CNOT2 Is Critically Involved in Atorvastatin Induced Apoptotic and Autophagic Cell Death in Non-Small Cell Lung Cancers


 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
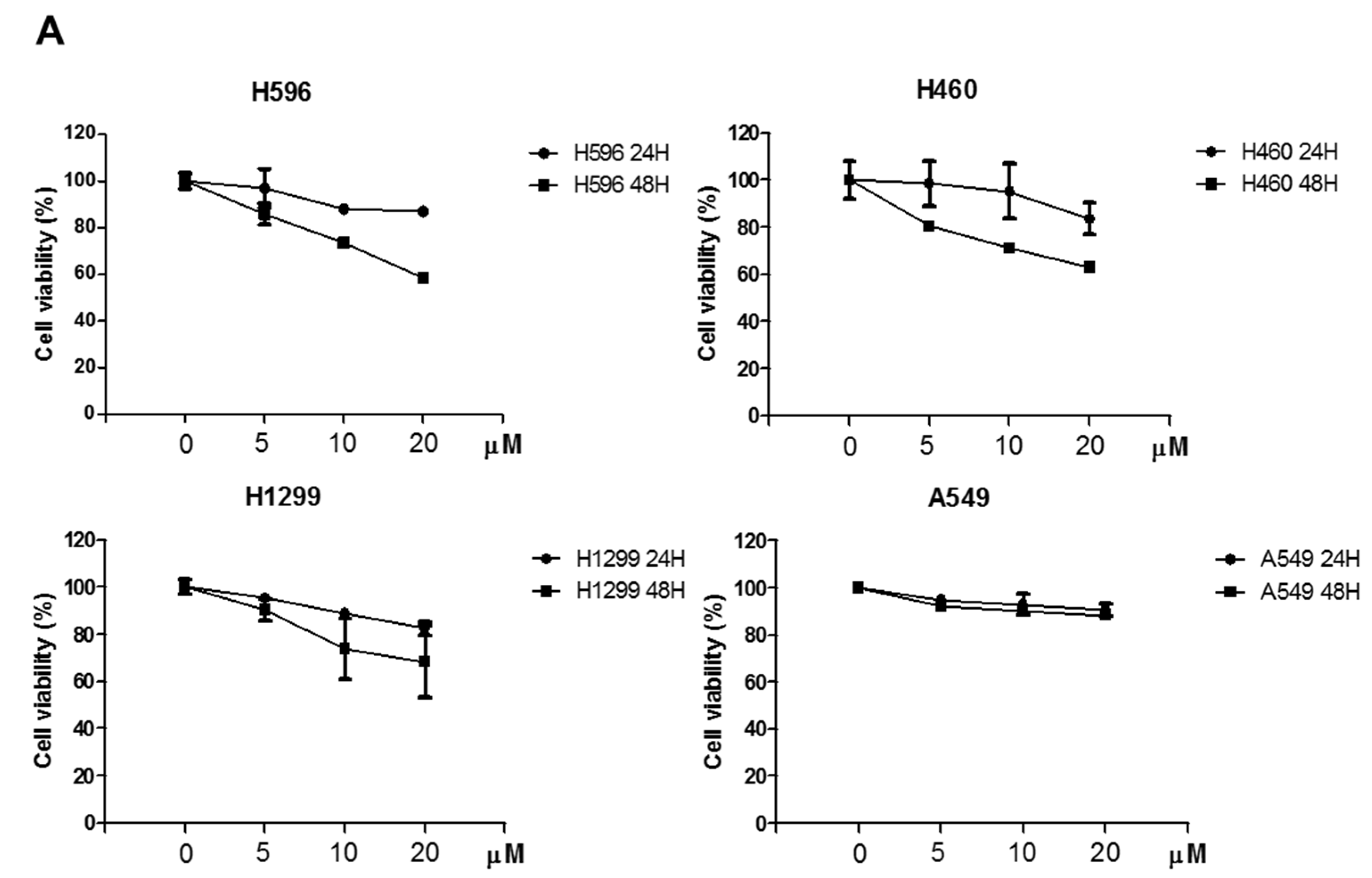
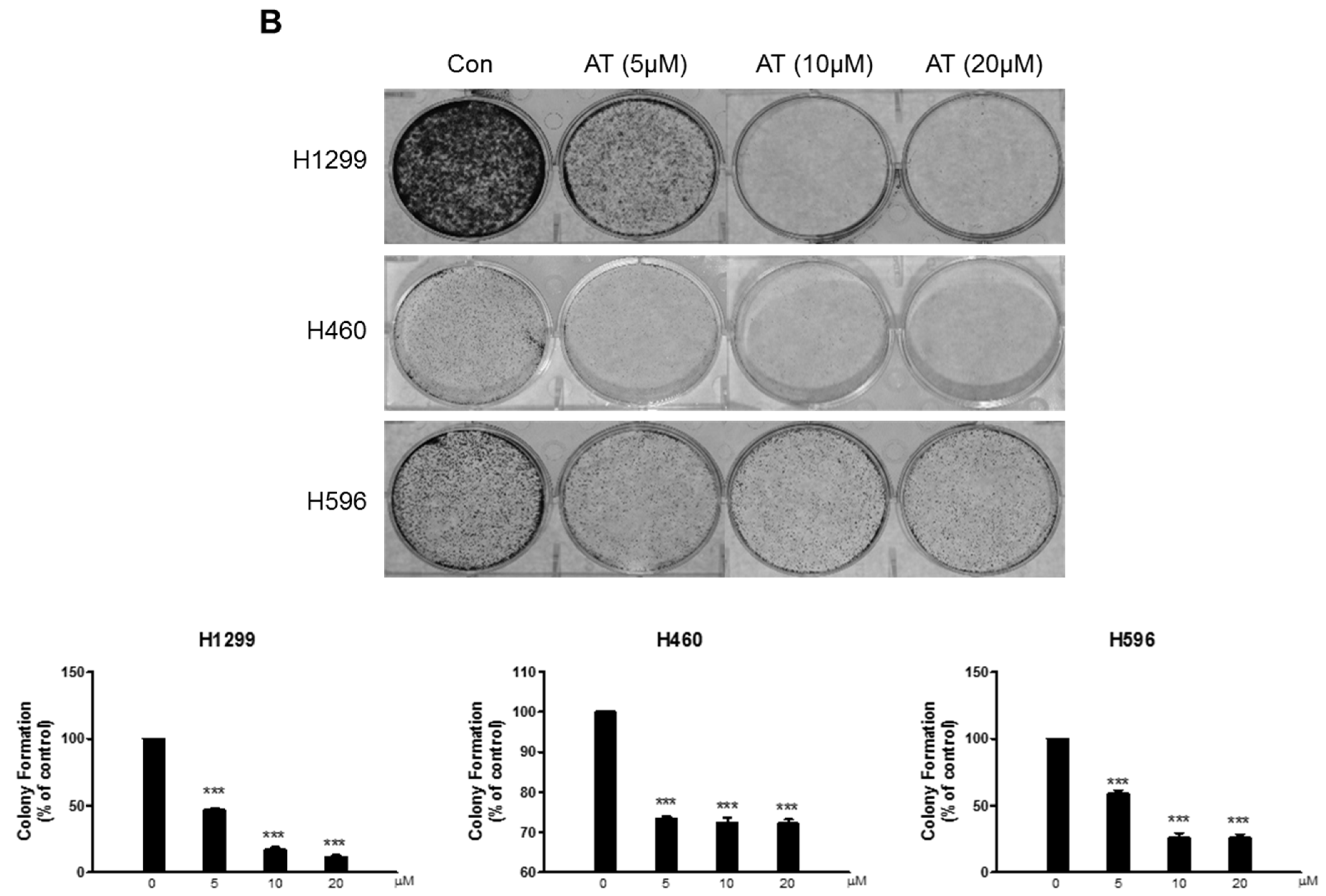
2.1. Effects of Atorvastatin on Cytotoxicity in H596, H460, and H1299 Cells
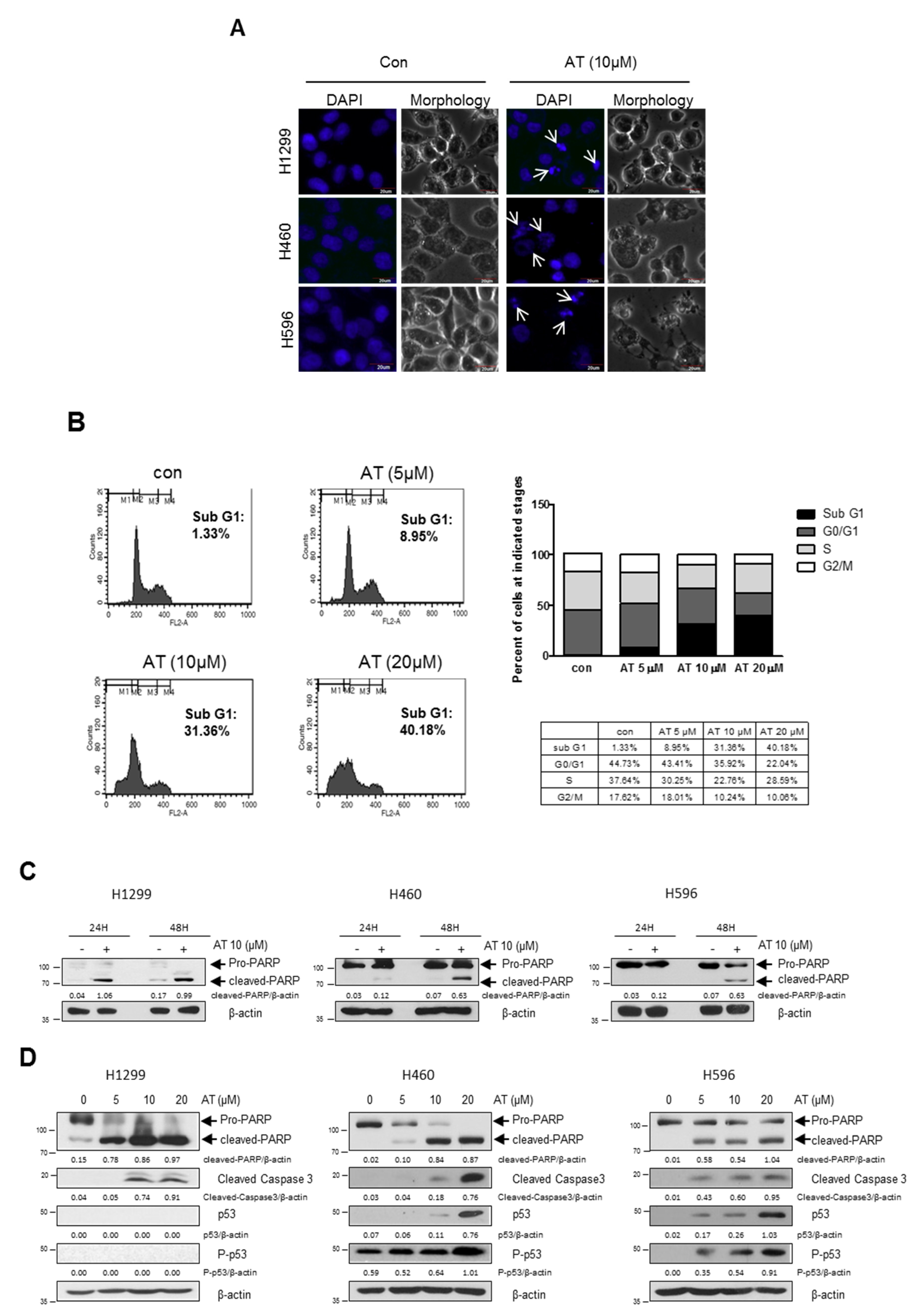
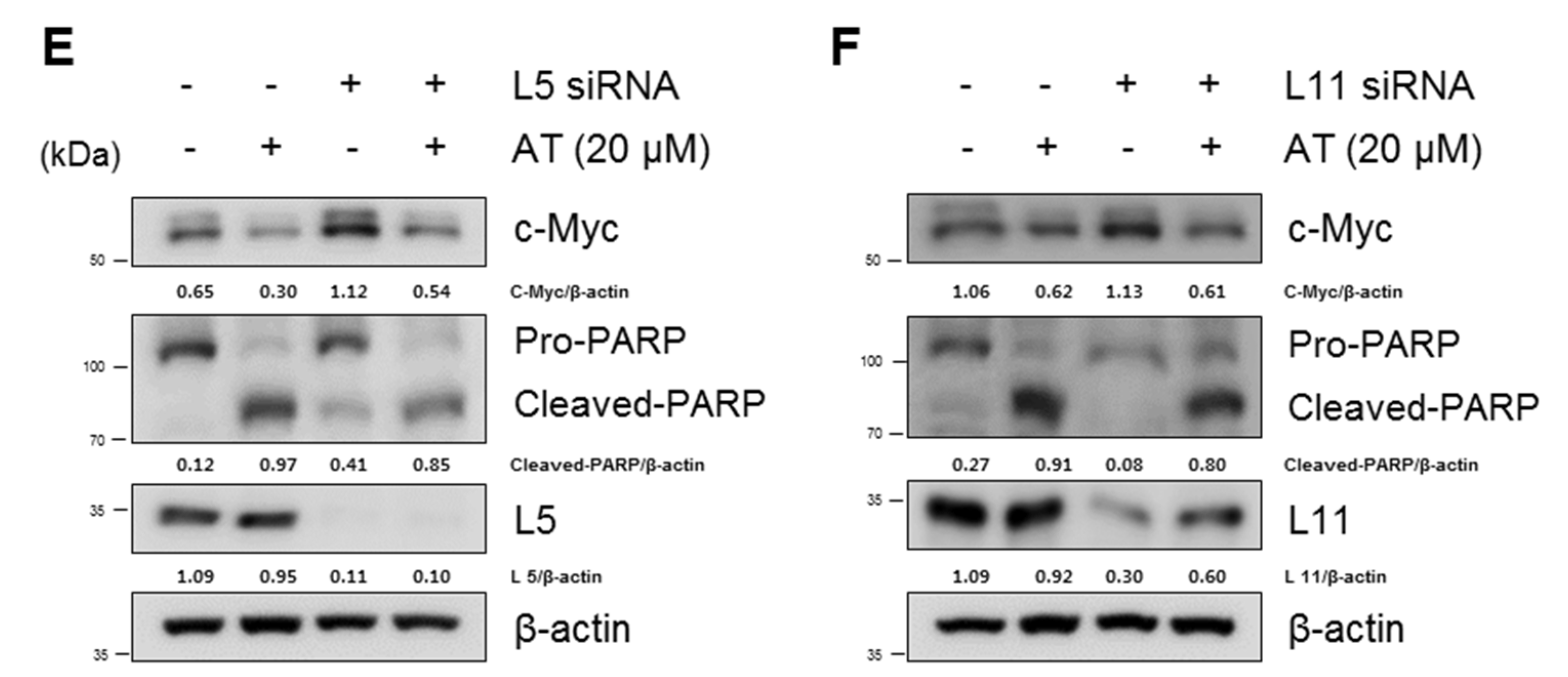
2.2. Atorvastatin Induced Apoptosis via Ribosomal Protein L5 and L11 in NSCLCs
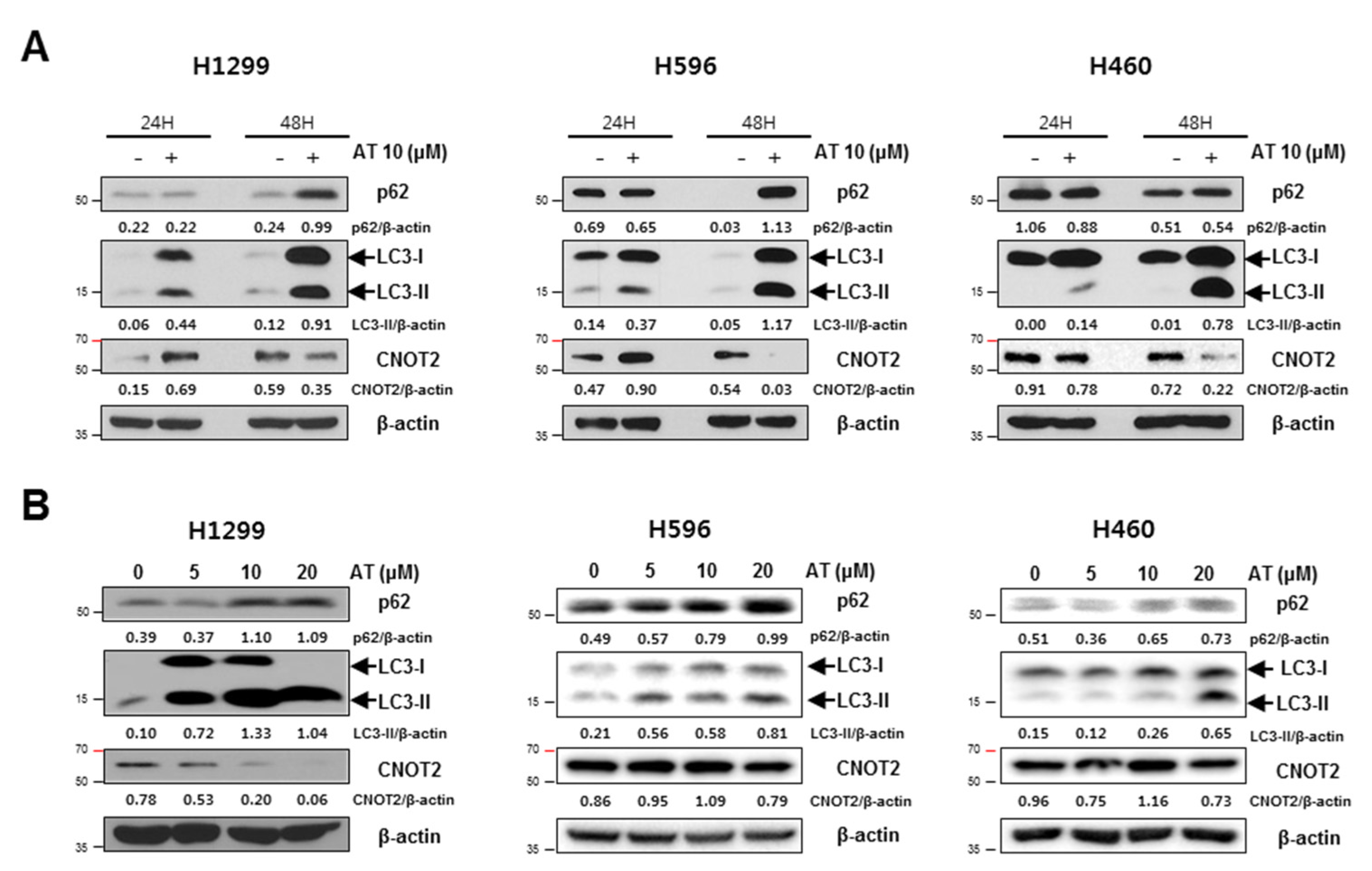
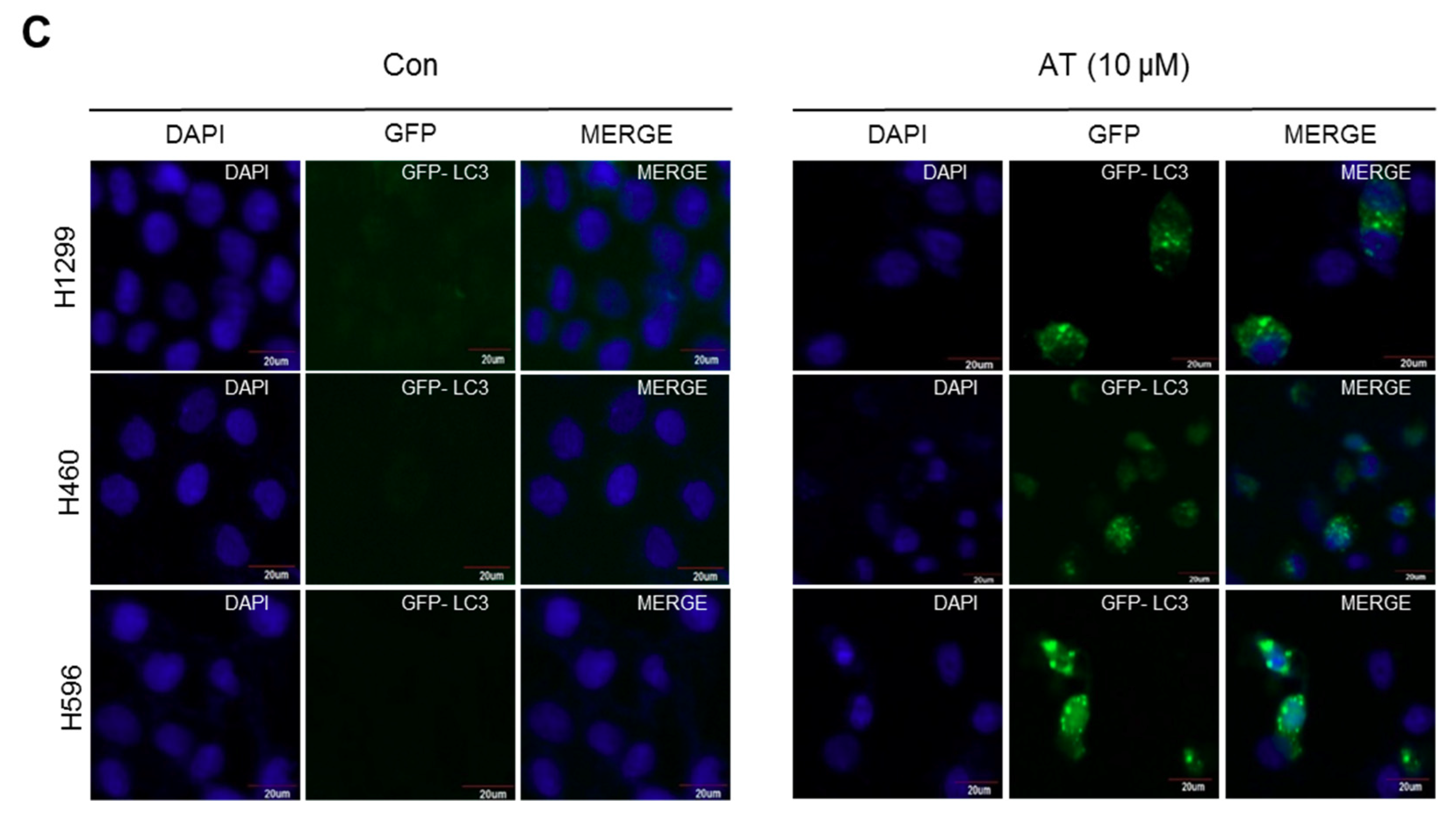
2.3. Atorvastatin Induced Autophagy in H596, H460, and H1299 Cells
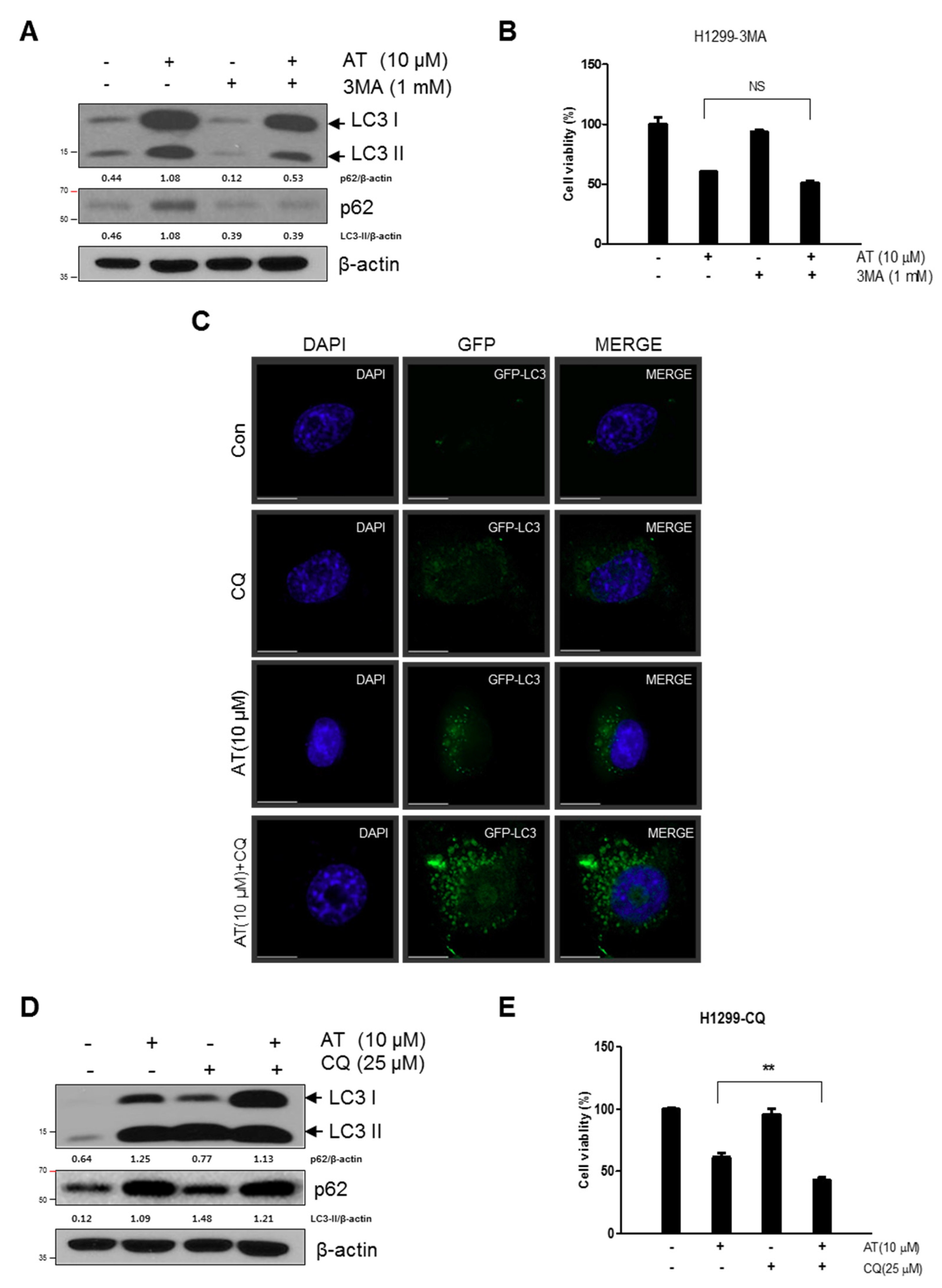
2.4. Late Stage Autophagy Inhibitor CQ, but not 3-MA, Enhanced Cytotoxicity and Decreased p62 and Activated LC3II in Atorvastatin Treated H1299 Cells
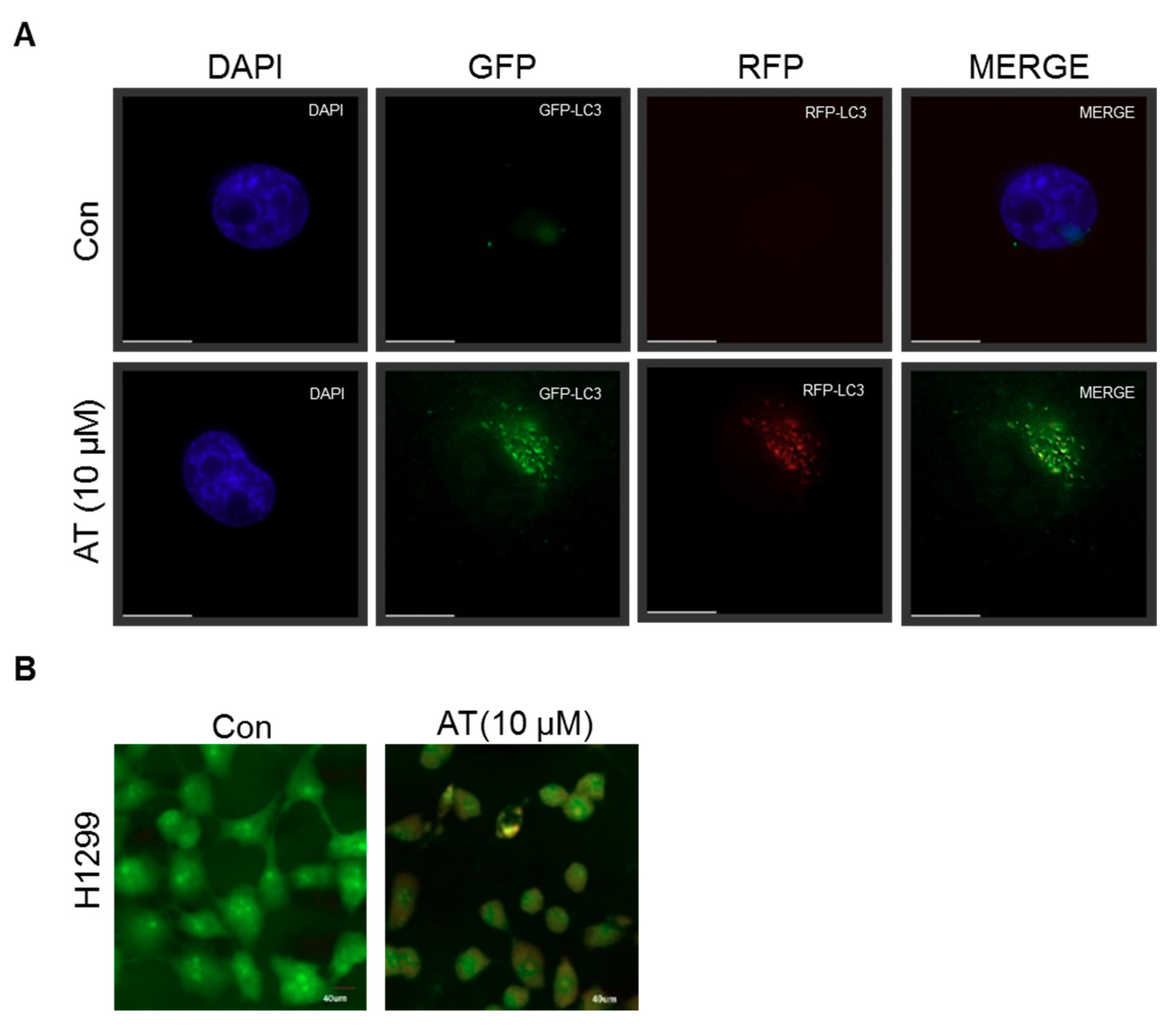
2.5. Atorvastatin Induced Impaired Autophagy in NSCLCs
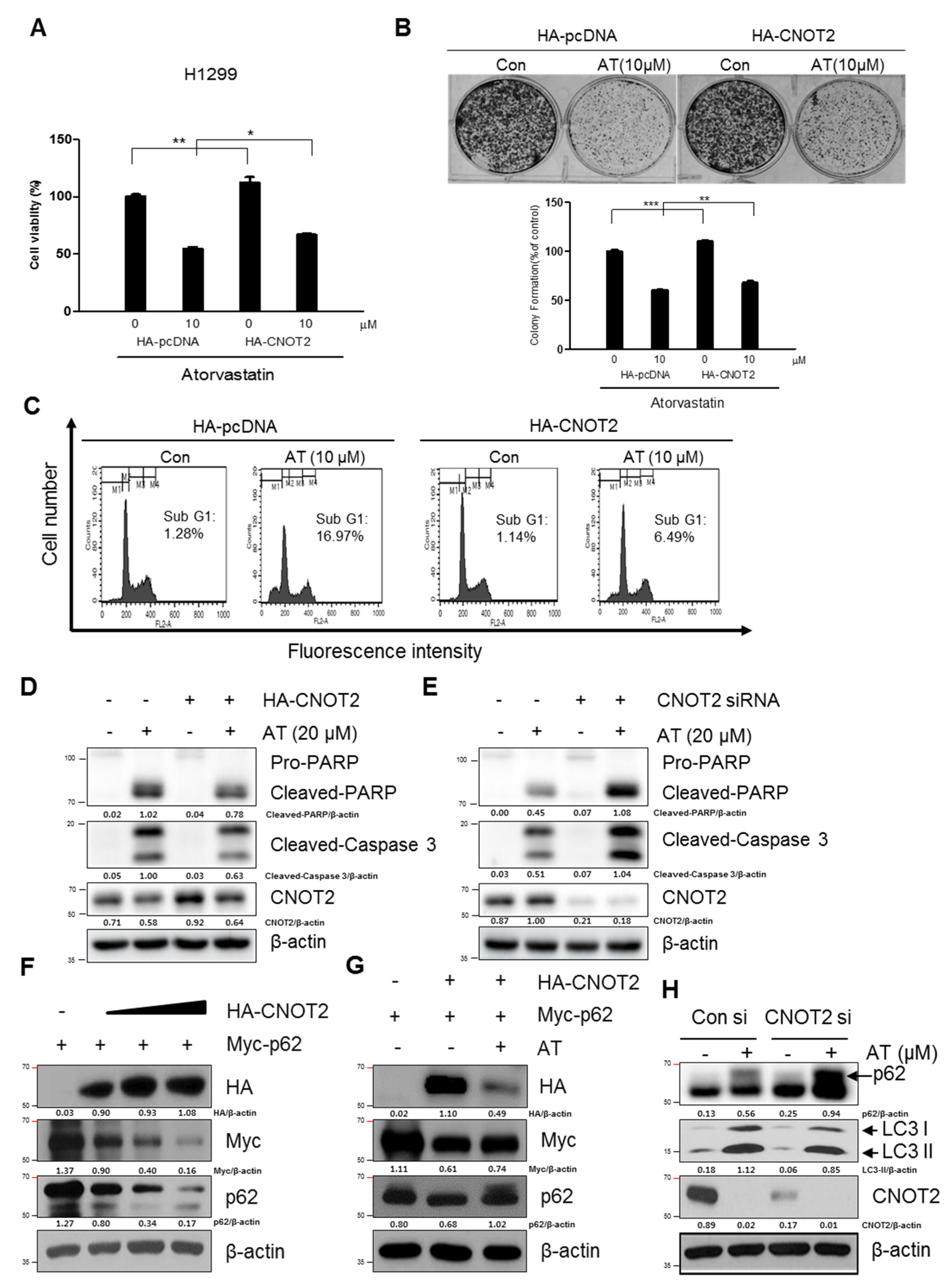
2.6. Pivotal Role of CNOT2 in Atorvastatin Induced Apoptotic and Apoptotic and Autophagic Cell Death in H1299 Cells
3. Discussion
4. Materials and Methods
4.1. Chemical and Reagents
4.2. Cell Culture
4.3. Cytotoxicity
4.4. Colony Formation Assay
4.5. Cell Cycle Analysis
4.6. Apoptosis Detection by DAPI Staining
4.7. Immunoblot Analysis
4.8. Detection of Acidic Vesicular Organelles
4.9. Autophagic Flux Assay
4.10. Immunofluorescence Assay
4.11. Detection of Acidic Vesicular Organelles
4.12. RNA Interference
