Isorhamnetin Induces Cell Cycle Arrest and Apoptosis Via Reactive Oxygen Species-Mediated AMP-Activated Protein Kinase Signaling Pathway Activation in Human Bladder Cancer Cells


 , ,
, ,  ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
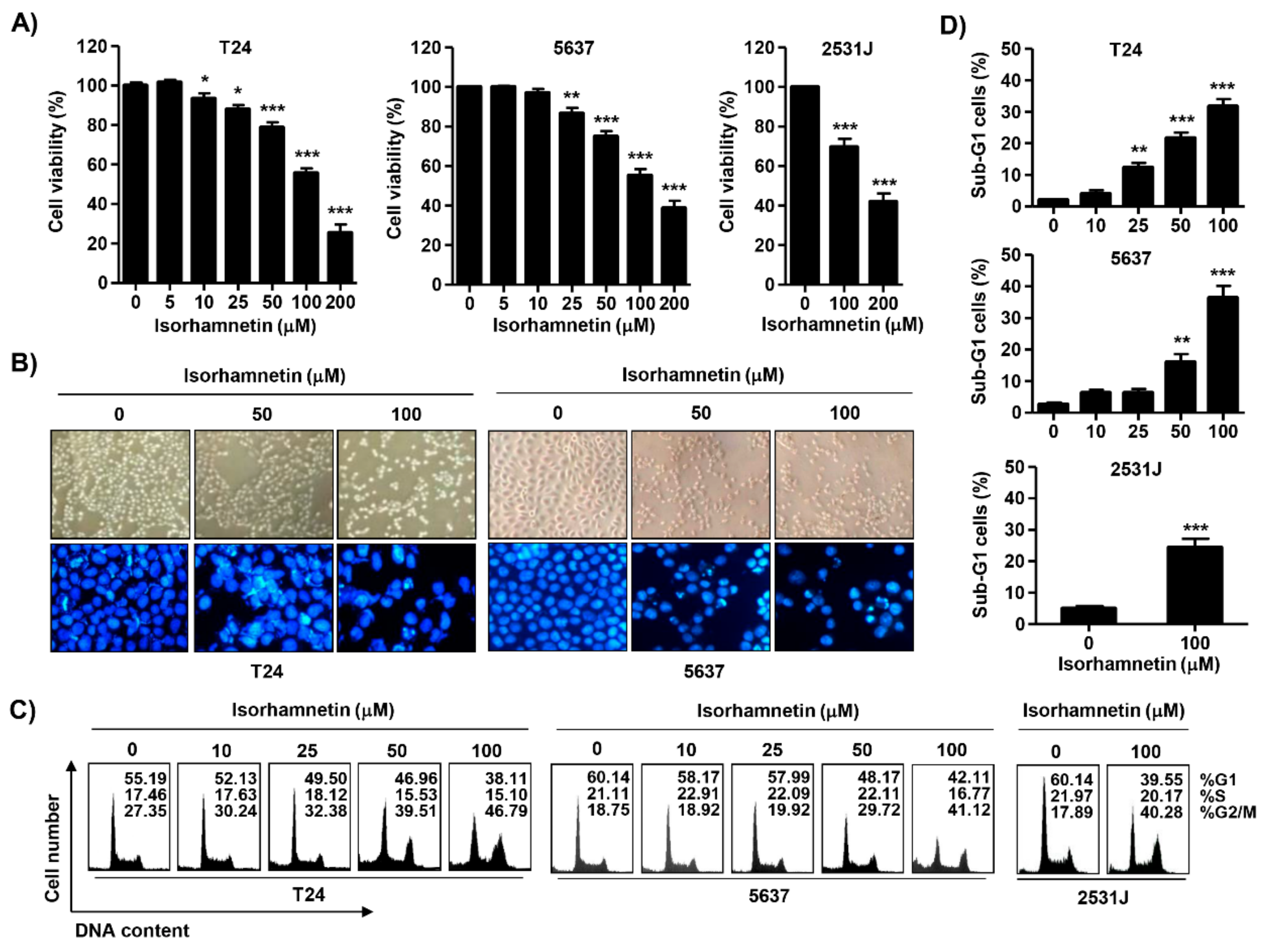
2.1. Isorhamnetin Inhibited Cell Viability in Bladder Cancer Cells
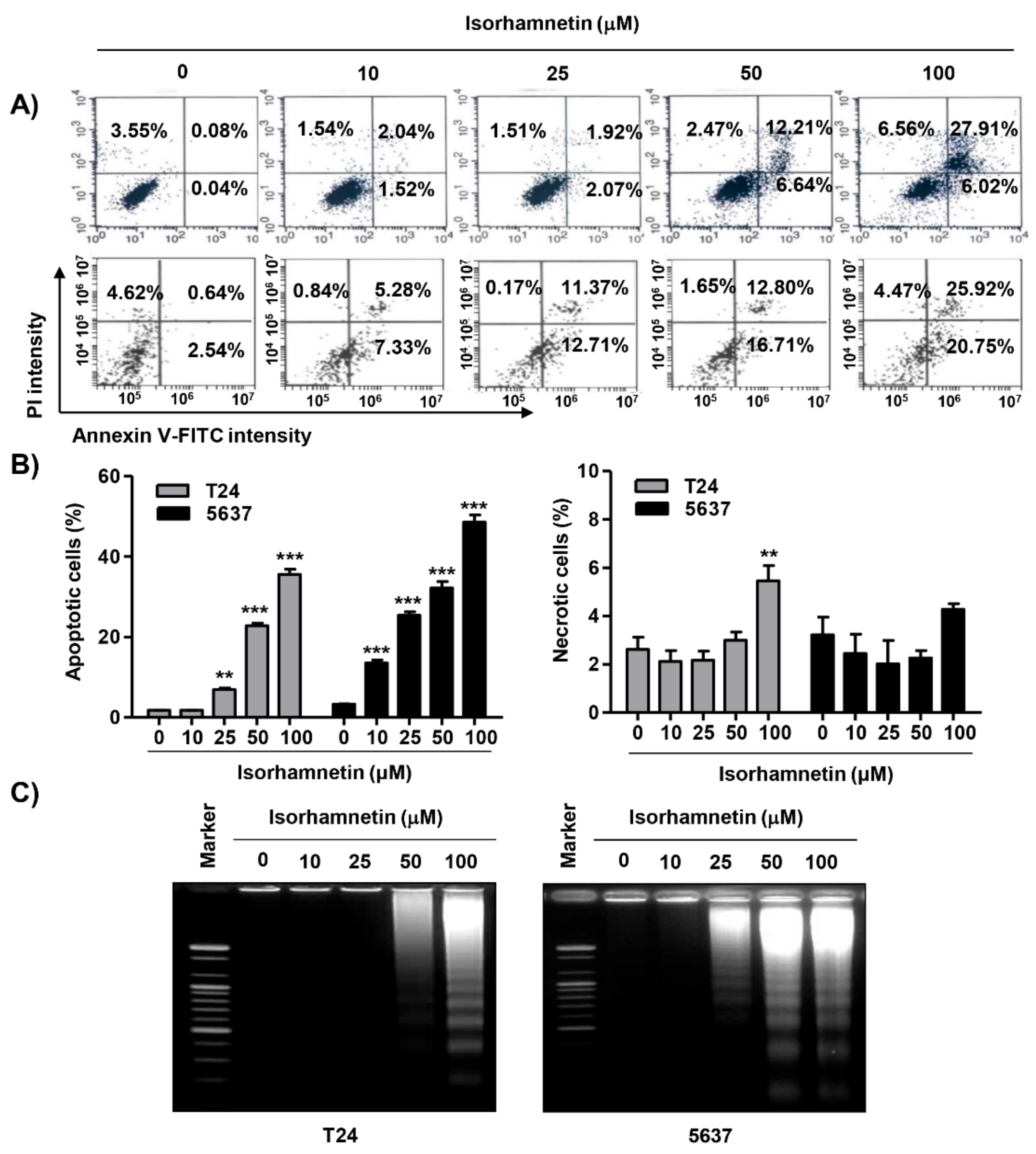
2.2. Isorhamnetin Induced G2/M Phase Arrest and Apoptosis in Bladder Cancer Cells
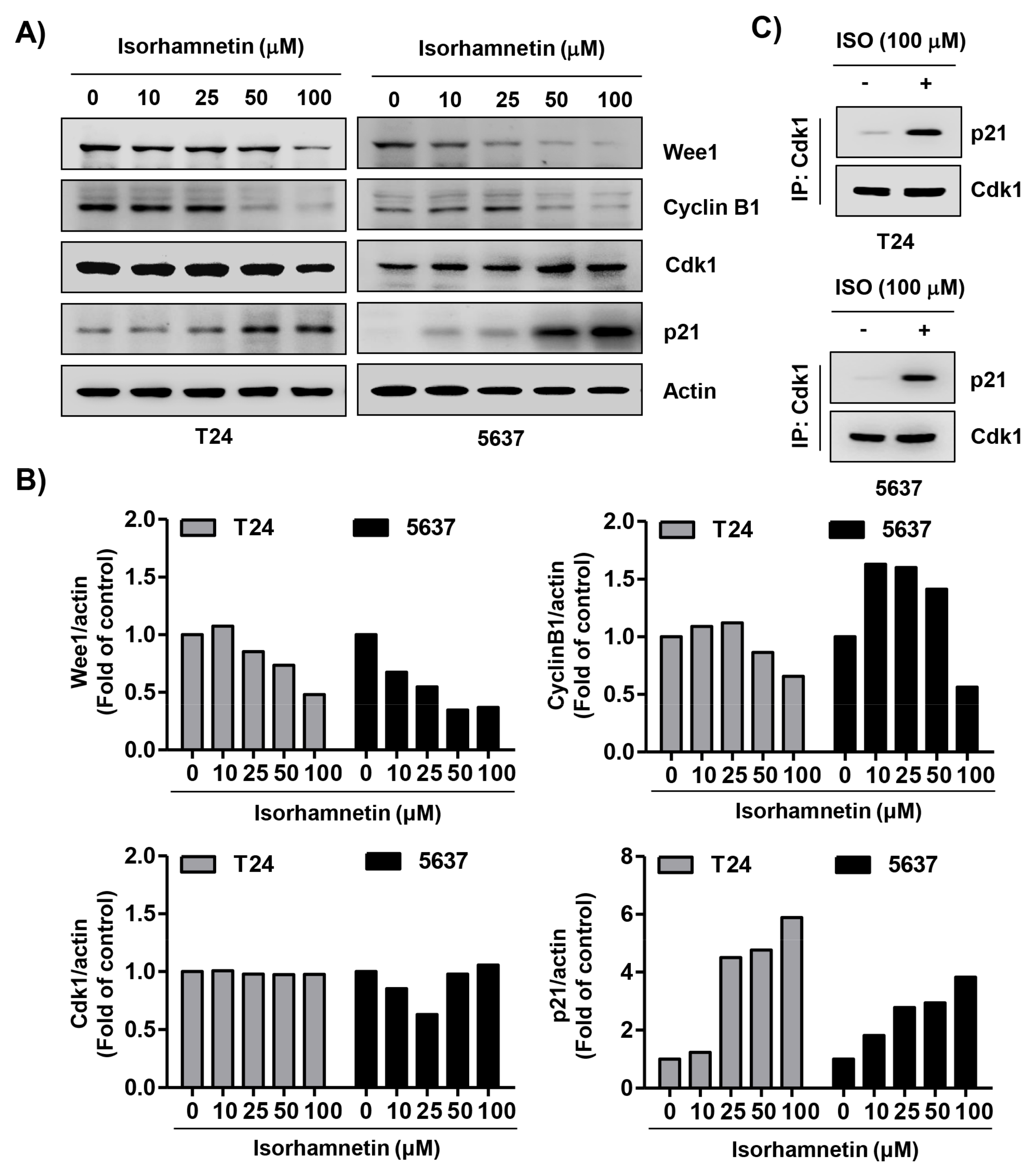
2.3. Isorhamnetin Regulated the Expression of G2/M Phase-Associated Proteins in Bladder Cancer Cells
2.4. Isorhamnetin Modulated the Expression of Apoptosis-Regulatory Proteins in Bladder Cancer Cells
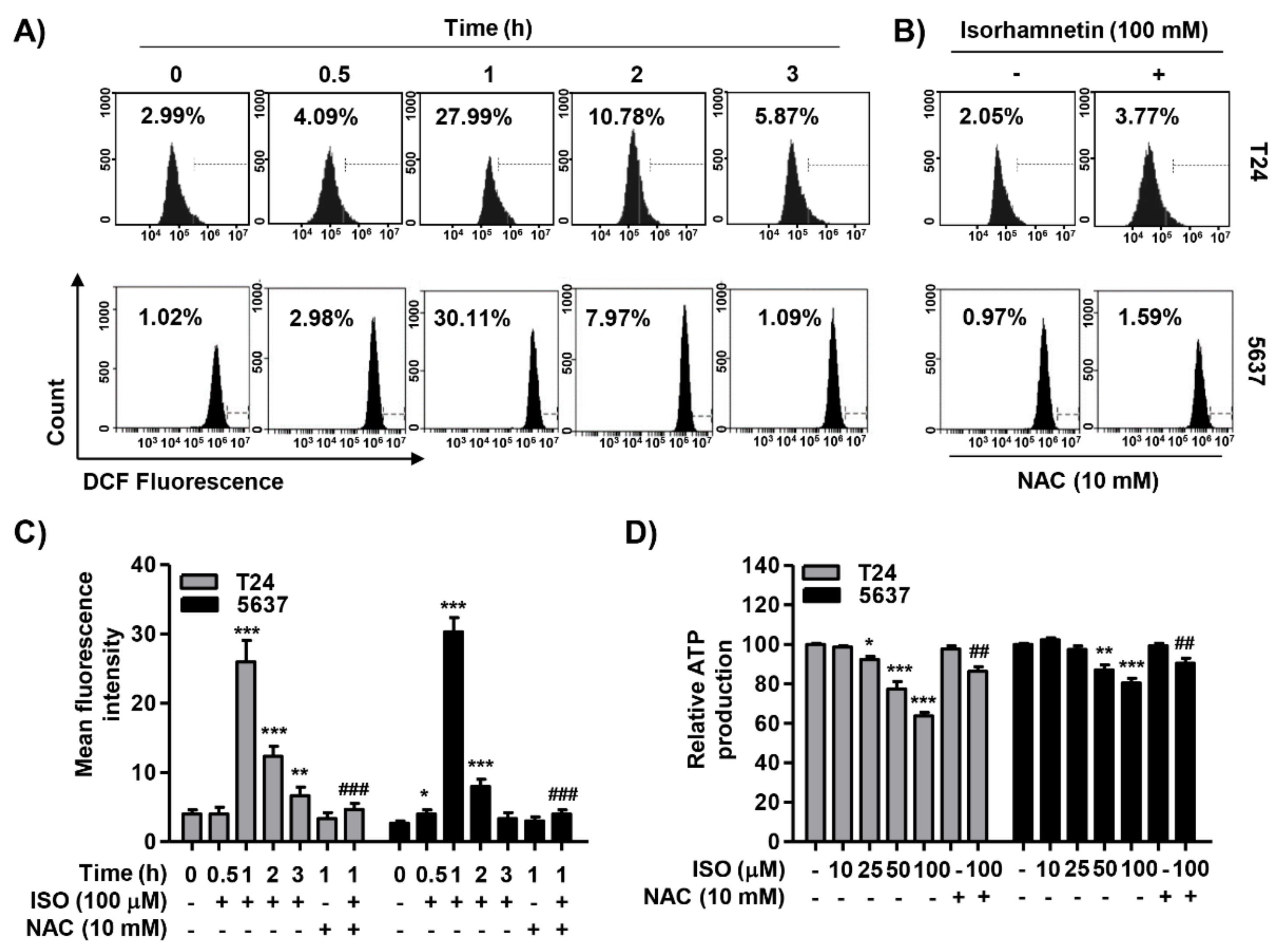
2.5. Isorhamnetin Increased ROS Generation but Decreased ATP Content in Cancer Cells
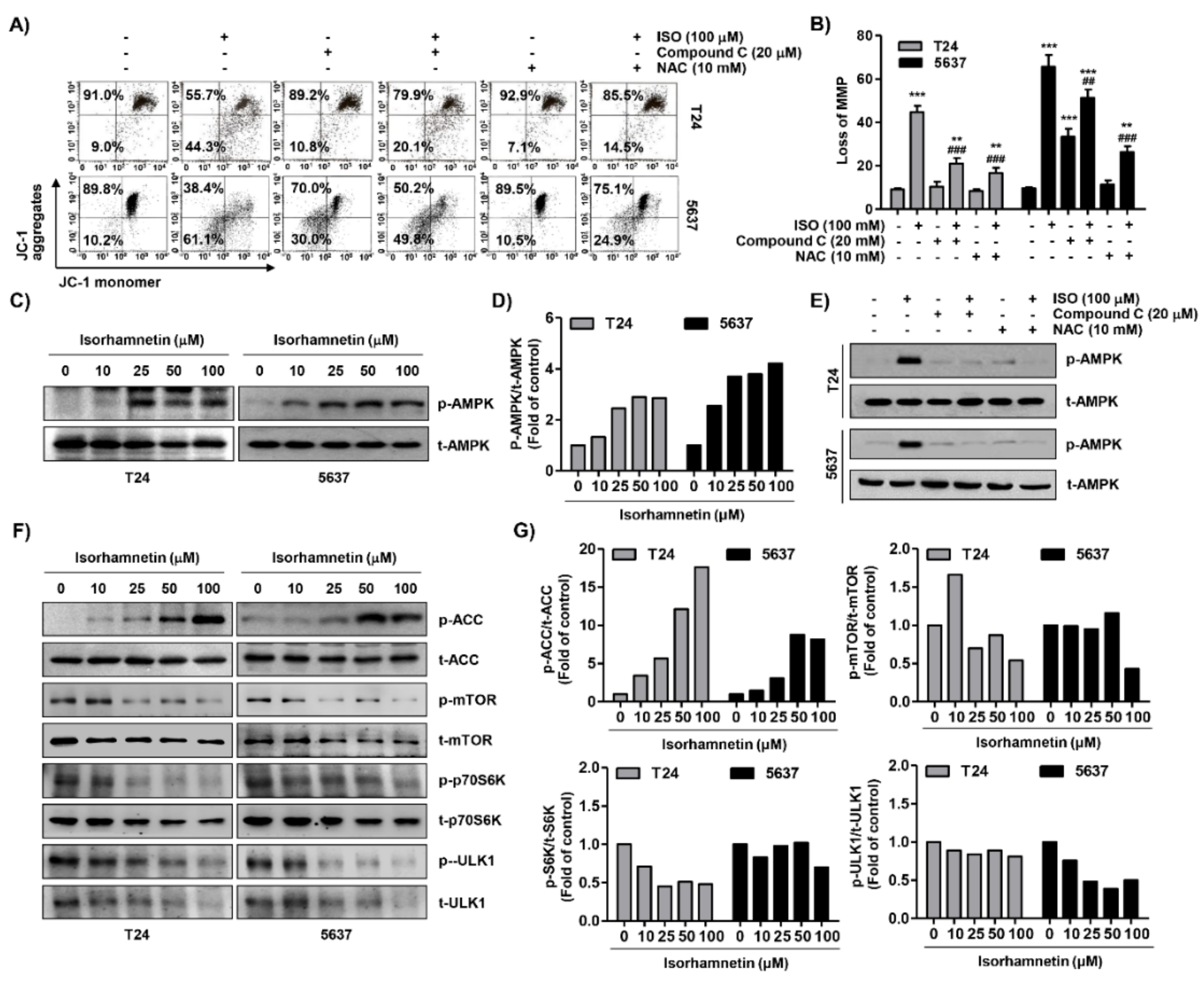
2.6. Isorhamnetin Reduced Mitochondrial Membrane Potential (MMP, ΔΨm) and Activated Adenosine 5’-Monophosphate-Activated Protein Kinase (AMPK) Signaling in Bladder Cancer Cells
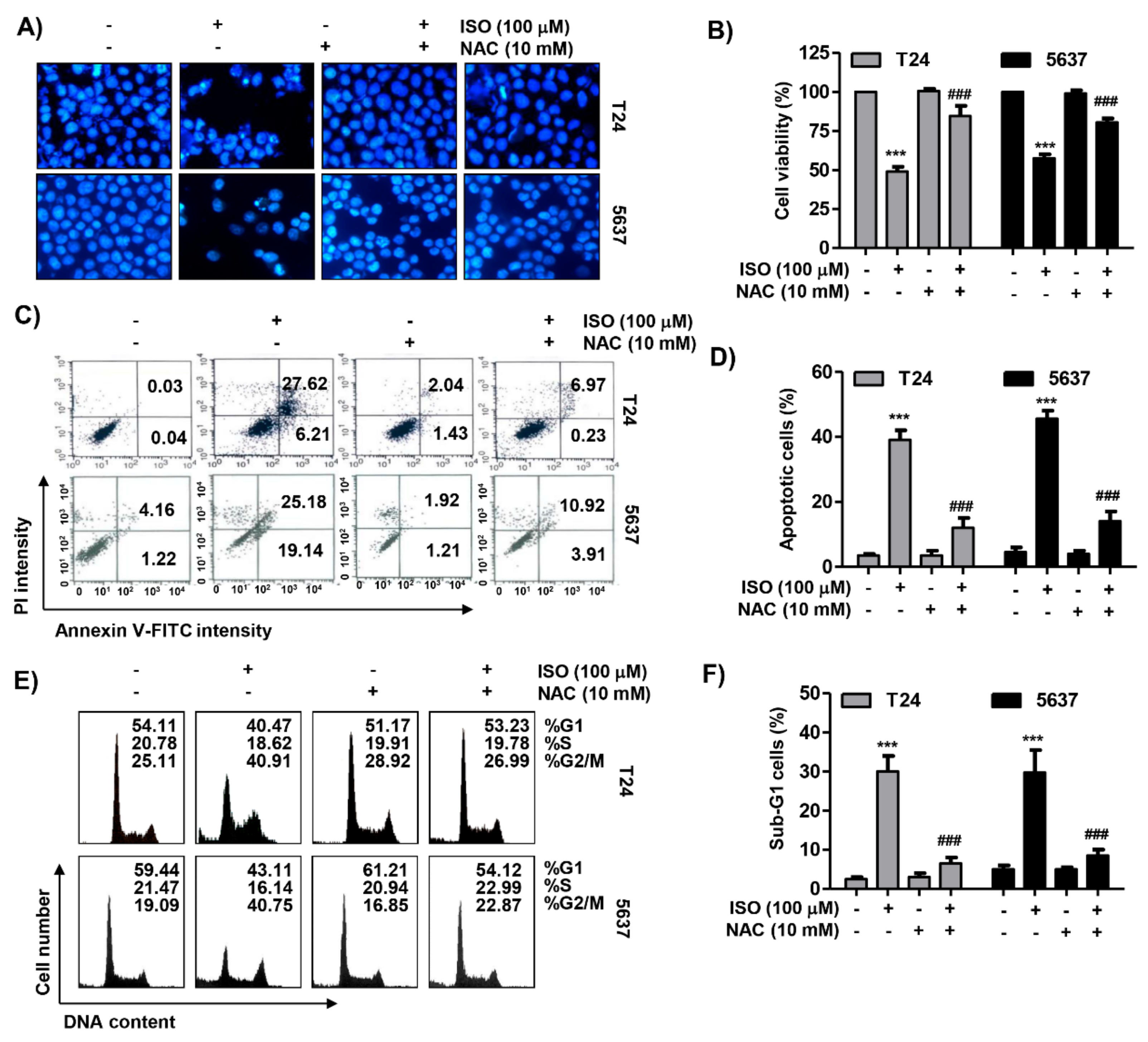
2.7. ROS Acted as an Upstream Regulator of Isorhamnetin-Mediated Apoptosis and Cell Cycle Blockade in Bladder Cancer Cells
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Isorhamnetin Treatment
4.2. Cell Viability Assay
4.3. Determination of Cell Cycle Distribution Using Flow Cytometric Analysis
4.4. Determination of Apoptotic Cell Death by Flow Cytometric Analysis
4.5. Nuclear Staining and Deoxyribonucleic Acid (DAN) Fragmentation Assay
4.6. Protein Extraction, Co-Immunoprecipitation, and Western Blot Analysis
4.7. Caspase Activity Assay
4.8. Measurement of ROS Production and MMP
4.9. Detection of ATP Levels
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Kumar, A.; Jaitak, V. Natural products as multidrug resistance modulators in cancer. Eur. J. Med. Chem. 2019, 176, 268–291. [Google Scholar] [CrossRef] [PubMed]
- Fu, B.; Wang, N.; Tan, H.Y.; Li, S.; Cheung, F.; Feng, Y. Multi-component herbal products in the prevention and treatment of chemotherapy-associated toxicity and side effects: A review on experimental and clinical evidences. Front. Pharmacol. 2018, 9, 1394. [Google Scholar] [CrossRef] [PubMed]
- Butler, M.S.; Robertson, A.A.; Cooper, M.A. Natural product and natural product derived drugs in clinical trials. Nat. Prod. Rep. 2014, 31, 1612–1661. [Google Scholar] [CrossRef] [PubMed]
