Unclassifiable Isolated Monoclonal Lymphocytosis: Comprehensive Description of a Retrospective Cohort

 , and
, and
Abstract
1. Introduction
2. Results
2.1. Clinical and Biological Characterization of the Cohort
2.2. Immunophenotypic Features
2.3. Cytogenetic Features
2.4. Gene Mutations
2.5. IGH Gene Repertoire
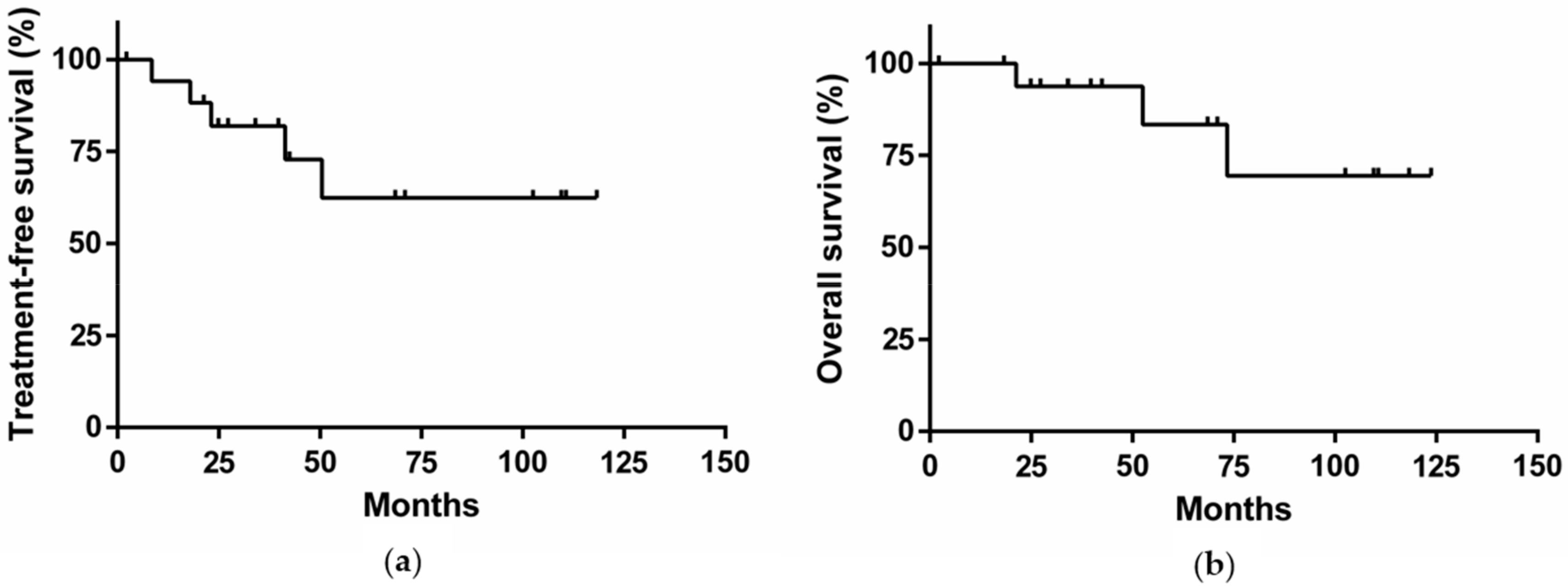
2.6. Clinical Outcome
3. Discussion
4. Materials and Methods
4.1. Patients
4.2. Morphological Analysis
4.3. Flow Cytometry Analysis
4.4. Cytogenetic Analysis
4.5. High-Throughput Sequencing
4.6. PCR Amplification and Sequence Analysis of Immunoglobulin Gene Rearrangements
4.7. Definitions of Treatment Responses
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Swerdlow, S.H.; Campo, E.; Harris, N.L.; Jaffe, E.S.; Pileri, S.A.; Stein, H.; Thiele, J. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues, 4th ed.; International Agency for Research on Cancer: Lyon, France, 2017. [Google Scholar]
- FDA. Available online: https://www.fda.gov/ (accessed on 23 June 2019).