4.13. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hirsch, F.R.; Scagliotti, G.V.; Mulshine, J.L.; Kwon, R.; Curran, W.J., Jr.; Wu, Y.L.; Paz-Ares, L. Lung cancer: current therapies and new targeted treatments. Lancet 2017, 389, 299–311. [Google Scholar] [CrossRef]
- Castellanos, E.; Feld, E.; Horn, L. Driven by Mutations: The Predictive Value of Mutation Subtype in EGFR-Mutated Non-Small Cell Lung Cancer. J. Thorac. Oncol. 2017, 12, 612–623. [Google Scholar] [CrossRef] [PubMed]
- Mayekar, M.K.; Bivona, T.G. Current Landscape of Targeted Therapy in Lung Cancer. Clinical pharmacology and therapeutics 2017, 102, 757–764. [Google Scholar] [CrossRef] [PubMed]
- Mishra, A.P.; Salehi, B.; Sharifi-Rad, M.; Pezzani, R.; Kobarfard, F.; Sharifi-Rad, J.; Nigam, M. Programmed Cell Death, from a Cancer Perspective: An Overview. Mol. Diagn. Ther. 2018, 22, 281–295. [Google Scholar] [CrossRef] [PubMed]
- Bhat, P.; Kriel, J.; Shubha Priya, B.; Basappa, S.; Shivananju, N.S.; Loos, B. Modulating autophagy in cancer therapy: Advancements and challenges for cancer cell death sensitization. Biochem. Pharmacol. 2018, 147, 170–182. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Kang, R.; Berghe, T.V.; Vandenabeele, P.; Kroemer, G. The molecular machinery of regulated cell death. Cell Res. 2019, 29, 347–364. [Google Scholar] [CrossRef]