- Nobili, S.; Lippi, D.; Witort, E.; Donnini, M.; Bausi, L.; Mini, E.; Capaccioli, S. Natural compounds for cancer treatment and prevention. Pharmacol. Res. 2009, 59, 365–378. [Google Scholar] [CrossRef]
- Efferth, T.; Fu, Y.J.; Zu, Y.G.; Schwarz, G.; Konkimalla, V.S.; Wink, M. Molecular target-guided tumor therapy with natural products derived from traditional Chinese medicine. Curr. Med. Chem. 2007, 14, 2024–2032. [Google Scholar] [CrossRef]
- Medema, R.H.; Macůrek, L. Checkpoint control and cancer. Oncogene 2012, 31, 2601–2613. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Bolhassani, A. Cancer chemoprevention by natural carotenoids as an efficient strategy. Anticancer Agents Med. Chem. 2015, 15, 1026–1231. [Google Scholar] [CrossRef]
- Schnekenburger, M.; Dicato, M.; Diederich, M. Plant-derived epigenetic modulators for cancer treatment and prevention. Biotechnol. Adv. 2014, 32, 1123–1132. [Google Scholar] [CrossRef]
- Gu, Q.; Duan, G.; Yu, X. Bioconversion of flavonoid glycosides from Hippophae rhamnoides leaves into flavonoid aglycones by Eurotium amstelodami. Microorganisms 2019, 7, 122. [Google Scholar] [CrossRef]
- Zhang, Q.; Cui, H. Simultaneous determination of quercetin, kaempferol, and isorhamnetin in phytopharmaceuticals of Hippophae rhamnoides L. by high-performance liquid chromatography with chemiluminescence detection. J. Sep. Sci. 2005, 28, 1171–1178. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Xu, X.M.; Chen, Y.; Yu, M.Y.; Wen, F.Y.; Zhang, H. Identification, quantification and antioxidant activity of acylated flavonol glycosides from sea buckthorn (Hippophae rhamnoides ssp. sinensis). Food Chem. 2013, 141, 1573–1579. [Google Scholar] [CrossRef] [PubMed]
- Castrillo, J.L.; Vanden Berghe, D.; Carrasco, L. 3-Methylquercetin is a potent and selective inhibitor of poliovirus RNA synthesis. Virology 1986, 152, 219–227. [Google Scholar] [CrossRef]
- Rösch, D.; Krumbein, A.; Mügge, C.; Kroh, L.W. Structural investigations of flavonol glycosides from sea buckthorn (Hippophaë rhamnoides) pomace by NMR spectroscopy and HPLC-ESI-MS(n). J. Agric. Food Chem. 2004, 52, 4039–4046. [Google Scholar] [CrossRef] [PubMed]
- Qi, F.; Sun, J.H.; Yan, J.Q.; Li, C.M.; Lv, X.C. Anti-inflammatory effects of isorhamnetin on LPS-stimulated human gingival fibroblasts by activating Nrf2 signaling pathway. Microb. Pathog. 2018, 120, 37–41. [Google Scholar] [CrossRef] [PubMed]
- Abdallah, H.M.; Esmat, A. Antioxidant and anti-inflammatory activities of the major phenolics from Zygophyllum simplex L. J. Ethnopharmacol. 2017, 205, 51–56. [Google Scholar] [CrossRef]
