Data Fusion Techniques for the Integration of Multi-Domain Genomic Data from Uveal Melanoma



Abstract
1. Introduction
2. Results
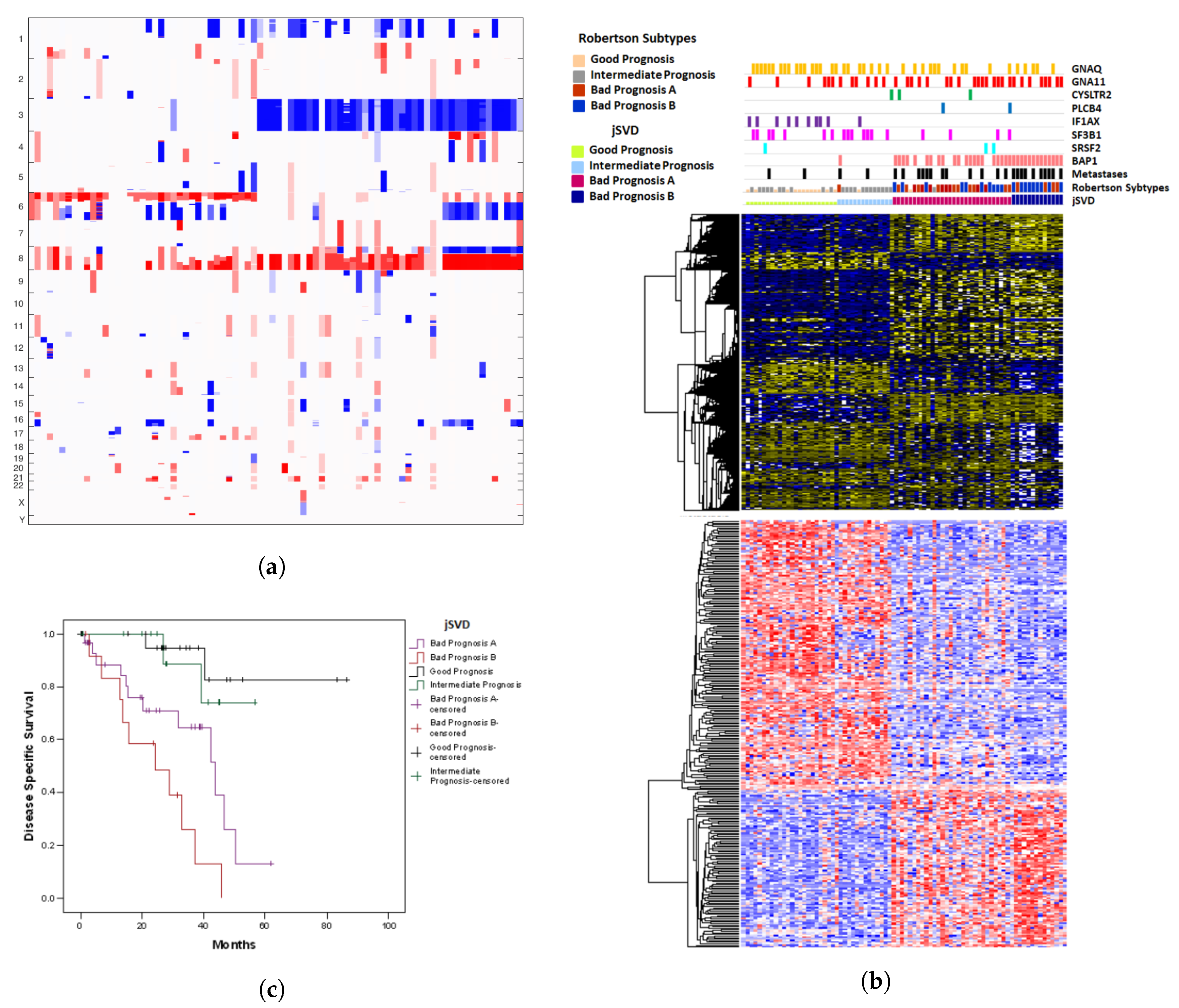
2.1. Results of jSVD
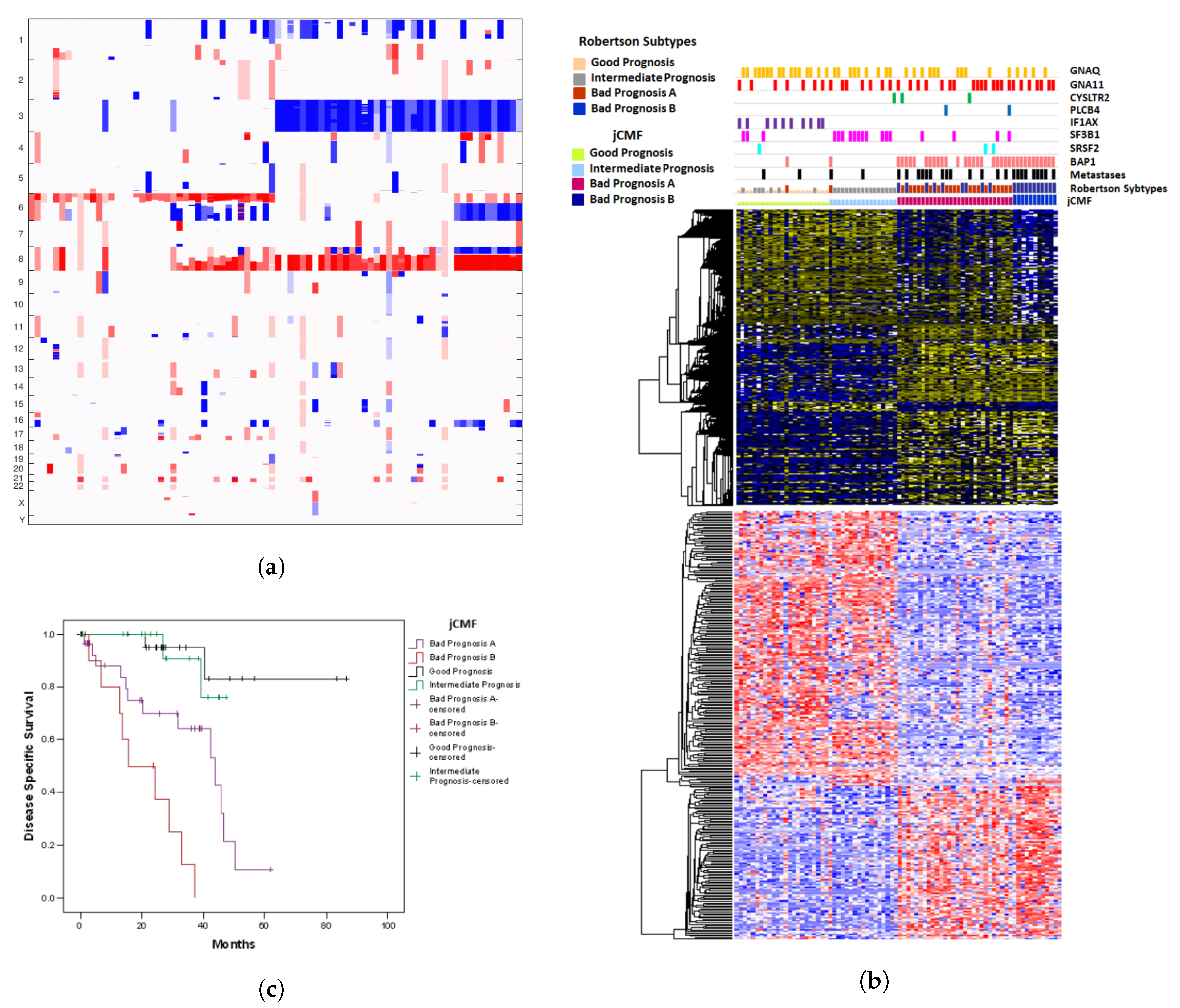
2.2. A Unified Approach: jCMF
2.3. A Comparison with SNF
2.4. Prognostic Misclassification
3. Discussion
4. Materials and Methods
4.1. Dataset
4.2. Algorithms
4.2.1. Preprocessing
4.2.2. Joint SVD
4.2.3. Joint Constrained Matrix Factorization
4.2.4. Similarity Network Fusion
4.2.5. Feature Extraction
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| CM | cutaneous melanoma |
| CNA | copy number alteration |
| GSVD | generalized singular value decomposition |
| jCMF | joint constrained matrix factorization |
| jNNMF | joint non negative matrix factorization |
| jSVD | joint singular value decomposition |
| NNMF | non negative matrix factorization |
| PCA | principal component analysis |
| SCA | simultaneous component analysis |
| SNF | similarity network fusion |
| SVD | singular value decomposition |
| TCGA | the Cancer Genome Atlas |
| UM | uveal melanoma |
References
- Chang, A.E.; Karnell, L.H.; Menck, H.R. The National Cancer Data Base report on cutaneous and noncutaneous melanoma: A summary of 84,836 cases from the past decade. The American College of Surgeons Commission on Cancer and the American Cancer Society. Cancer 1998, 83, 1664–1678. [Google Scholar] [CrossRef]
- Virgili, G.; Gatta, G.; Ciccolallo, L.; Capocaccia, R.; Biggeri, A.; Crocetti, E.; Lutz, J.M.; Paci, E. Incidence of uveal melanoma in Europe. Ophthalmology 2007, 114, 2309–2315. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.D.; Borden, E.C. Metastatic uveal melanoma. Ophthalmol. Clin. N. Am. 2005, 18, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Diener-West, M.; Reynolds, S.M.; Agugliaro, D.J.; Caldwell, R.; Cumming, K.; Earle, J.D.; Hawkins, B.S.; Hayman, J.A.; Jaiyesimi, I.; Jampol, L.M.; et al. Development of metastatic disease after enrollment in the COMS trials for treatment of choroidal melanoma: Collaborative Ocular Melanoma Study Group Report No. 26. Arch. Ophthalmol. 2005, 123, 1639–1643. [Google Scholar] [PubMed]
