Reading Cancer: Chromatin Readers as Druggable Targets for Cancer Treatment




Abstract
1. Introduction
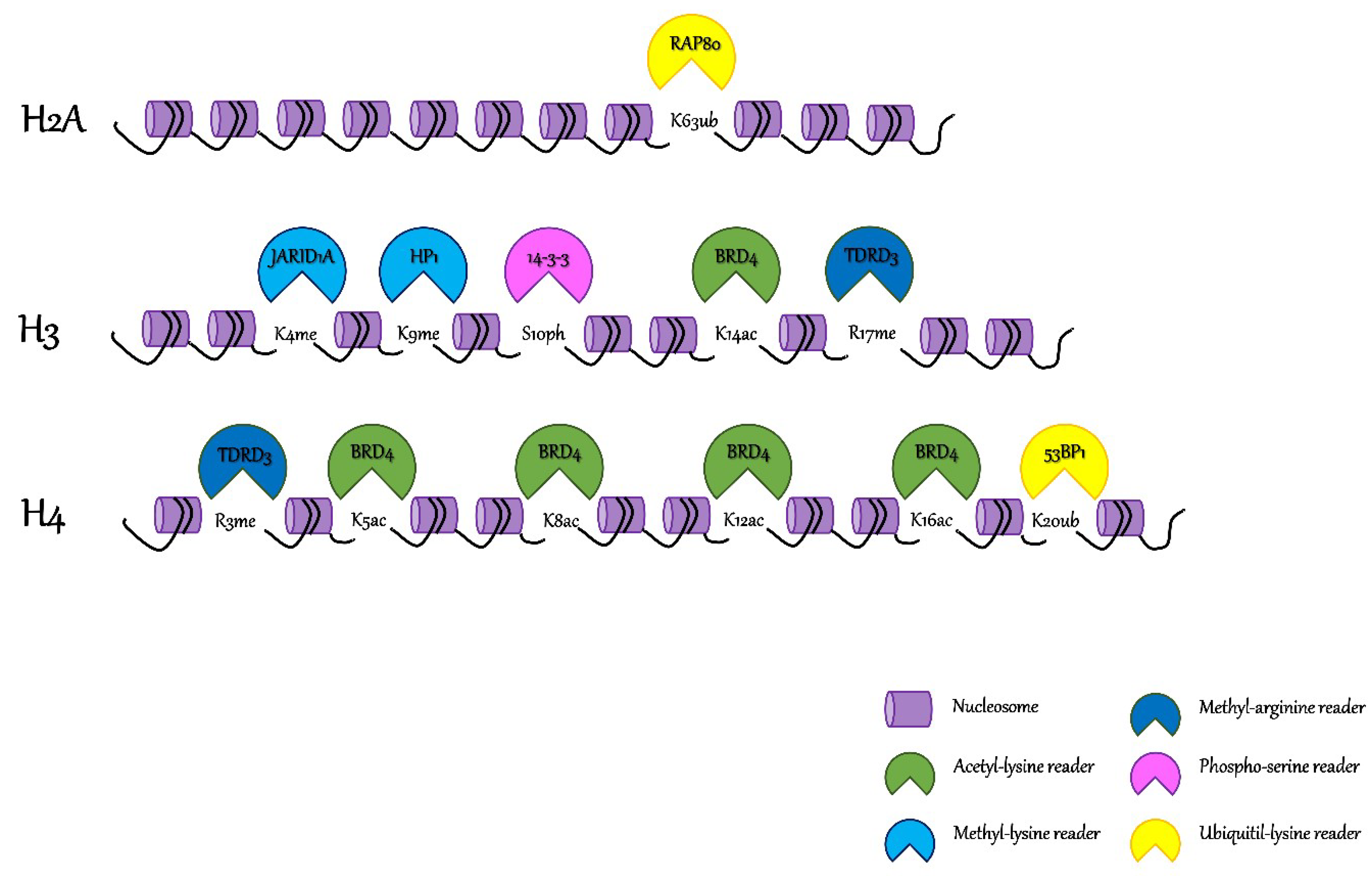
2. Dissecting Chromatin Orthography
3. Interpreting the “Chromatin Tale”
4. Epigenetic Alterations are a Common Place in Cancer
5. Druggable Epigenome
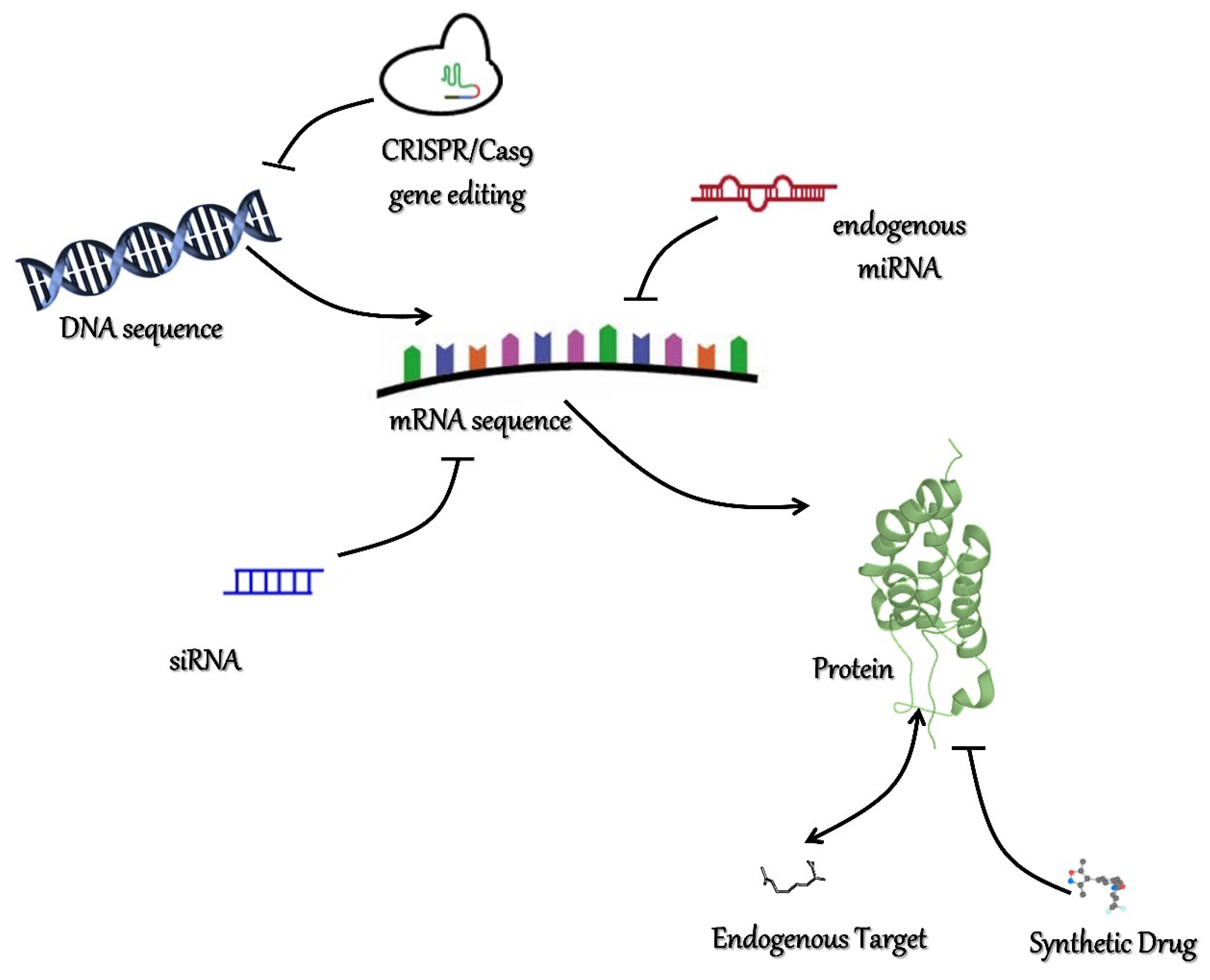
6. New Challenges
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- El Kennani, S.; Crespo, M.; Govin, J.; Pflieger, D. Proteomic Analysis of Histone Variants and Their PTMs: Strategies and Pitfalls. Proteomes 2018, 6, 29. [Google Scholar] [CrossRef] [PubMed]
- Abedin, S.M.; Boddy, C.S.; Munshi, H.G. BET inhibitors in the treatment of hematologic malignancies: Current insights and future prospects. Oncotargets 2016, 9, 5943–5953. [Google Scholar]
- Bowman, G.D.; Poirier, M.G. Post-Translational Modifications of Histones That Influence Nucleosome Dynamics. Chem. Rev. 2015, 115, 2274–2295. [Google Scholar] [CrossRef]
- Celano, M.; Mio, C.; Sponziello, M.; Verrienti, A.; Bulotta, S.; Durante, C.; Damante, G.; Russo, D. Targeting post-translational histone modifications for the treatment of non-medullary thyroid cancer. Mol. Cell. Endocrinol. 2018, 409, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Bartke, T.; Borgel, J.; DiMaggio, P.A. Proteomics in epigenetics: New perspectives for cancer research. Brief. Funct. Genom. 2013, 12, 205–218. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Tollefsbol, T.O. Age-related epigenetic drift and phenotypic plasticity loss: Implications in prevention of age-related human diseases. Epigenomics 2016, 8, 1637–1651. [Google Scholar] [CrossRef] [PubMed]
- Dawson, M.A. The cancer epigenome: Concepts, challenges, and therapeutic opportunities. Science 2017, 355, 1147–1152. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Cooper, S.; Brockdorff, N. The interplay of histone modifications—Writers that read. EMBO Rep. 2015, 16, 1467–1481. [Google Scholar] [CrossRef] [PubMed]
- Taverna, S.D.; Li, H.; Ruthenburg, A.J.; Allis, C.D.; Patel, D.J. How chromatin-binding modules interpret histone modifications: Lessons from professional pocket pickers. Nat. Struct. Mol. Biol. 2007, 14, 1025–1040. [Google Scholar] [CrossRef] [PubMed]
- Sadakierska-Chudy, A.; Filip, M. A Comprehensive View of the Epigenetic Landscape. Part II: Histone Post-translational Modification, Nucleosome Level, and Chromatin Regulation by ncRNAs. Neurotox. Res. 2015, 27, 172–197. [Google Scholar] [CrossRef]