- Barth, S.; Glick, D.; Macleod, K.F. Autophagy: assays and artifacts. J. Pathol. 2010, 221, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Turcios, L.; Chacon, E.; Garcia, C.; Eman, P.; Cornea, V.; Jiang, J.; Spear, B.; Liu, C.; Watt, D.S.; Marti, F.; et al. Autophagic flux modulation by Wnt/beta-catenin pathway inhibition in hepatocellular carcinoma. PLoS ONE 2019, 14, e0212538. [Google Scholar] [CrossRef] [PubMed]
- Russo, M.; Russo, G.L. Autophagy inducers in cancer. Biochem. Pharmacol. 2018, 153, 51–61. [Google Scholar] [CrossRef]
- Yun, S.M.; Jung, J.H.; Jeong, S.J.; Sohn, E.J.; Kim, B.; Kim, S.H. Tanshinone IIA induces autophagic cell death via activation of AMPK and ERK and inhibition of mTOR and p70 S6K in KBM-5 leukemia cells. Phytother. Res. 2014, 28, 458–464. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Klionsky, D.J. Regulation mechanisms and signaling pathways of autophagy. Annu. Rev. Genet. 2009, 43, 67–93. [Google Scholar] [CrossRef]
- Ishii, T.; Itoh, K.; Takahashi, S.; Sato, H.; Yanagawa, T.; Katoh, Y.; Bannai, S.; Yamamoto, M. Transcription factor Nrf2 coordinately regulates a group of oxidative stress-inducible genes in macrophages. J. Biol. Chem. 2000, 275, 16023–16029. [Google Scholar] [CrossRef] [PubMed]
- Bialik, S.; Dasari, S.K.; Kimchi, A. Autophagy-dependent cell death-where, how and why a cell eats itself to death. J. Cell Sci. 2018, 131. [Google Scholar] [CrossRef] [PubMed]