- Seo, S.; Seo, K.; Ki, S.H.; Shin, S.M. Isorhamnetin inhibits reactive oxygen species-dependent hypoxia inducible factor (HIF)-1α accumulation. Biol. Pharm. Bull. 2016, 39, 1830–1838. [Google Scholar] [CrossRef]
- Yang, J.H.; Kim, S.C.; Kim, K.M.; Jang, C.H.; Cho, S.S.; Kim, S.J.; Ku, S.K.; Cho, I.J.; Ki, S.H. Isorhamnetin attenuates liver fibrosis by inhibiting TGF-β/Smad signaling and relieving oxidative stress. Eur. J. Pharmacol. 2016, 783, 92–102. [Google Scholar] [CrossRef]
- Luo, Y.; Sun, G.; Dong, X.; Wang, M.; Qin, M.; Yu, Y.; Sun, X. Isorhamnetin attenuates atherosclerosis by inhibiting macrophage apoptosis via PI3K/AKT activation and HO-1 induction. PLoS ONE 2015, 10, e0120259. [Google Scholar] [CrossRef]
- Zhang, H.W.; Hu, J.J.; Fu, R.Q.; Liu, X.; Zhang, Y.H.; Li, J.; Liu, L.; Li, Y.N.; Deng, Q.; Luo, Q.S.; et al. Flavonoids inhibit cell proliferation and induce apoptosis and autophagy through downregulation of Ⅲ9Kγ mediated PI3K/AKT/mTOR/p70S6K/ULK signaling pathway in human breast cancer cells. Sci. Rep. 2018, 8, 11255. [Google Scholar] [CrossRef]
- Wei, J.; Su, H.; Bi, Y.; Li, J.; Feng, L.; Sheng, W. Anti-proliferative effect of isorhamnetin on HeLa cells through inducing G2/M cell cycle arrest. Exp. Ther. Med. 2018, 15, 3917–3923. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Chen, X.; Dan, J.; Cao, Y.; Gao, S.; Guo, Z.; Zerbe, P.; Chai, Y.; Diao, Y.; Zhang, L. Characterization of anti-leukemia components from Indigo naturalis using comprehensive two-dimensional K562/cell membrane chromatography and in silico target identification. Sci. Rep. 2016, 6, 25491. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Yang, X.; Chen, C.; Cai, S.; Hu, J. Isorhamnetin suppresses colon cancer cell growth through the PI3K-Akt-mTOR pathway. Mol. Med. Rep. 2014, 9, 935–940. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.Y.; Wang, Y.M.; Gong, H.; Zhao, H.; Lv, X.Y.; Yuan, G.H.; Han, S.R. Isorhamnetin flavonoid synergistically enhances the anticancer activity and apoptosis induction by cisplatin and carboplatin in non-small cell lung carcinoma (NSCLC). Int. J. Clin. Exp. Pathol. 2015, 8, 25–37. [Google Scholar] [PubMed]