- Russo, D.; Durante, C.; Bulotta, S.; Puppin, C.; Puxeddu, E.; Filetti, S.; Damante, G. Targeting histone deacetylase in thyroid cancer. Expert Opin. Targets 2013, 17, 179–193. [Google Scholar] [CrossRef] [PubMed]
- Javaid, N.; Choi, S. Acetylation- and Methylation-Related Epigenetic Proteins in the Context of Their Targets. Genes 2017, 8, 196. [Google Scholar] [CrossRef] [PubMed]
- Simó-Riudalbas, L.; Esteller, M. Targeting the histone orthography of cancer: Drugs for writers, erasers and readers. Br. J. Pharm. 2015, 172, 2716–2732. [Google Scholar] [CrossRef] [PubMed]
- Ferri, E.; Petosa, C.; McKenna, C.E. Bromodomains: Structure, function and pharmacology of inhibition. Biochem. Pharm. 2016, 106, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Lori, L.; Pasquo, A.; Lori, C.; Petrosino, M.; Chiaraluce, R.; Tallant, C.; Knapp, S.; Consalvi, V. Effect of BET Missense Mutations on Bromodomain Function, Inhibitor Binding and Stability. PLoS ONE 2016, 11, e0159180. [Google Scholar] [CrossRef] [PubMed]
- Muller, S.; Filippakopoulos, P.; Knapp, S. Bromodomains as therapeutic targets. Expert Rev. Mol. Med. 2011, 13. [Google Scholar] [CrossRef] [PubMed]
- Greschik, H.; Schüle, R.; Günther, T. Selective targeting of epigenetic reader domains. Expert Opin. Drug Discov. 2017, 12, 449–463. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.; Li, Y.; Xiong, X.; Chen, Z.; Li, H. YEATS Domain—A Histone Acylation Reader in Health and Disease. J. Mol. Biol. 2017, 429, 1994–2002. [Google Scholar] [CrossRef]
- Filippakopoulos, P.; Picaud, S.; Mangos, M.; Keates, T.; Lambert, J.-P.; Barsyte-Lovejoy, D.; Felletar, I.; Volkmer, R.; Müller, S.; Pawson, T.; et al. Histone Recognition and Large-Scale Structural Analysis of the Human Bromodomain Family. Cell 2012, 149, 214–231. [Google Scholar] [CrossRef]
- Brownlee, P.M.; Chambers, A.L.; Oliver, A.W.; Downs, J.A. Cancer and the bromodomains of BAF180. Biochem. Soc. Trans. 2012, 40, 364–369. [Google Scholar] [CrossRef]
- Wagner, T.; Robaa, D.; Sippl, W.; Jung, M. Mind the Methyl: Methyllysine Binding Proteins in Epigenetic Regulation. ChemMedChem 2014, 9, 466–483. [Google Scholar] [CrossRef] [PubMed]