- Amaro, A.; Gangemi, R.; Piaggio, F.; Angelini, G.; Barisione, G.; Ferrini, S.; Pfeffer, U. The biology of uveal melanoma. Cancer Metastasis Rev. 2017, 36, 109–140. [Google Scholar] [CrossRef] [PubMed]
- Coupland, S.E.; Damato, B.E. Molecular analysis of uveal melanoma. Ophthalmology 2013, 120, e50. [Google Scholar] [CrossRef] [PubMed]
- Robertson, A.G.; Shih, J.; Yau, C.; Gibb, E.A.; Oba, J.; Mungall, K.L.; Hess, J.M.; Uzunangelov, V.; Walter, V.; Danilova, L.; et al. Integrative Analysis Identifies Four Molecular and Clinical Subsets in Uveal Melanoma. Cancer Cell 2018, 33, 151. [Google Scholar] [CrossRef] [PubMed]
- Dono, M.; Angelini, G.; Cecconi, M.; Amaro, A.; Esposito, A.I.; Mirisola, V.; Maric, I.; Lanza, F.; Nasciuti, F.; Viaggi, S.; et al. Mutation frequencies of GNAQ, GNA11, BAP1, SF3B1, EIF1AX and TERT in uveal melanoma: Detection of an activating mutation in the TERT gene promoter in a single case of uveal melanoma. Br. J. Cancer 2014, 110, 1058–1065. [Google Scholar] [CrossRef] [PubMed]
- Harbour, J.W.; Onken, M.D.; Roberson, E.D.; Duan, S.; Cao, L.; Worley, L.A.; Council, M.L.; Matatall, K.A.; Helms, C.; Bowcock, A.M. Frequent mutation of BAP1 in metastasizing uveal melanomas. Science 2010, 330, 1410–1413. [Google Scholar] [CrossRef]
- Harbour, J.W.; Roberson, E.D.; Anbunathan, H.; Onken, M.D.; Worley, L.A.; Bowcock, A.M. Recurrent mutations at codon 625 of the splicing factor SF3B1 in uveal melanoma. Nat. Genet. 2013, 45, 133–135. [Google Scholar] [CrossRef] [PubMed]
- Martin, M.; Masshofer, L.; Temming, P.; Rahmann, S.; Metz, C.; Bornfeld, N.; van de Nes, J.; Klein-Hitpass, L.; Hinnebusch, A.G.; Horsthemke, B.; et al. Exome sequencing identifies recurrent somatic mutations in EIF1AX and SF3B1 in uveal melanoma with disomy 3. Nat. Genet. 2013, 45, 933–936. [Google Scholar] [CrossRef] [PubMed]