- Wang, J.L.; Quan, Q.; Ji, R.; Guo, X.Y.; Zhang, J.M.; Li, X.; Liu, Y.G. Isorhamnetin suppresses PANC-1 pancreatic cancer cell proliferation through S phase arrest. Biomed. Pharmacother. 2018, 108, 925–933. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Kroon, P.A.; Shao, H.; Needs, P.W.; Yang, X. Differential effects of quercetin and two of its derivatives, isorhamnetin and isorhamnetin-3-glucuronide, in inhibiting the proliferation of human breast-cancer MCF-7 cells. J. Agric. Food Chem. 2018, 66, 7181–7189. [Google Scholar] [CrossRef]
- Sak, K.; Lust, H.; Kase, M.; Jaal, J. Cytotoxic action of methylquercetins in human lung adenocarcinoma cells. Oncol. Lett. 2018, 15, 1973–1978. [Google Scholar] [CrossRef]
- Huang, S.P.; Ho, T.M.; Yang, C.W.; Chang, Y.J.; Chen, J.F.; Shaw, N.S.; Horng, J.C.; Hsu, S.L.; Liao, M.Y.; Wu, L.C.; et al. Chemopreventive potential of ethanolic extracts of luobuma leaves (Apocynum venetum L.) in androgen insensitive prostate cancer. Nutrients 2017, 9, 948. [Google Scholar] [CrossRef]
- Li, Q.; Ren, F.Q.; Yang, C.L.; Zhou, L.M.; Liu, Y.Y.; Xiao, J.; Zhu, L.; Wang, Z.G. Anti-proliferation effects of isorhamnetin on lung cancer cells in vitro and in vivo. Asian Pac. J. Cancer. Prev. 2015, 16, 3035–3042. [Google Scholar] [CrossRef]
- Antunes-Ricardo, M.; Moreno-García, B.E.; Gutiérrez-Uribe, J.A.; Aráiz-Hernández, D.; Alvarez, M.M.; Serna-Saldivar, S.O. Induction of apoptosis in colon cancer cells treated with isorhamnetin glycosides from Opuntia ficusindica pads. Plant Foods Hum. Nutr. 2014, 69, 331–336. [Google Scholar] [CrossRef]
- Lee, H.J.; Lee, H.J.; Lee, E.O.; Ko, S.G.; Bae, H.S.; Kim, C.H.; Ahn, K.S.; Lu, J.; Kim, S.H. Mitochondria-cytochrome c-caspase-9 cascade mediates isorhamnetin-induced apoptosis. Cancer Lett. 2008, 270, 342–353. [Google Scholar] [CrossRef]
- Hu, S.; Huang, L.; Meng, L.; Sun, H.; Zhang, W.; Xu, Y. Isorhamnetin inhibits cell proliferation and induces apoptosis in breast cancer via Akt and mitogen-activated protein kinase kinase signaling pathways. Mol. Med. Rep. 2015, 12, 6745–6751. [Google Scholar] [CrossRef] [PubMed]
- Choi, K.C.; Chung, W.T.; Kwon, J.K.; Yu, J.Y.; Jang, Y.S.; Park, S.M.; Lee, S.Y.; Lee, J.C. Inhibitory effects of quercetin on aflatoxin B1-induced hepatic damage in mice. Food Chem. Toxicol. 2010, 48, 2747–2753. [Google Scholar] [CrossRef] [PubMed]
- Hibasami, H.; Mitani, A.; Katsuzaki, H.; Imai, K.; Yoshioka, K.; Komiya, T. Isolation of five types of flavonol from seabuckthorn (Hippophae rhamnoides) and induction of apoptosis by some of the flavonols in human promyelotic leukemia HL-60 cells. Int. J. Mol. Med. 2005, 15, 805–809. [Google Scholar] [CrossRef] [PubMed]