- James, L.I.; Barsyte-Lovejoy, D.; Zhong, N.; Krichevsky, L.; Korboukh, V.K.; Herold, M.J.; MacNevin, C.J.; Norris, J.L.; Sagum, C.A.; Tempel, W.; et al. Discovery of a chemical probe for the L3MBTL3 methyl-lysine reader domain. Nat. Chem. Biol. 2013, 9, 184–191. [Google Scholar] [CrossRef] [PubMed]
- Goulet, I.; Boisvenue, S.; Mokas, S.; Mazroui, R.; Côté, J. TDRD3, a novel Tudor domain-containing protein, localizes to cytoplasmic stress granules. Hum. Mol. Genet. 2008, 17, 3055–3074. [Google Scholar] [CrossRef] [PubMed]
- Sawicka, A.; Seiser, C. Sensing core histone phosphorylation—A matter of perfect timing. Biochim. Biophys. Acta 2014, 1839, 711–718. [Google Scholar] [CrossRef]
- Smeenk, G.; Mailand, N. Writers, Readers, and Erasers of Histone Ubiquitylation in DNA Double-Strand Break Repair. Front. Genet. 2016, 7, 122. [Google Scholar] [CrossRef] [PubMed]
- Lombardi, P.M.; Matunis, M.J.; Wolberger, C. RAP80, ubiquitin and SUMO in the DNA damage response. J. Mol. Med. 2017, 95, 799–807. [Google Scholar] [CrossRef] [PubMed]
- Appikonda, S.; Thakkar, K.N.; Barton, M.C. Regulation of gene expression in human cancers by TRIM24. Drug Discov. Today Technol. 2016, 19, 57–63. [Google Scholar] [CrossRef]
- Zaware, N.; Zhou, M.-M. Chemical modulators for epigenome reader domains as emerging epigenetic therapies for cancer and inflammation. Curr. Opin. Chem. Biol. 2017, 39, 116–125. [Google Scholar] [CrossRef]
- Dawson, M.A.; Kouzarides, T.; Huntly, B.J.P. Targeting Epigenetic Readers in Cancer. N. Engl. J. Med. 2012, 367, 647–657. [Google Scholar] [CrossRef]
- Gough, S.M.; Lee, F.; Yang, F.; Walker, R.L.; Zhu, Y.J.; Pineda, M.; Onozawa, M.; Chung, Y.J.; Bilke, S.; Wagner, E.K.; et al. NUP98-PHF23 is a chromatin modifying oncoprotein that causes a wide array of leukemias sensitive to inhibition of PHD domain histone reader function. Cancer Discov. 2014, 4, 564–577. [Google Scholar] [CrossRef]
- Brugarolas, J. PBRM1 and BAP1 as Novel Targets for Renal Cell Carcinoma. Cancer J. Sudbury Mass 2013, 19, 324–332. [Google Scholar] [CrossRef] [PubMed]
- Schenk, T.; Chen, W.C.; Göllner, S.; Howell, L.; Jin, L.; Hebestreit, K.; Klein, H.-U.; Popescu, A.C.; Burnett, A.; Mills, K.; et al. Inhibition of the LSD1 (KDM1A) demethylase reactivates the all-trans-retinoic acid differentiation pathway in acute myeloid leukemia. Nat. Med. 2012, 18, 605–611. [Google Scholar] [CrossRef] [PubMed]