- Matutes, E.; Owusu-Ankomah, K.; Morilla, R.; Garcia Marco, J.; Houlihan, A.; Que, T.H.; Catovsky, D. The immunological profile of B-cell disorders and proposal of a scoring system for the diagnosis of CLL. Leukemia 1994, 8, 1640–1645. [Google Scholar] [PubMed]
- Moreau, E.J.; Matutes, E.; A’Hern, R.P.; Morilla, A.M.; Morilla, R.M.; Owusu-Ankomah, K.A.; Seon, B.K.; Catovsky, D. Improvement of the chronic lymphocytic leukemia scoring system with the monoclonal antibody SN8 (CD79b). Am. J. Clin. Pathol. 1997, 108, 378–382. [Google Scholar] [CrossRef] [PubMed]
- Huh, Y.O.; Schweighofer, C.D.; Ketterling, R.P.; Knudson, R.A.; Vega, F.; Kim, J.E.; Luthra, R.; Keating, M.J.; Medeiros, L.J.; Abruzzo, L.V. Chronic lymphocytic leukemia with t(14;19)(q32;q13) is characterized by atypical morphologic and immunophenotypic features and distinctive genetic features. Am. J. Clin. Pathol. 2011, 135, 686–696. [Google Scholar] [CrossRef] [PubMed]
- Forbes, S.A.; Beare, D.; Boutselakis, H.; Bamford, S.; Bindal, N.; Tate, J.; Cole, C.G.; Ward, S.; Dawson, E.; Ponting, L.; et al. COSMIC: Somatic cancer genetics at high-resolution. Nucleic Acids Res. 2017, 45, D777–D783. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, J.M.; Cooper, D.N.; Schuelke, M.; Seelow, D. MutationTaster2: Mutation prediction for the deep-sequencing age. Nat. Methods 2014, 11, 361–362. [Google Scholar] [CrossRef] [PubMed]
- Sim, N.-L.; Kumar, P.; Hu, J.; Henikoff, S.; Schneider, G.; Ng, P.C. SIFT web server: Predicting effects of amino acid substitutions on proteins. Nucleic Acids Res. 2012, 40, W452–W457. [Google Scholar] [CrossRef]
- Bikos, V.; Darzentas, N.; Hadzidimitriou, A.; Davis, Z.; Hockley, S.; Traverse-Glehen, A.; Algara, P.; Santoro, A.; Gonzalez, D.; Mollejo, M.; et al. Over 30% of patients with splenic marginal zone lymphoma express the same immunoglobulin heavy variable gene: Ontogenetic implications. Leukemia 2012, 26, 1638–1646. [Google Scholar] [CrossRef] [PubMed]
- Agathangelidis, A.; Darzentas, N.; Hadzidimitriou, A.; Brochet, X.; Murray, F.; Yan, X.-J.; Davis, Z.; van Gastel-Mol, E.J.; Tresoldi, C.; Chu, C.C.; et al. Stereotyped B-cell receptors in one-third of chronic lymphocytic leukemia: A molecular classification with implications for targeted therapies. Blood 2012, 119, 4467–4475. [Google Scholar] [CrossRef]
- Döhner, H.; Stilgenbauer, S.; Benner, A.; Leupolt, E.; Kröber, A.; Bullinger, L.; Döhner, K.; Bentz, M.; Lichter, P. Genomic aberrations and survival in chronic lymphocytic leukemia. N. Engl. J. Med. 2000, 343, 1910–1916. [Google Scholar] [CrossRef]
- Puente, X.S.; Beà, S.; Valdés-Mas, R.; Villamor, N.; Gutiérrez-Abril, J.; Martín-Subero, J.I.; Munar, M.; Rubio-Pérez, C.; Jares, P.; Aymerich, M.; et al. Non-coding recurrent mutations in chronic lymphocytic leukaemia. Nature 2015, 526, 519–524. [Google Scholar] [CrossRef]
- Matutes, E.; Oscier, D.; Montalban, C.; Berger, F.; Callet-Bauchu, E.; Dogan, A.; Felman, P.; Franco, V.; Iannitto, E.; Mollejo, M.; et al. Splenic marginal zone lymphoma proposals for a revision of diagnostic, staging and therapeutic criteria. Leukemia 2007, 22, 487–495. [Google Scholar] [CrossRef]