- Zeschnigk, M.; Lohmann, D.R. Prognostic Testing in Uveal Melanoma. In Cancer Genomics: Molecular Classification, Prognosis and Response Prediction; Pfeffer, U., Ed.; Springer: Dordrecht, The Netherlands, 2013; pp. 79–96. [Google Scholar]
- Tschentscher, F.; Husing, J.; Holter, T.; Kruse, E.; Dresen, I.G.; Jockel, K.H.; Anastassiou, G.; Schilling, H.; Bornfeld, N.; Horsthemke, B.; et al. Tumor classification based on gene expression profiling shows that uveal melanomas with and without monosomy 3 represent two distinct entities. Cancer Res. 2003, 63, 2578–2584. [Google Scholar] [PubMed]
- Onken, M.D.; Worley, L.A.; Ehlers, J.P.; Harbour, J.W. Gene expression profiling in uveal melanoma reveals two molecular classes and predicts metastatic death. Cancer Res. 2004, 64, 7205–7209. [Google Scholar] [CrossRef] [PubMed]
- Harbour, J.W. A prognostic test to predict the risk of metastasis in uveal melanoma based on a 15-gene expression profile. Methods Mol. Biol. 2014, 1102, 427–440. [Google Scholar] [PubMed]
- Prescher, G.; Bornfeld, N.; Hirche, H.; Horsthemke, B.; Jockel, K.H.; Becher, R. Prognostic implications of monosomy 3 in uveal melanoma. Lancet 1996, 347, 1222–1225. [Google Scholar] [PubMed]
- Gupta, M.P.; Lane, A.M.; DeAngelis, M.M.; Mayne, K.; Crabtree, M.; Gragoudas, E.S.; Kim, I.K. Clinical Characteristics of Uveal Melanoma in Patients With Germline BAP1 Mutations. JAMA Ophthalmol. 2015, 133, 881–887. [Google Scholar] [CrossRef] [PubMed]
- Field, M.G.; Decatur, C.L.; Kurtenbach, S.; Gezgin, G.; van der Velden, P.A.; Jager, M.J.; Kozak, K.N.; Harbour, J.W. PRAME as an Independent Biomarker for Metastasis in Uveal Melanoma. Clin. Cancer Res. 2016, 22, 1234–1242. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wu, F.X.; Ngom, A. A review on machine learning principles for multi-view biological data integration. Brief. Bioinform. 2018, 19, 325–340. [Google Scholar] [CrossRef] [PubMed]