- Sahai, V.; Redig, A.J.; Collier, K.A.; Eckerdt, F.D.; Munshi, H.G.; Sahai, V.; Redig, A.J.; Collier, K.A.; Eckerdt, F.D.; Munshi, H.G. Targeting BET bromodomain proteins in solid tumors. Oncotarget 2016, 7, 53997–54009. [Google Scholar] [CrossRef] [PubMed]
- Tao, K.; Yang, J.; Hu, Y.; Deng, A. Knockdown of YEATS4 inhibits colorectal cancer cell proliferation and induces apoptosis. Am. J. Transl. Res. 2015, 7, 616–623. [Google Scholar] [PubMed]
- Kaminskas, E.; Farrell, A.T.; Wang, Y.-C.; Sridhara, R.; Pazdur, R. FDA Drug Approval Summary: Azacitidine (5-azacytidine, VidazaTM) for Injectable Suspension. Oncologist 2005, 10, 176–182. [Google Scholar] [CrossRef] [PubMed]
- Mann, B.S.; Johnson, J.R.; Cohen, M.H.; Justice, R.; Pazdur, R. FDA Approval Summary: Vorinostat for Treatment of Advanced Primary Cutaneous T-Cell Lymphoma. Oncologist 2007, 12, 1247–1252. [Google Scholar] [CrossRef] [PubMed]
- LeRoy, G.; DiMaggio, P.A.; Chan, E.Y.; Zee, B.M.; Blanco, M.A.; Bryant, B.; Flaniken, I.Z.; Liu, S.; Kang, Y.; Trojer, P.; et al. A quantitative atlas of histone modification signatures from human cancer cells. Epigenetics Chromatin 2013, 6, 20. [Google Scholar] [CrossRef]
- Sponziello, M.; Durante, C.; Boichard, A.; Dima, M.; Puppin, C.; Verrienti, A.; Tamburrano, G.; Di Rocco, G.; Redler, A.; Lacroix, L.; et al. Epigenetic-related gene expression profile in medullary thyroid cancer revealed the overexpression of the histone methyltransferases EZH2 and SMYD3 in aggressive tumours. Mol. Cell. Endocrinol. 2014, 392, 8–13. [Google Scholar] [CrossRef]
- Lu, R.; Wang, G.G. Pharmacologic Targeting of Chromatin Modulators as Therapeutics of Acute Myeloid Leukemia. Front. Oncol. 2017, 7, 241. [Google Scholar] [CrossRef]
- Filippakopoulos, P.; Qi, J.; Picaud, S.; Shen, Y.; Smith, W.B.; Fedorov, O.; Morse, E.M.; Keates, T.; Hickman, T.T.; Felletar, I.; et al. Selective inhibition of BET bromodomains. Nature 2010, 468, 1067–1073. [Google Scholar] [CrossRef]
- Mio, C.; Lavarone, E.; Conzatti, K.; Baldan, F.; Toffoletto, B.; Puppin, C.; Filetti, S.; Durante, C.; Russo, D.; Orlacchio, A.; et al. MCM5 as a target of BET inhibitors in thyroid cancer cells. Endocr. Relat. Cancer 2016, 23, 335–347. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Zhang, Y.; Shan, W.; Hu, Z.; Yuan, J.; Pi, J.; Wang, Y.; Fan, L.; Tang, Z.; Li, C.; et al. Repression of BET activity sensitizes homologous recombination–proficient cancers to PARP inhibition. Sci. Transl. Med. 2017, 9, eaal1645. [Google Scholar] [CrossRef]