- Prasain, J.K.; Rajbhandari, R.; Keeton, A.B.; Piazza, G.A.; Barnes, S. Metabolism and growth inhibitory activity of cranberry derived flavonoids in bladder cancer cells. Food Funct. 2016, 7, 4012–4019. [Google Scholar] [CrossRef]
- Ebrahimi, S.; Hosseini, M.; Shahidsales, S.; Maftouh, M.; Ferns, G.A.; Ghayour-Mobarhan, M.; Hassanian, S.M.; Avan, A. Targeting the Akt/PI3K Signaling Pathway as a Potential Therapeutic Strategy for the Treatment of Pancreatic Cancer. Curr. Med. Chem. 2017, 24, 1321–1331. [Google Scholar] [CrossRef] [PubMed]
- Hassan, M.; Watari, H.; AbuAlmaaty, A.; Ohba, Y.; Sakuragi, N. Apoptosis and molecular targeting therapy in cancer. Biomed. Res. Int. 2014, 2014, 150845. [Google Scholar] [CrossRef]
- Bai, J.; Li, Y.; Zhang, G. Cell cycle regulation and anticancer drug discovery. Cancer Biol. Med. 2017, 14, 348–362. [Google Scholar]
- Sánchez-Martínez, C.; Gelbert, L.M.; Lallena, M.J.; De Dios, A. Cyclin dependent kinase (CDK) inhibitors as anticancer drugs. Bioorg. Med. Chem. Lett. 2015, 25, 3420–3435. [Google Scholar] [CrossRef]
- Matheson, C.J.; Backos, D.S.; Reigan, P. Targeting WEE1 kinase in cancer. Trends Pharmacol. Sci. 2016, 37, 872–881. [Google Scholar] [CrossRef]
- Bulavin, D.V.; Demidenko, Z.N.; Phillips, C.; Moody, S.A.; Fornace, A.J., Jr. Phosphorylation of Xenopus Cdc25C at Ser285 interferes with ability to activate a DNA damage replication checkpoint in pre-midblastula embryos. Cell Cycle 2003, 2, 263–266. [Google Scholar] [CrossRef]
- Karimian, A.; Ahmadi, Y.; Yousefi, B. Multiple functions of p21 in cell cycle, apoptosis and transcriptional regulation after DNA damage. DNA Repair 2016, 42, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Reinhardt, H.C.; Schumacher, B. The p53 network: Cellular and systemic DNA damage responses in aging and cancer. Trends Genet. 2012, 28, 128–136. [Google Scholar] [CrossRef] [PubMed]
- Hydbring, P.; Malumbres, M.; Sicinski, P. Non-canonical functions of cell cycle cyclins and cyclin-dependent kinases. Nat. Rev. Mol. Cell Biol. 2016, 17, 280–292. [Google Scholar] [CrossRef] [PubMed]
- Cooper, M.J.; Haluschak, J.J.; Johnson, D.; Schwartz, S.; Morrison, L.J.; Lippa, M.; Hatzivassiliou, G.; Tan, J. p53 mutations in bladder carcinoma cell lines. Oncol. Res. 1994, 6, 569–579. [Google Scholar] [PubMed]
- Pfeffer, C.M.; Singh, A.T.K. Apoptosis: A target for anticancer therapy. Int. J. Mol. Sci. 2018, 19, 448. [Google Scholar] [CrossRef]
