol. Oncol. 2016, 9, 47. [Google Scholar] [CrossRef] [PubMed]
- Makino, K.; Day, C.P.; Wang, S.C.; Li, Y.M.; Hung, M.C. Upregulation of IKKalpha/IKKbeta by Integrin-Linked kinase is required for HER2/neu-induced NF-kappaB antiapoptotic pathway. Oncogene 2004, 23, 3883–3887. [Google Scholar] [CrossRef][Green Version]
- Biswas, D.K.; Iglehart, J.D. Linkage between EGFR family receptors and nuclear factor kappaB (NF-kappaB) signaling in breast cancer. J. Cell. Physiol. 2006, 209, 645–652. [Google Scholar] [CrossRef]
- Merkhofer, E.C.; Cogswell, P.; Baldwin, A.S. Her2 activates NF-kappaB and induces invasion through the canonical pathway involving IKKalpha. Oncogene 2010, 29, 1238–1248. [Google Scholar] [CrossRef]
- Montoliu, L.; Blendy, J.A.; Cole, T.J.; Schutz, G. Analysis of the cAMP response on Liver-Specific gene expression in transgenic mice. Fundam. Clin. Pharmacol. 1994, 8, 138–146. [Google Scholar] [CrossRef]
- Lim, W.; Song, G. Characteristics, Tissue-Specific expression, and hormonal regulation of expression of tyrosine aminotransferase in the avian female reproductive tract. Domest. Anim. Endocrinol. 2016, 57, 10–20. [Google Scholar] [CrossRef]
- Mondanelli, G.; Ugel, S.; Grohmann, U.; Bronte, V. The immune regulation in cancer by the amino acid metabolizing enzymes ARG and IDO. Curr. Opin. Pharmacol. 2017, 35, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Mayne, R.; Brewton, R.G.; Mayne, P.M.; Baker, J.R. Isolation and characterization of the chains of type V/type XI collagen present in bovine vitreous. J. Biol. Chem. 1993, 268, 9381–9386. [Google Scholar] [PubMed]
- Wilson, R.; Norris, E.L.; Brachvogel, B.; Angelucci, C.; Zivkovic, S.; Gordon, L.; Bernardo, B.C.; Stermann, J.; Sekiguchi, K.; Gorman, J.J.; et al. Changes in the chondrocyte and extracellular matrix proteome during post-natal mouse cartilage development. Mol. Cell. Proteomics 2012, 11, M111 014159. [Google Scholar] [CrossRef] [PubMed]
- Nasser, S.; Kurdolgu, A.A.; Izatt, T.; Aldrich, J.; Russell, M.L.; Christoforides, A.; Tembe, W.; Keifer, J.A.; Corneveaux, J.J.; Byron, S.A.; et al. An integrated framework for reporting clinically relevant biomarkers from paired tumor/normal genomic and transcriptomic sequencing data in support of clinical trials in personalized medicine. In Proceedings of the Pacific Symposium on Biocomputing 2015, Kohala Coast, HI, USA, 4–8 January 2015; pp. 56–67. [Google Scholar]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef]
- Christoforides, A.; Carpten, J.D.; Weiss, G.J.; Demeure, M.J.; Von Hoff, D.D.; Craig, D.W. Identification of somatic mutations in cancer through Bayesian-Based analysis of sequenced genome pairs. BMC Genom. 2013, 14, 302. [Google Scholar] [CrossRef]
- Saunders, C.J.; Miller, N.A.; Soden, S.E.; Dinwiddie, D.L.; Noll, A.; Alnadi, N.A.; Andraws, N.; Patterson, M.L.; Krivohlavek, L.A.; Fellis, J.; et al. Rapid Whole-Genome sequencing for genetic disease diagnosis in neonatal intensive care units. Sci. Transl. Med. 2012, 4, 154ra135. [Google Scholar] [CrossRef]