- Mio, C.; Gerratana, L.; Bolis, M.; Caponnetto, F.; Zanello, A.; Barbina, M.; Loreto, C.D.; Garattini, E.; Damante, G.; Puglisi, F. BET proteins regulate homologous recombination-mediated DNA repair: BRCAness and implications for cancer therapy. Int. J. Cancer 2018, 144, 755–766. [Google Scholar] [CrossRef] [PubMed]
- Lovén, J.; Hoke, H.A.; Lin, C.Y.; Lau, A.; Orlando, D.A.; Vakoc, C.R.; Bradner, J.E.; Lee, T.I.; Young, R.A. Selective Inhibition of Tumor Oncogenes by Disruption of Super-Enhancers. Cell 2013, 153, 320–334. [Google Scholar] [CrossRef] [PubMed]
- Audia, J.E.; Campbell, R.M. Histone Modifications and Cancer. Cold Spring Harb. Perspect. Biol. 2016, 8, a019521. [Google Scholar] [CrossRef] [PubMed]
- Doroshow, D.B.; Eder, J.P.; LoRusso, P.M. BET inhibitors: A novel epigenetic approach. Ann. Oncol. 2017, 28, 1776–1787. [Google Scholar] [CrossRef]
- Conticello, C.; Martinetti, D.; Adamo, L.; Buccheri, S.; Giuffrida, R.; Parrinello, N.; Lombardo, L.; Anastasi, G.; Amato, G.; Cavalli, M.; et al. Disulfiram, an old drug with new potential therapeutic uses for human hematological malignancies. Int. J. Cancer 2012, 131, 2197–2203. [Google Scholar] [CrossRef]
- Igoe, N.; Bayle, E.D.; Fedorov, O.; Tallant, C.; Savitsky, P.; Rogers, C.; Owen, D.R.; Deb, G.; Somervaille, T.C.P.; Andrews, D.M.; et al. Design of a Biased Potent Small Molecule Inhibitor of the Bromodomain and PHD Finger-Containing (BRPF) Proteins Suitable for Cellular and In Vivo Studies. J. Med. Chem. 2017, 60, 668–680. [Google Scholar] [CrossRef]
- Palmer, W.S. Development of small molecule inhibitors of BRPF1 and TRIM24 bromodomains. Drug Discov. Today Technol. 2016, 19, 65–71. [Google Scholar] [CrossRef]
- Ronnekleiv-Kelly, S.M.; Sharma, A.; Ahuja, N. Epigenetic therapy and chemosensitization in solid malignancy. Cancer Treat. Rev. 2017, 55, 200–208. [Google Scholar] [CrossRef]
- Aftimos, P.G.; Bechter, O.; Awada, A.; Jungels, C.; Dumez, H.; Huyvaert, N.; Costermans, J.; Lee, C.; Meeus, M.-A.; Burkard, U.; et al. Phase I first-in-man trial of a novel bromodomain and extra-terminal domain (BET) inhibitor (BI 894999) in patients (Pts) with advanced solid tumors. J. Clin. Oncol. 2017, 35, 2504. [Google Scholar] [CrossRef]