- Kantari, C.; Walczak, H. Caspase-8 and bid: Caught in the act between death receptors and mitochondria. Biochim. Biophys. Acta 2011, 1813, 558–563. [Google Scholar] [CrossRef] [PubMed]
- Edlich, F. BCL-2 proteins and apoptosis: Recent insights and unknowns. Biochem. Biophys. Res. Commun. 2018, 500, 26–34. [Google Scholar] [CrossRef]
- Birkinshaw, R.W.; Czabotar, P.E. The BCL-2 family of proteins and mitochondrial outer membrane permeabilisation. Semin. Cell Dev. Biol. 2017, 72, 152–162. [Google Scholar] [CrossRef]
- Kiraz, Y.; Adan, A.; Kartal Yandim, M.; Baran, Y. Major apoptotic mechanisms and genes involved in apoptosis. Tumour Biol. 2016, 37, 8471–8486. [Google Scholar] [CrossRef]
- Schultz, D.R.; Harrington, W.J., Jr. Apoptosis: Programmed cell death at a molecular level. Semin. Arthritis Rheum. 2003, 32, 345–369. [Google Scholar] [CrossRef] [PubMed]
- Ruan, Y.; Hu, K.; Chen, H. Autophagy inhibition enhances isorhamnetin-induced mitochondria-dependent apoptosis in non-small cell lung cancer cells. Mol. Med. Rep. 2015, 12, 5796–5806. [Google Scholar] [CrossRef] [PubMed]
- Badrinath, N.; Yoo, S.Y. Mitochondria in cancer: In the aspects of tumorigenesis and targeted therapy. Carcinogenesis 2018, 39, 1419–1430. [Google Scholar] [CrossRef] [PubMed]
- Moloney, J.N.; Cotter, T.G. ROS signalling in the biology of cancer. Semin. Cell Dev. Biol. 2018, 80, 50–64. [Google Scholar] [CrossRef] [PubMed]
- Galadari, S.; Rahman, A.; Pallichankandy, S.; Thayyullathil, F. Reactive oxygen species and cancer paradox: To promote or to suppress? Free Radic. Biol. Med. 2017, 104, 144–164. [Google Scholar] [CrossRef] [PubMed]
- Thirupathi, A.; De Souza, C.T. Multi-regulatory network of ROS: The interconnection of ROS, PGC-1 alpha, and AMPK-SIRT1 during exercise. J. Physiol. Biochem. 2017, 73, 487–494. [Google Scholar] [CrossRef]
- Wu, S.B.; Wu, Y.T.; Wu, T.P.; Wei, Y.H. Role of AMPK-mediated adaptive responses in human cells with mitochondrial dysfunction to oxidative stress. Biochim. Biophys. Acta 2014, 1840, 1331–1344. [Google Scholar] [CrossRef] [PubMed]
- Zou, M.H.; Kirkpatrick, S.S.; Davis, B.J.; Nelson, J.S.; Wiles, W.G., 4th; Schlattner, U.; Neumann, D.; Brownlee, M.; Freeman, M.B.; Goldman, M.H. Activation of the AMP-activated protein kinase by the anti-diabetic drug metformin in vivo. Role of mitochondrial reactive nitrogen species. J. Biol. Chem. 2004, 279, 43940–43951. [Google Scholar] [CrossRef]