- Zwartjes, C.G.; Jayne, S.; van den Berg, D.L.; Timmers, H.T. Repression of promoter activity by CNOT2, a subunit of the transcription regulatory Ccr4-not complex. J. Biol. Chem. 2004, 279, 10848–10854. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.; Lamark, T.; Sjottem, E.; Larsen, K.B.; Awuh, J.A.; Overvatn, A.; McMahon, M.; Hayes, J.D.; Johansen, T. p62/SQSTM1 is a target gene for transcription factor NRF2 and creates a positive feedback loop by inducing antioxidant response element-driven gene transcription. J. Biol. Chem. 2010, 285, 22576–22591. [Google Scholar] [CrossRef] [PubMed]
- Ito, K.; Inoue, T.; Yokoyama, K.; Morita, M.; Suzuki, T.; Yamamoto, T. CNOT2 depletion disrupts and inhibits the CCR4-NOT deadenylase complex and induces apoptotic cell death. Genes Cells 2011, 16, 368–379. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Hou, J.; Zhang, J.; An, Y.; Zhang, X.; Yue, L.; Liu, J.; Li, X. Atorvastatin synergizes with IFN-gamma in treating human non-small cell lung carcinomas via potent inhibition of RhoA activity. Eur. J. Pharmacol. 2012, 682, 161–170. [Google Scholar] [CrossRef]
- Elmadhun, N.Y.; Lassaletta, A.D.; Chu, L.M.; Liu, Y.; Feng, J.; Sellke, F.W. Atorvastatin increases oxidative stress and modulates angiogenesis in Ossabaw swine with the metabolic syndrome. J. Thorac. Cardiovasc. Surg. 2012, 144, 1486–1493. [Google Scholar] [CrossRef]
- Kang, M.; Jeong, C.W.; Ku, J.H.; Kwak, C.; Kim, H.H. Inhibition of autophagy potentiates atorvastatin-induced apoptotic cell death in human bladder cancer cells in vitro. Int. J. Mol. Sci. 2014, 15, 8106–8121. [Google Scholar] [CrossRef] [PubMed]