- Cramer, S.A.; Adjei, I.M.; Labhasetwar, V. Advancements in the delivery of epigenetic drugs. Expert Opin. Drug Deliv. 2015, 12, 1501–1512. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.C.; Min, Y.; Palm, R.C.; Fiordalisi, J.J.; Wagner, K.T.; Hyder, N.; Cox, A.D.; Caster, J.M.; Tian, X.; Wang, A.Z. Nanoparticle formulations of histone deacetylase inhibitors for effective chemoradiotherapy in solid tumors. Biomaterials 2015, 51, 208–215. [Google Scholar] [CrossRef] [PubMed]
- Lam, F.C.; Morton, S.W.; Wyckoff, J.; Vu Han, T.-L.; Hwang, M.K.; Maffa, A.; Balkanska-Sinclair, E.; Yaffe, M.B.; Floyd, S.R.; Hammond, P.T. Enhanced efficacy of combined temozolomide and bromodomain inhibitor therapy for gliomas using targeted nanoparticles. Nat. Commun. 2018, 9, 1991. [Google Scholar] [CrossRef]
- Lee, C.S.; Bishop, E.S.; Zhang, R.; Yu, X.; Farina, E.M.; Yan, S.; Zhao, C.; Zeng, Z.; Shu, Y.; Wu, X.; et al. Adenovirus-mediated gene delivery: Potential applications for gene and cell-based therapies in the new era of personalized medicine. Genes Dis. 2017, 4, 43–63. [Google Scholar] [CrossRef] [PubMed]
- Ehrke-Schulz, E.; Schiwon, M.; Leitner, T.; Dávid, S.; Bergmann, T.; Liu, J.; Ehrhardt, A. CRISPR/Cas9 delivery with one single adenoviral vector devoid of all viral genes. Sci. Rep. 2017, 7, 17113. [Google Scholar] [CrossRef] [PubMed]
- Maggio, I.; Holkers, M.; Liu, J.; Janssen, J.M.; Chen, X.; Gonçalves, M.A.F.V. Adenoviral vector delivery of RNA-guided CRISPR/Cas9 nuclease complexes induces targeted mutagenesis in a diverse array of human cells. Sci. Rep. 2014, 4, 5105. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Lage, M.; Puig-Serra, P.; Menendez, P.; Torres-Ruiz, R.; Rodriguez-Perales, S. CRISPR/Cas9 for Cancer Therapy: Hopes and Challenges. Biomedicines 2018, 6, 105. [Google Scholar] [CrossRef] [PubMed]
- Burnett, J.C.; Rossi, J.J.; Tiemann, K. Current Progress of siRNA/shRNA Therapeutics in Clinical Trials. Biotechnol. J. 2011, 6, 1130–1146. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, C.; Sharma, A.R.; Sharma, G.; Doss, C.G.P.; Lee, S.-S. Therapeutic miRNA and siRNA: Moving from Bench to Clinic as Next Generation Medicine. Mol. Nucleic Acids 2017, 8, 132–143. [Google Scholar] [CrossRef] [PubMed]