- Rappoport, N.; Shamir, R. Multi-omic and multi-view clustering algorithms: Review and cancer benchmark. Nucleic Acids Res. 2018, 46, 10546–10562. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Zhou, F.; Ren, J.; Li, X.; Jiang, Y.; Ma, S. A Selective Review of Multi-Level Omics Data Integration Using Variable Selection. High-Throughput 2019, 8, 4. [Google Scholar] [CrossRef]
- Smilde, A.K.; Måge, I.; Næs, T.; Hankemeier, T.; Lips, M.A.; Kiers, H.A.L.; Acar, E.; Bro, R. Common and distinct components in data fusion. J. Chemom. 2017, 31, e2900. [Google Scholar] [CrossRef]
- Acar, E.; Bro, R.; Smilde, A.K. Data Fusion in Metabolomics Using Coupled Matrix and Tensor Factorizations. Proc. IEEE 2015, 103, 1602–1620. [Google Scholar] [CrossRef]
- Meng, C.; Zeleznik, O.A.; Thallinger, G.G.; Kuster, B.; Gholami, A.M.; Culhane, A.C. Dimension reduction techniques for the integrative analysis of multi-omics data. Brief. Bioinform. 2016, 17, 628–641. [Google Scholar] [CrossRef] [PubMed]
- Sorber, L.; Van Barel, M.; De Lathauwer, L. Structured Data Fusion. EEE J. Sel. Top. Signal Process. 2015, 9, 586–600. [Google Scholar] [CrossRef]
- Wang, B.; Mezlini, A.M.; Demir, F.; Fiume, M.; Tu, Z.; Brudno, M.; Haibe-Kains, B.; Goldenberg, A. Similarity network fusion for aggregating data types on a genomic scale. Nat. Methods 2014, 11, 333–337. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.D.; Seung, H.S. Algorithms for Non-negative Matrix Factorization. In Advances in Neural Information Processing Systems 13; Leen, T.K., Dietterich, T.G., Tresp, V., Eds.; MIT Press: Cambridge, MA, USA, 2001; pp. 556–562. [Google Scholar]
- Li, W.; Zhang, S.; Liu, C.C.; Zhou, X.J. Identifying multi-layer gene regulatory modules from multi-dimensional genomic data. Bioinformatics 2012, 28, 2458–2466. [Google Scholar] [CrossRef]
- Lin, D.; Zhang, J.; Li, J.; Calhoun, V.D.; Deng, H.W.; Wang, Y.P. Group sparse canonical correlation analysis for genomic data integration. BMC Bioinform. 2013, 14, 245. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Chaudhary, K.; Garmire, L. More Is Better: Recent Progress in Multi-Omics Data Integration Methods. Front. Genet. 2017, 8, 84. [Google Scholar] [CrossRef] [PubMed]