- Faraji, F.; Hu, Y.; Yang, H.H.; Lee, M.P.; Winkler, G.S.; Hafner, M.; Hunter, K.W. Post-transcriptional Control of Tumor Cell Autonomous Metastatic Potential by CCR4-NOT Deadenylase CNOT7. PLoS Genet. 2016, 12, e1005820. [Google Scholar] [CrossRef]
- Ruan, Y.; Hu, K.; Chen, H. Autophagy inhibition enhances isorhamnetininduced mitochondriadependent apoptosis in nonsmall cell lung cancer cells. Mol. Med. Rep. 2015, 12, 5796–5806. [Google Scholar] [CrossRef] [PubMed]
- Botchkarev, V.A.; Flores, E.R. p53/p63/p73 in the epidermis in health and disease. Cold Spring Harb. Perspect Med. 2014, 4. [Google Scholar] [CrossRef] [PubMed]
- Vousden, K.H.; Lane, D.P. p53 in health and disease. Nat. Rev. Mol. Cell Biol. 2007, 8, 275–283. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Lu, H. Signaling to p53: ribosomal proteins find their way. Cancer Cell 2009, 16, 369–377. [Google Scholar] [CrossRef]
- Larsen, B.D.; Sorensen, C.S. The caspase-activated DNase: apoptosis and beyond. FEBS J. 2017, 284, 1160–1170. [Google Scholar] [CrossRef]
- Schuler, M.; Green, D.R. Mechanisms of p53-dependent apoptosis. Biochem. Soc. Trans. 2001, 29, 684–688. [Google Scholar] [CrossRef]
- Qian, Y.; Qiu, M.; Wu, Q.; Tian, Y.; Zhang, Y.; Gu, N.; Li, S.; Xu, L.; Yin, R. Enhanced cytotoxic activity of cetuximab in EGFR-positive lung cancer by conjugating with gold nanoparticles. Sci. Rep. 2014, 4, 7490. [Google Scholar] [CrossRef]