- Delgado, J.; Matutes, E.; Morilla, A.M.; Morilla, R.M.; Owusu-Ankomah, K.A.; Rafiq-Mohammed, F.; del Giudice, I.; Catovsky, D. Diagnostic Significance of CD20 and FMC7 Expression in B-Cell Disorders. Am. J. Clin. Pathol. 2003, 120, 754–759. [Google Scholar] [CrossRef]
- Salido, M.; Baró, C.; Oscier, D.; Stamatopoulos, K.; Dierlamm, J.; Matutes, E.; Traverse-Glehen, A.; Berger, F.; Felman, P.; Thieblemont, C.; et al. Cytogenetic aberrations and their prognostic value in a series of 330 splenic marginal zone B-cell lymphomas: A multicenter study of the Splenic B-Cell Lymphoma Group. Blood 2010, 116, 1479–1488. [Google Scholar] [CrossRef]
- Van den Brand, M.; van Krieken, J.H.J.M. Recognizing nodal marginal zone lymphoma: Recent advances and pitfalls. A systematic review. Haematologica 2013, 98, 1003–1013. [Google Scholar] [CrossRef] [PubMed]
- João, C.; Farinha, P.; da Silva, M.G.; Martins, C.; Crespo, M.; Cabeçadas, J. Cytogenetic abnormalities in MALT lymphomas and their precursor lesions from different organs. A fluorescence in situ hybridization (FISH) study. Histopathology 2007, 50, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Balatti, V.; Bottoni, A.; Palamarchuk, A.; Alder, H.; Rassenti, L.Z.; Kipps, T.J.; Pekarsky, Y.; Croce, C.M. NOTCH1 mutations in CLL associated with trisomy 12. Blood 2012, 119, 329–331. [Google Scholar] [CrossRef] [PubMed]
- Landau, D.A.; Tausch, E.; Taylor-Weiner, A.N.; Stewart, C.; Reiter, J.G.; Bahlo, J.; Kluth, S.; Bozic, I.; Lawrence, M.; Böttcher, S.; et al. Mutations driving CLL and their evolution in progression and relapse. Nature 2015, 526, 525–530. [Google Scholar] [CrossRef] [PubMed]
- Ngo, V.N.; Young, R.M.; Schmitz, R.; Jhavar, S.; Xiao, W.; Lim, K.-H.; Kohlhammer, H.; Xu, W.; Yang, Y.; Zhao, H.; et al. Oncogenically active MYD88 mutations in human lymphoma. Nature 2011, 470, 115–119. [Google Scholar] [CrossRef] [PubMed]
- Parry, M.; Rose-Zerilli, M.J.J.; Ljungström, V.; Gibson, J.; Wang, J.; Walewska, R.; Parker, H.; Parker, A.; Davis, Z.; Gardiner, A.; et al. Genetics and Prognostication in Splenic Marginal Zone Lymphoma: Revelations from Deep Sequencing. Clin. Cancer Res. 2015, 21, 4174–4183. [Google Scholar] [CrossRef] [PubMed]
- Spina, V.; Khiabanian, H.; Messina, M.; Monti, S.; Cascione, L.; Bruscaggin, A.; Spaccarotella, E.; Holmes, A.B.; Arcaini, L.; Lucioni, M.; et al. The genetics of nodal marginal zone lymphoma. Blood 2016, 128, 1362–1373. [Google Scholar] [CrossRef] [PubMed]
- Treon, S.P.; Xu, L.; Yang, G.; Zhou, Y.; Liu, X.; Cao, Y.; Sheehy, P.; Manning, R.J.; Patterson, C.J.; Tripsas, C.; et al. MYD88 L265P Somatic Mutation in Waldenström’s Macroglobulinemia. N. Engl. J. Med. 2012, 367, 826–833. [Google Scholar] [CrossRef] [PubMed]
- Xochelli, A.; Kalpadakis, C.; Gardiner, A.; Baliakas, P.; Vassilakopoulos, T.P.; Mould, S.; Davis, Z.; Stalika, E.; Kanellis, G.; Angelopoulou, M.K.; et al. Clonal B-cell lymphocytosis exhibiting immunophenotypic features consistent with a marginal-zone origin: Is this a distinct entity? Blood 2014, 123, 1199–1206. [Google Scholar] [CrossRef] [PubMed]