- Cibulskis, K.; Lawrence, M.S.; Carter, S.L.; Sivachenko, A.; Jaffe, D.; Sougnez, C.; Gabriel, S.; Meyerson, M.; Lander, E.S.; Getz, G. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat. Biotechnol. 2013, 31, 213–219. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Halperin, R.F.; Liang, W.S.; Kulkarni, S.; Tassone, E.E.; Adkins, J.; Enriquez, D.; Tran, N.L.; Hank, N.C.; Newell, J.; Kodira, C.; et al. Leveraging Spatial Variation in Tumor Purity for Improved Somatic Variant Calling of Archival Tumor Only Samples. Front. Oncol. 2019, 9, 119. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tumor ID | Age | Tumor Size (cm) | Tumor Grade | Lymph Node (+/total) | HER2 Scores | ER (%) | Neoadjuvant Trastuzumab (Duration) |
|---|---|---|---|---|---|---|---|
| S2 | 58 | 2.2 | III | 5/19 | 3+ | 40 | Y (3 months) |
| S3 | 55 | 1.5 | II | 2/14 | 3+ | 90 | Y (4 months) |
| S4 | 38 | 1.0 | III | 0/13 | 3+ | 0 | Y (2 months) |
| S5 | 61 | 1.5 | III | 0/0 | 3+ | 80 | Y (3 weeks) |
| S6 | 48 | 0.2 | NP | 0/8 | 3+ | 0 | Y (1 year) |
| S7 | 59 | NP | III | 5/21 | 3+ | 0 | Y (3 months) |
| S10 | 36 | NP | NP | NP | 3+ | 0 | Y (3 months) |
| S11 | 43 | 4.5 | III | 1/6 | 3+ | 0 | Y (NP) |
| Tumor ID | NF-κB p65 | CD4 | CD8 | PD-1 | # SNV/CNV | Putative Driver Mutations |
|---|---|---|---|---|---|---|
| S2 | ++ | – | ++ | – | 8 | N/A |
| S3 | ++ | ++ | ++ | – | 20 | HER2 copy gain; PTEN HD |
| S4 | ++ | + | ++ | + | 8 | HER2 overexpression |
| S5 | ++ | ++ | ++ | + | 16 | HER2 copy gain |
| S6 | ND | ND | ND | – | 3 | N/A |
| S7 | ++ | + | + | – | 7 | PIK3CA aSNV |
| S10 | ++ | ++ | ++ | ++ | 17 | HER2 copy gain/SV; TP53 iSNV |
| S11 | ++ | ++ | ++ | + | 25 | PIK3CA aSNV, TP53 iSNV |
| BC Subtype | n | Group 1 | Group 2 |
|---|---|---|---|
| All | 1090 | 250 (23%) | 309 (28%) |
| ER+HER2− | 590 | 131 (22%) | 159 (27%) |
| HER2+ | 181 | 36 (20%) | 53 (29%) |
| TNBC | 160 | 32 (20%) | 51 (32%) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hendricks, W.P.D.; Briones, N.; Halperin, R.F.; Facista, S.; Heaton, P.R.; Mahadevan, D.; Kim, S. PD-1-Associated Gene Expression Signature of Neoadjuvant Trastuzumab-Treated Tumors Correlates with Patient Survival in HER2-Positive Breast Cancer. Cancers 2019, 11, 1566. https://doi.org/10.3390/cancers11101566
Hendricks WPD, Briones N, Halperin RF, Facista S, Heaton PR, Mahadevan D, Kim S. PD-1-Associated Gene Expression Signature of Neoadjuvant Trastuzumab-Treated Tumors Correlates with Patient Survival in HER2-Positive Breast Cancer. Cancers. 2019; 11(10):1566. https://doi.org/10.3390/cancers11101566
Chicago/Turabian StyleHendricks, William P. D., Natalia Briones, Rebecca F. Halperin, Salvatore Facista, Paul R. Heaton, Daruka Mahadevan, and Suwon Kim. 2019. "PD-1-Associated Gene Expression Signature of Neoadjuvant Trastuzumab-Treated Tumors Correlates with Patient Survival in HER2-Positive Breast Cancer" Cancers 11, no. 10: 1566. https://doi.org/10.3390/cancers11101566
APA StyleHendricks, W. P. D., Briones, N., Halperin, R. F., Facista, S., Heaton, P. R., Mahadevan, D., & Kim, S. (2019). PD-1-Associated Gene Expression Signature of Neoadjuvant Trastuzumab-Treated Tumors Correlates with Patient Survival in HER2-Positive Breast Cancer. Cancers, 11(10), 1566. https://doi.org/10.3390/cancers11101566