- Liao, J.M.; Zhou, X.; Gatignol, A.; Lu, H. Ribosomal proteins L5 and L11 co-operatively inactivate c-Myc via RNA-induced silencing complex. Oncogene 2014, 33, 4916–4923. [Google Scholar] [CrossRef]
- Derenzini, M.; Montanaro, L.; Trere, D. Ribosome biogenesis and cancer. Acta. Histochemica 2017, 119, 190–197. [Google Scholar] [CrossRef]
- Pelletier, J.; Thomas, G.; Volarevic, S. Ribosome biogenesis in cancer: new players and therapeutic avenues. Nat. Rev. Cancer 2018, 18, 51–63. [Google Scholar] [CrossRef]
- Zhou, X.; Hao, Q.; Zhang, Q.; Liao, J.M.; Ke, J.W.; Liao, P.; Cao, B.; Lu, H. Ribosomal proteins L11 and L5 activate TAp73 by overcoming MDM2 inhibition. Cell Death Differ. 2015, 22, 755–766. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.; Tackmann, N.R.; Liu, S.; Yang, J.; Dong, J.; Wu, C.; Cox, A.D.; Zhang, Y. RPL23 Links Oncogenic RAS Signaling to p53-Mediated Tumor Suppression. Cancer Res. 2016, 76, 5030–5039. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.H.; Liao, J.M.; Zhang, Q.; Zeng, S.; Nguyen, D.; Hao, Q.; Zhou, X.; Cao, B.; Kim, S.H.; Lu, H. Inauhzin(c) inactivates c-Myc independently of p53. Cancer Biol. Ther. 2015, 16, 412–419. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.H.; Lee, H.; Kim, J.H.; Sim, D.Y.; Ahn, H.; Kim, B.; Chang, S.; Kim, S.H. p53-Dependent Apoptotic Effect of Puromycin via Binding of Ribosomal Protein L5 and L11 to MDM2 and its Combination Effect with RITA or Doxorubicin. Cancers 2019, 11. [Google Scholar] [CrossRef]