- Choi, S.L.; Kim, S.J.; Lee, K.T.; Kim, J.; Mu, J.; Birnbaum, M.J.; Soo Kim, S.; Ha, J. The regulation of AMP activated protein kinase by H2O2. Biochem. Biophys. Res. Commun. 2001, 287, 92–97. [Google Scholar] [CrossRef]
- Wu, S.B.; Wei, Y.H. AMPK-mediated Increase of glycolysis as an adaptive response to oxidative stress in human cells: Implication of the cell survival in mitochondrial diseases. Biochim. Biophys. Acta 2012, 1822, 233–247. [Google Scholar] [CrossRef]
- Cao, C.; Lu, S.; Jiang, Q.; Wang, W.J.; Song, X.; Kivlin, R.; Wallin, B.; Bagdasarian, A.; Tamakloe, T.; Chu, W.M.; et al. EGFR activation confers protections against UV-induced apoptosis in cultured mouse skin dendritic cells. Cell Signal 2018, 20, 1830–1838. [Google Scholar] [CrossRef]
- Corton, J.M.; Gillespie, J.G.; Hardi, D.G. Role of the AMP-activated protein kinase in the cellular stress response. Curr. Biol. 1994, 4, 315–324. [Google Scholar] [CrossRef]
- Tavazzi, B.; Di Pierro, D.; Amorini, A.M.; Fazzina, G.; Tuttobene, M.; Giardina, B.; Lazzarino, G. Energy metabolism and lipid peroxidation of human erythrocytes as a function of increased oxidative stress. Eur. J. Biochem. 2000, 267, 684–689. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Xu, B.; Liu, L.; Luo, Y.; Yin, J.; Zhou, H.; Chen, W.; Shen, T.; Han, X.; Huang, S. Hydrogen peroxide inhibits mTOR signaling by activation of AMPK alpha leading to apoptosis of neuronal cells. Lab. Investig. 2010, 90, 762–773. [Google Scholar] [CrossRef] [PubMed]
- Vara-Ciruelos, D.; Russell, F.M.; Hardie, D.G. The strange case of AMPK and cancer: Dr Jekyll or Mr Hyde? Open Biol. 2019, 9, 190099. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Egan, D.F.; Shackelford, D.B.; Mihaylova, M.M.; Gelino, S.; Kohnz, R.A.; Mair, W.; Vasquez, D.S.; Joshi, A.; Gwinn, D.M.; Taylor, R.; et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science 2011, 331, 456–461. [Google Scholar] [CrossRef]
- Wong, C.H.; Iskandar, K.B.; Yadav, S.K.; Hirpara, J.L.; Loh, T.; Pervaiz, S. Simultaneous induction of non-canonical autophagy and apoptosis in cancer cells by ROS-dependent ERK and JNK activation. PLoS ONE 2010, 5, e9996. [Google Scholar] [CrossRef] [PubMed]
- Ci, Y.; Shi, K.; An, J.; Yang, Y.; Hui, K.; Wu, P.; Shi, L.; Xu, C. ROS inhibit autophagy by downregulating ULK1 mediated by the phosphorylation of p53 in selenite-treated NB4 cells. Cell Death Dis. 2014, 5, e1542. [Google Scholar] [CrossRef]