- Bennett, J.M.; Catovsky, D.; Daniel, M.T.; Flandrin, G.; Galton, D.A.; Gralnick, H.R.; Sultan, C. Proposals for the classification of chronic (mature) B and T lymphoid leukaemias. French-American-British (FAB) Cooperative Group. J. Clin. Pathol. 1989, 42, 567–584. [Google Scholar] [CrossRef] [PubMed]
- Criel, A.; Michaux, L.; De Wolf-Peeters, C. The concept of typical and atypical chronic lymphocytic leukaemia. Leuk. Lymphoma 1999, 33, 33–45. [Google Scholar] [CrossRef]
- Matutes, E.; Oscier, D.; Garcia-Marco, J.; Ellis, J.; Copplestone, A.; Gillingham, R.; Hamblin, T.; Lens, D.; Swansbury, G.J.; Catovsky, D. Trisomy 12 defines a group of CLL with atypical morphology: Correlation between cytogenetic, clinical and laboratory features in 544 patients. Br. J. Haematol. 1996, 92, 382–388. [Google Scholar] [CrossRef] [PubMed]
- Hallek, M. Chronic lymphocytic leukemia: 2017 update on diagnosis, risk stratification, and treatment. Am. J. Hematol. 2017, 92, 946–965. [Google Scholar] [CrossRef] [PubMed]
- Baseggio, L.; Traverse-Glehen, A.; Callet-Bauchu, E.; Morel, D.; Magaud, J.P.; Berger, F.; Salles, G.; Felman, P. Relevance of a scoring system including CD11c expression in the identification of splenic diffuse red pulp small B-cell lymphoma (SRPL). Hematol. Oncol. 2011, 29, 47–51. [Google Scholar] [CrossRef]
- Stelzer, G.T.; Marti, G.; Hurley, A.; McCoy, P.; Lovett, E.J.; Schwartz, A. U.S.-Canadian consensus recommendations on the immunophenotypic analysis of hematologic neoplasia by flow cytometry: Standardization and validation of laboratory procedures. Cytometry 1997, 30, 214–230. [Google Scholar] [CrossRef]
- Callet-Bauchu, E.; Rimokh, R.; Tigaud, I.; Pagès, J.; Gazzo, S.; Bastion, Y.; Sebban, C.; Magaud, J.P.; Coiffier, B.; Felman, P. dic(4;17)(p11;p11): A new recurrent chromosomal abnormality in chronic B-lymphoid disorders. Genes Chromosomes Cancer 1996, 17, 185–190. [Google Scholar] [CrossRef]
- McGowan-Jordan, J.; Simons, A.; Schmid, M. ISCN 2016: An International System for Human Cytogenomic Nomenclature (2016); Karger Medical Scientific: Basel, Switzerland, 2016. [Google Scholar]
- Van Dongen, J.J.M.; Langerak, A.W.; Brüggemann, M.; Evans, P.A.S.; Hummel, M.; Lavender, F.L.; Delabesse, E.; Davi, F.; Schuuring, E.; García-Sanz, R.; et al. Design and standardization of PCR primers and protocols for detection of clonal immunoglobulin and T-cell receptor gene recombinations in suspect lymphoproliferations: Report of the BIOMED-2 Concerted Action BMH4-CT98-3936. Leukemia 2003, 17, 2257–2317. [Google Scholar] [CrossRef]
- Lefranc, M.-P.; Giudicelli, V.; Ginestoux, C.; Jabado-Michaloud, J.; Folch, G.; Bellahcene, F.; Wu, Y.; Gemrot, E.; Brochet, X.; Lane, J.; et al. IMGT, the international ImMunoGeneTics information system. Nucleic Acids Res. 2009, 37, D1006–D1012. [Google Scholar] [CrossRef] [PubMed]
- Brochet, X.; Lefranc, M.-P.; Giudicelli, V. IMGT/V-QUEST: The highly customized and integrated system for IG and TR standardized V-J and V-D-J sequence analysis. Nucleic Acids Res. 2008, 36, W503–W508. [Google Scholar] [CrossRef] [PubMed]