- Chun, Y.; Kim, J. Autophagy: An Essential Degradation Program for Cellular Homeostasis and Life. Cells 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Yoshii, S.R.; Mizushima, N. Monitoring and Measuring Autophagy. Int. J. Mol. Sci. 2017, 18. [Google Scholar] [CrossRef]
- Denton, D.; Kumar, S. Autophagy-dependent cell death. Cell Death Differ. 2018, 26, 605–616. [Google Scholar] [CrossRef]
- Vegliante, R.; Ciriolo, M.R. Autophagy and Autophagic Cell Death: Uncovering New Mechanisms Whereby Dehydroepiandrosterone Promotes Beneficial Effects on Human Health. Vitam. Horm. 2018, 108, 273–307. [Google Scholar]
- Lou, J.S.; Bi, W.C.; Chan, G.K.L.; Jin, Y.; Wong, C.W.; Zhou, Z.Y.; Wang, H.Y.; Yao, P.; Dong, T.T.X.; Tsim, K.W.K. Ginkgetin induces autophagic cell death through p62/SQSTM1-mediated autolysosome formation and redox setting in non-small cell lung cancer. Oncotarget 2017, 8, 93131–93148. [Google Scholar] [CrossRef]
- Zhao, X.; Wei, S.; Li, Z.; Lin, C.; Zhu, Z.; Sun, D.; Bai, R.; Qian, J.; Gao, X.; Chen, G.; et al. Autophagic flux blockage in alveolar epithelial cells is essential in silica nanoparticle-induced pulmonary fibrosis. Cell Death Dis. 2019, 10, 127. [Google Scholar] [CrossRef]
- Zheng, X.; Dumitru, R.; Lackford, B.L.; Freudenberg, J.M.; Singh, A.P.; Archer, T.K.; Jothi, R.; Hu, G. Cnot1, Cnot2, and Cnot3 maintain mouse and human ESC identity and inhibit extraembryonic differentiation. Stem Cells 2012, 30, 910–922. [Google Scholar] [CrossRef] [PubMed]