- Lombardo, G.E.; Maggisano, V.; Celano, M.; Cosco, D.; Mignogna, C.; Baldan, F.; Lepore, S.M.; Allegri, L.; Moretti, S.; Durante, C.; et al. Anti-hTERT siRNA-Loaded Nanoparticles Block the Growth of Anaplastic Thyroid Cancer Xenograft. Mol. Cancer 2018, 17, 1187–1195. [Google Scholar] [CrossRef] [PubMed]
- Tonouchi, E.; Gen, Y.; Muramatsu, T.; Hiramoto, H.; Tanimoto, K.; Inoue, J.; Inazawa, J. miR-3140 suppresses tumor cell growth by targeting BRD4 via its coding sequence and downregulates the BRD4-NUT fusion oncoprotein. Sci. Rep. 2018, 8, 4482. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Meng, D.; Zhang, S.-H.; Zhang, Y.; Deng, Z.; Kong, L.-B. microRNA-608 inhibits human hepatocellular carcinoma cell proliferation via targeting the BET family protein BRD4. Biochem. Biophys. Res. Commun. 2018, 501, 1060–1067. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Ji, H.; Tang, H.; Xu, Z. microRNA-124a suppresses PHF19 over-expression, EZH2 hyper-activation, and aberrant cell proliferation in human glioma. Biochem. Biophys. Res. Commun. 2018, 503, 1610–1617. [Google Scholar] [CrossRef] [PubMed]
- Lepore, I.; Dell’Aversana, C.; Pilyugin, M.; Conte, M.; Nebbioso, A.; De Bellis, F.; Tambaro, F.P.; Izzo, T.; Garcia-Manero, G.; Ferrara, F.; et al. HDAC Inhibitors Repress BARD1 Isoform Expression in Acute Myeloid Leukemia Cells via Activation of miR-19a and/or b. PLoS ONE 2013, 8, e83018. [Google Scholar] [CrossRef] [PubMed]
- Ghislin, S.; Deshayes, F.; Middendorp, S.; Boggetto, N.; Alcaide-Loridan, C. PHF19 and Akt control the switch between proliferative and invasive states in melanoma. Cell Cycle 2012, 11, 1634–1645. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Liu, L.; Shan, W.; Yang, Z.-Q. An integrated genomic analysis of Tudor domain–containing proteins identifies PHD finger protein 20-like 1 (PHF20L1) as a candidate oncogene in breast cancer. Mol. Oncol. 2016, 10, 292–302. [Google Scholar] [CrossRef]
- Ge, Z.; Song, E.J.; Kawasawa, Y.I.; Li, J.; Dovat, S.; Song, C. WDR5 high expression and its effect on tumorigenesis in leukemia. Oncotarget 2016, 7, 37740–37754. [Google Scholar] [CrossRef]
- Carugo, A.; Genovese, G.; Seth, S.; Nezi, L.; Rose, J.L.; Bossi, D.; Cicalese, A.; Shah, P.K.; Viale, A.; Pettazzoni, P.F.; et al. In Vivo Functional Platform Targeting Patient-Derived Xenografts Identifies WDR5-Myc Association as a Critical Determinant of Pancreatic Cancer. Cell Rep. 2016, 16, 133–147. [Google Scholar] [CrossRef]