- Hallek, M.; Cheson, B.D.; Catovsky, D.; Caligaris-Cappio, F.; Dighiero, G.; Döhner, H.; Hillmen, P.; Keating, M.J.; Montserrat, E.; Rai, K.R.; et al. Guidelines for the diagnosis and treatment of chronic lymphocytic leukemia: A report from the International Workshop on Chronic Lymphocytic Leukemia updating the National Cancer Institute–Working Group 1996 guidelines. Blood 2008, 111, 5446–5456. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| UPN | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 | 16 | 17 | 18 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Clinical features | Sex | F | F | M | F | M | M | F | F | F | F | M | M | M | M | F | M | F | F |
| Age (years) | 65 | 74 | 61 | 79 | 70 | 86 | 84 | 92 | 62 | 82 | 66 | 69 | 78 | 81 | 86 | 55 | 81 | 90 | |
| ECOG score | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | |
| Blood count | Lymphocytes (G/L) | 8.39 | 7.61 | 8.41 | 21.32 | 25.69 | 11.61 | 13.23 | 9.66 | 5.87 | 15.63 | 5.64 | 9.60 | 7.45 | 9.50 | 11.13 | 5.30 | 7.80 | 14.20 |
| Cytology | CLL-like | CLL-like | At. | CLL-like | / | CLL-like | CLL-like | At. | CLL-like | CLL-like | CLL-like | CLL-like | At. | CLL-like | CLL-like | CLL-like | CLL-like | At. | |
| Neutrophils (G/L) | 4.46 | 6.30 | 6.95 | 7.11 | 7.01 | 5.52 | 4.72 | 4.61 | 3.95 | 6.76 | 4.98 | 4.50 | 5.23 | 2.38 | 6.91 | 9.70 | 5.45 | 2.96 | |
| Hemoglobin (g/L) | 130 | 140 | 154 | 130 | 143 | 136 | 141 | 132 | 122 | 142 | 158 | 150 | 152 | 129 | 124 | 146 | 131 | 131 | |
| Platelets (G/L) | 257 | 206 | 184 | 386 | 154 | 208 | 246 | 207 | 215 | 240 | 155 | 277 | 247 | 130 | 345 | 468 | 326 | 166 | |
| RMH score | RMH score | 2 | 2 | 1 | 1 | 1 | 1 | 2 | 1 | 2 | 2 | 1 | 2 | 1 | 1 | 2 | 2 | 2 | 2 |
| CD5 | |||||||||||||||||||
| CD23 | |||||||||||||||||||
| Chain | λ | κ | λ | κ | λ | λ | λ | κ | λ | κ | κ | λ | κ | κ | λ | κ | κ | κ | |
| Fmc7 | |||||||||||||||||||
| CD22 | |||||||||||||||||||
| Other immunophenotyping markers | CD79b | ||||||||||||||||||
| CD43 | |||||||||||||||||||
| CD20 | |||||||||||||||||||
| Cytogenetic features | 11q deletion | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − |
| Trisomy 12 | + | + | + | + | − | − | − | + | + | + | − | − | + | + | + | + | + | + | |
| 13q deletion | + | + | + | − | + | + | + | + | − | − | + | + | − | − | − | − | − | − | |
| 17p deletion | − | − | − | − | + | − | + | − | − | − | − | − | − | − | − | − | − | − | |
| Clinical outcome | Treatment | No | No | RC | aR-CHOP | Splen. | No | RC | No | No | No | No | No | No | C | No | No | No | No |
| Response | / | / | CR | PR | PR | / | CR | / | / | / | / | / | / | / | / | / | / | / | |
| Death | No | No | No | No | No | No | Yes | No | No | No | No | No | No | Yes | No | Yes | No | No | |
| Follow-up (months) | 118 | 110 | 124 | 18 | 34 | 69 | 73 | 2 | 103 | 111 | 71 | 40 | 42 | 53 | 34 | 21 | 27 | 25 |
| UPN | ATM | BCOR | BIRC3 | CARD11 | LYN | MYD88 | SF3B1 | TP53 | TRAF2 |
|---|---|---|---|---|---|---|---|---|---|
| 1 | |||||||||
| 2 | |||||||||
| 3 | |||||||||
| 5 | |||||||||
| 6 | |||||||||
| 7 | |||||||||
| 10 | |||||||||
| 14 | |||||||||
| UPN | Gene | NM | VAF (%) | cDNA Change | Protein Change | Mutation Type | COSMIC Database | SIFT Prediction | Mutation Taster Prediction |