- Sohn, E.J.; Jung, D.B.; Lee, H.; Han, I.; Lee, J.; Lee, H.; Kim, S.H. CNOT2 promotes proliferation and angiogenesis via VEGF signaling in MDA-MB-231 breast cancer cells. Cancer Lett. 2018, 412, 88–98. [Google Scholar] [CrossRef] [PubMed]









© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, J.; Jung, J.H.; Hwang, J.; Park, J.E.; Kim, J.-H.; Park, W.Y.; Suh, J.Y.; Kim, S.-H. CNOT2 Is Critically Involved in Atorvastatin Induced Apoptotic and Autophagic Cell Death in Non-Small Cell Lung Cancers. Cancers 2019, 11, 1470. https://doi.org/10.3390/cancers11101470
Lee J, Jung JH, Hwang J, Park JE, Kim J-H, Park WY, Suh JY, Kim S-H. CNOT2 Is Critically Involved in Atorvastatin Induced Apoptotic and Autophagic Cell Death in Non-Small Cell Lung Cancers. Cancers. 2019; 11(10):1470. https://doi.org/10.3390/cancers11101470
Chicago/Turabian StyleLee, Jihyun, Ji Hoon Jung, Jisung Hwang, Ji Eon Park, Ju-Ha Kim, Woon Yi Park, Jin Young Suh, and Sung-Hoon Kim. 2019. "CNOT2 Is Critically Involved in Atorvastatin Induced Apoptotic and Autophagic Cell Death in Non-Small Cell Lung Cancers" Cancers 11, no. 10: 1470. https://doi.org/10.3390/cancers11101470
APA StyleLee, J., Jung, J. H., Hwang, J., Park, J. E., Kim, J.-H., Park, W. Y., Suh, J. Y., & Kim, S.-H. (2019). CNOT2 Is Critically Involved in Atorvastatin Induced Apoptotic and Autophagic Cell Death in Non-Small Cell Lung Cancers. Cancers, 11(10), 1470. https://doi.org/10.3390/cancers11101470