- Sato, H. Joint singular value decomposition algorithm based on the Riemannian trust-region method. JSIAM Lett. 2015, 7, 13–16. [Google Scholar] [CrossRef]
- Lee, C.H.; Alpert, B.O.; Sankaranarayanan, P.; Alter, O. GSVD Comparison of Patient-Matched Normal and Tumor aCGH Profiles Reveals Global Copy-Number Alterations Predicting Glioblastoma Multiforme Survival. PLoS ONE 2012, 7, 1–11. [Google Scholar] [CrossRef]
- Hunyadi, B.; Camps, D.; Sorber, L.; Van Paesschen, W.; De Vos, M.; Van Huffel, S.; De Lathauwer, L. Block term decomposition for modelling epileptic seizures. EURASIP J. Adv. Signal Process. 2014, 2014, 139. [Google Scholar] [CrossRef]
- Horsman, D.E.; Sroka, H.; Rootman, J.; White, V.A. Monosomy 3 and isochromosome 8q in a uveal melanoma. Cancer Genet. Cytogenet. 1990, 45, 249–253. [Google Scholar] [CrossRef]
- Onken, M.D.; Worley, L.A.; Tuscan, M.D.; Harbour, J.W. An accurate, clinically feasible multi-gene expression assay for predicting metastasis in uveal melanoma. J. Mol. Diagn. 2010, 12, 461–468. [Google Scholar] [CrossRef]
- Yavuzyigitoglu, S.; Koopmans, A.E.; Verdijk, R.M.; Vaarwater, J.; Eussen, B.; van Bodegom, A.; Paridaens, D.; Kilic, E.; de Klein, A. Uveal Melanomas with SF3B1 Mutations: A Distinct Subclass Associated with Late-Onset Metastases. Ophthalmology 2016, 123, 1118–1128. [Google Scholar] [CrossRef] [PubMed]
- Pfeffer, U.; Romeo, F.; Noonan, D.M.; Albini, A. Prediction of breast cancer metastasis by genomic profiling: Where do we stand? Clin. Exp. Metastasis 2009, 26, 547–558. [Google Scholar] [CrossRef] [PubMed]
- Boumal, N.; Mishra, B.; Absil, P.A.; Sepulchre, R. Manopt, a Matlab Toolbox for Optimization on Manifolds. J. Mach. Learn. Res. 2014, 15, 1455–1459. [Google Scholar]
- Vervliet, N.; Debals, O.; Sorber, L.; Van Barel, M.; De Lathauwer, L. Tensorlab v3.0. Available online: http://www.tensorlab.net/ (accessed on 15 January 2019).