| 1 | BCOR | NM_001123385.1 | 49,7 | c.3863C>G | p.S1288C | Missense | Not described | Deleterious | Disease causing |
| 3 | TRAF2 | NM_021138.3 | 7,2 | c.1210G>A | p.G404R | Missense | Not described | Deleterious | Disease causing |
| 5 | TP53 | NM_000546.4 | 44,5 | c.701A>G | p.Y234C | Missense | Described | Deleterious | Disease causing |
| 5 | TP53 | NM_000546.4 | 7,2 | c.733G>T | p.G245C | Missense | Described | Deleterious | Disease causing |
| 6 | TP53 | NM_000546.4 | 5,4 | c.742C>T | p.R248W | Missense | Described | Deleterious | Disease causing |
| 6 | SF3B1 | NM_012433.2 | 17,3 | c.2584G>A | p.E862K | Missense | Described | Deleterious | Disease causing |
| 6 | MYD88 | NM_002468.4 | 19,3 | c.656C>G | p.S219C | Missense | Described | Deleterious | Disease causing |
| 6 | LYN | NM_002350.3 | 5,0 | c.602_603del | p.F201Sfs*8 | Frameshift | Not described | Frameshift | Frameshift |
| 7 | TP53 | NM_000546.4 | 39,5 | c.536A>C | p.H179P | Missense | Described | Deleterious | Disease causing |
| 7 | TP53 | NM_000546.4 | 12,7 | c.637C>G | p.R213G | Missense | Described | Deleterious | Disease causing |
| 10 | ATM | NM_000051.3 | 17,7 | c.8560C>T | p.R2854C | Missense | Described | Deleterious | Disease causing |
| 10 | BIRC3 | NM_001165.3 | 6,8 | c.1284_1288del | p.E429Gfs*7 | Frameshift | Described | Frameshift | Frameshift |
| 10 | BIRC3 | NM_001165.3 | 3,0 | c.1639del | p.Q547Nfs*21 | Frameshift | Described | Frameshift | Frameshift |
| 10 | CARD11 | NM_032415.4 | 43,2 | c.2060C>T | p.A687V | Missense | Described | Tolerated | Disease causing |
| 14 | MYD88 | NM_002468.4 | 27,7 | c.656C>G | p.S219C | Missense | Described | Deleterious | Disease causing |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Degaud, M.; Baseggio, L.; Grange, B.; Manzoni, D.; Huet, S.; Callet-Bauchu, E.; Traverse-Glehen, A.; Davi, F.; Ghesquières, H.; Salles, G.; et al. Unclassifiable Isolated Monoclonal Lymphocytosis: Comprehensive Description of a Retrospective Cohort. Cancers 2019, 11, 1495. https://doi.org/10.3390/cancers11101495
Degaud M, Baseggio L, Grange B, Manzoni D, Huet S, Callet-Bauchu E, Traverse-Glehen A, Davi F, Ghesquières H, Salles G, et al. Unclassifiable Isolated Monoclonal Lymphocytosis: Comprehensive Description of a Retrospective Cohort. Cancers. 2019; 11(10):1495. https://doi.org/10.3390/cancers11101495
Chicago/Turabian StyleDegaud, Michaël, Lucile Baseggio, Béatrice Grange, Delphine Manzoni, Sarah Huet, Evelyne Callet-Bauchu, Alexandra Traverse-Glehen, Frédéric Davi, Hervé Ghesquières, Gilles Salles, and et al. 2019. "Unclassifiable Isolated Monoclonal Lymphocytosis: Comprehensive Description of a Retrospective Cohort" Cancers 11, no. 10: 1495. https://doi.org/10.3390/cancers11101495
APA StyleDegaud, M., Baseggio, L., Grange, B., Manzoni, D., Huet, S., Callet-Bauchu, E., Traverse-Glehen, A., Davi, F., Ghesquières, H., Salles, G., & Sujobert, P. (2019). Unclassifiable Isolated Monoclonal Lymphocytosis: Comprehensive Description of a Retrospective Cohort. Cancers, 11(10), 1495. https://doi.org/10.3390/cancers11101495