- Park, C.; Jeong, N.Y.; Kim, G.Y.; Han, M.H.; Chung, I.M.; Kim, W.J.; Yoo, Y.H.; Choi, Y.H. Momilactone B induces apoptosis and G1 arrest of the cell cycle in human monocytic leukemia U937 cells through downregulation of pRB phosphorylation and induction of the cyclin-dependent kinase inhibitor p21Waf1/Cip1. Oncol. Rep. 2014, 31, 1653–1660. [Google Scholar] [CrossRef] [PubMed]
- Koh, E.M.; Lee, E.K.; Song, C.H.; Song, J.; Chung, H.Y.; Chae, C.H.; Jung, K.J. Ferulate, an active component of wheat germ, ameliorates oxidative stress-induced PTK/PTP imbalance and PP2A inactivation. Toxicol. Res. 2018, 34, 333–341. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.Y.; Kim, J.H.; Lee, J.C.; Won, M.H.; Yang, S.R.; Kim, H.C.; Wie, M.B. Zinc oxide nanoparticles exhibit both cyclooxygenase- and lipoxygenase-mediated apoptosis in human bone marrow-derived mesenchymal stem cells. Toxicol. Res. 2019, 35, 83–91. [Google Scholar] [CrossRef] [PubMed]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, C.; Cha, H.-J.; Choi, E.O.; Lee, H.; Hwang-Bo, H.; Ji, S.Y.; Kim, M.Y.; Kim, S.Y.; Hong, S.H.; Cheong, J.; et al. Isorhamnetin Induces Cell Cycle Arrest and Apoptosis Via Reactive Oxygen Species-Mediated AMP-Activated Protein Kinase Signaling Pathway Activation in Human Bladder Cancer Cells. Cancers 2019, 11, 1494. https://doi.org/10.3390/cancers11101494
Park C, Cha H-J, Choi EO, Lee H, Hwang-Bo H, Ji SY, Kim MY, Kim SY, Hong SH, Cheong J, et al. Isorhamnetin Induces Cell Cycle Arrest and Apoptosis Via Reactive Oxygen Species-Mediated AMP-Activated Protein Kinase Signaling Pathway Activation in Human Bladder Cancer Cells. Cancers. 2019; 11(10):1494. https://doi.org/10.3390/cancers11101494
Chicago/Turabian StylePark, Cheol, Hee-Jae Cha, Eun Ok Choi, Hyesook Lee, Hyun Hwang-Bo, Seon Yeong Ji, Min Yeong Kim, So Young Kim, Su Hyun Hong, JaeHun Cheong, and et al. 2019. "Isorhamnetin Induces Cell Cycle Arrest and Apoptosis Via Reactive Oxygen Species-Mediated AMP-Activated Protein Kinase Signaling Pathway Activation in Human Bladder Cancer Cells" Cancers 11, no. 10: 1494. https://doi.org/10.3390/cancers11101494
APA StylePark, C., Cha, H.-J., Choi, E. O., Lee, H., Hwang-Bo, H., Ji, S. Y., Kim, M. Y., Kim, S. Y., Hong, S. H., Cheong, J., Kim, G.-Y., Yun, S. J., Hwang, H. J., Kim, W.-J., & Choi, Y. H. (2019). Isorhamnetin Induces Cell Cycle Arrest and Apoptosis Via Reactive Oxygen Species-Mediated AMP-Activated Protein Kinase Signaling Pathway Activation in Human Bladder Cancer Cells. Cancers, 11(10), 1494. https://doi.org/10.3390/cancers11101494