- Ng, A.Y.; Jordan, M.I.; Weiss, Y. On Spectral Clustering: Analysis and an algorithm. In Advances in Neural Information Processing Systems 14; Dietterich, T.G., Becker, S., Ghahramani, Z., Eds.; MIT Press: Cambridge, MA, USA, 2002; pp. 849–856. [Google Scholar]
- Tusher, V.G.; Tibshirani, R.; Chu, G. Significance analysis of microarrays applied to the ionizing radiation response. Proc. Natl. Acad. Sci. USA 2001, 98, 5116–5121. [Google Scholar] [CrossRef]
- Zhang, J.; Carey, V.; Gentleman, R. An extensible application for assembling annotation for genomic data. Bioinformatics 2003, 19, 155–156. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Observed | Predicted | n | Pearson Chi-Square * | Odds Ratio | 95% Confidence Interval | ||||
|---|---|---|---|---|---|---|---|---|---|
| low | interm. | high | |||||||
| Robertson et al. | Chr3 status | low | 38 | − | 16 | 80 | 3.2–31.0 | ||
| high | 5 | − | 21 | ||||||
| DNA- meth. | low | 21 | 10 | 23 | 69 | 2.7–176.5 | |||
| high | 1 | 1 | 24 | ||||||
| CNA | low | 14 | 21 | 19 | 57 | 2.0–140.9 | |||
| high | 1 | 2 | 23 | ||||||
| Pfeffer et al. | jSVD | low | 21 | 12 | 21 | 66 | 2.3–52.8 | ||
| high | 2 | 2 | 22 | ||||||
| jCMF | low | 21 | 15 | 18 | 63 | 2.6–62.2 | |||
| high | 2 | 2 | 22 | ||||||
| SNF | low | 17 | 19 | 18 | 59 | 2.6–179.0 | |||
| high | 1 | 2 | 23 | ||||||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pfeffer, M.; Uschmajew, A.; Amaro, A.; Pfeffer, U. Data Fusion Techniques for the Integration of Multi-Domain Genomic Data from Uveal Melanoma. Cancers 2019, 11, 1434. https://doi.org/10.3390/cancers11101434
Pfeffer M, Uschmajew A, Amaro A, Pfeffer U. Data Fusion Techniques for the Integration of Multi-Domain Genomic Data from Uveal Melanoma. Cancers. 2019; 11(10):1434. https://doi.org/10.3390/cancers11101434
Chicago/Turabian StylePfeffer, Max, André Uschmajew, Adriana Amaro, and Ulrich Pfeffer. 2019. "Data Fusion Techniques for the Integration of Multi-Domain Genomic Data from Uveal Melanoma" Cancers 11, no. 10: 1434. https://doi.org/10.3390/cancers11101434
APA StylePfeffer, M., Uschmajew, A., Amaro, A., & Pfeffer, U. (2019). Data Fusion Techniques for the Integration of Multi-Domain Genomic Data from Uveal Melanoma. Cancers, 11(10), 1434. https://doi.org/10.3390/cancers11101434