- Paolino, D.; Cosco, D.; Gaspari, M.; Celano, M.; Wolfram, J.; Voce, P.; Puxeddu, E.; Filetti, S.; Celia, C.; Ferrari, M.; et al. Targeting the thyroid gland with thyroid-stimulating hormone (TSH)-nanoliposomes. Biomaterials 2014, 35, 7101–7109. [Google Scholar] [CrossRef]
- Xiao, Y.; Shi, K.; Qu, Y.; Chu, B.; Qian, Z. Engineering Nanoparticles for Targeted Delivery of Nucleic Acid Therapeutics in Tumor. Mol. Methods Clin. Dev. 2018, 12, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Qiu, W.; Chen, R.; Chen, X.; Zhang, H.; Song, L.; Cui, W.; Zhang, J.; Ye, D.; Zhang, Y.; Wang, Z. Oridonin-loaded and GPC1-targeted gold nanoparticles for multimodal imaging and therapy in pancreatic cancer. Int. J. Nanomed. 2018, 13, 6809–6827. [Google Scholar] [CrossRef] [PubMed]
- Bai, Z.-T.; Bai, B.; Zhu, J.; Di, C.-X.; Li, X.; Zhou, W.-C. Epigenetic actions of environmental factors and promising drugs for cancer therapy. Oncol. Lett. 2018, 15, 2049–2056. [Google Scholar] [CrossRef] [PubMed]
- Smith, H.J.; Straughn, J.M.; Buchsbaum, D.J.; Arend, R.C. Epigenetic therapy for the treatment of epithelial ovarian cancer: A clinical review. Gynecol. Oncol. Rep. 2017, 20, 81–86. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Enzymes | Residues | Families | Components |
|---|---|---|---|
| Methylation | |||
| Writers | Lysine | Methyltransferases (HMTs) | EZH1/2, MLL1-5, SET1/7/9, SUV39h1/2 SUV40h1/2, G9a, EHMT1, NSD1/2, SMYD2, DOT1L |
| Arginine | Methyltransferases (HMTs) | PRMT1/2/4/5/6/7, CARM1 | |
| Erasers | Lysine | Demethylases (KDMs) | UTX, JMJD3, KDM1A/B, KDM2A/B, KDM4A/B/C/D, KDM5A/B/C/D, PHF2/8 JHDM1a/b, JHDM2a/b, JMJD2A/B/C/D |
| Arginine | Demethylases (RDMs) | JMJD6 | |
| Readers | Lysine | Chromodomains Tudor domains PWWP domains Ankyrin repeats | MOF, MRG15 MBT, PHF1/19, TDRD7 BRPF1, NSD1-3 G9a/GLP |
| Arginine | Tudor domains | WDR5, TDRD3, SMN1 | |
| Acetylation | |||
| Writers | Lysine | Acetyltransferases (HATs) | KAT2A/B, KAT3A/B, KAT6/5/7/8, Tip60, CREBBP, EP300, PCAF |
| Erasers | Lysine | Deacetylases | HDAC1/2/3/4/5/7/8/9/11, SIRT1/2/6/7 |
| Readers | Lysine | Bromodomains | BRD2/3/4/T |
| Phosphorylation | |||
| Writers | Serine | Kinases | CDK1/2, MSK1/2, Mst1, ATR, ATM, RSK2, AMPK, IKK-alpha, AuroraB |
| Threonine | Kinases | Haspin/Gsg2, Dlk/Zip | |
| Tyrosine | Kinases | Mst1, WSTF | |
| Erasers | Serine | Phosphatases | PP2A1, PP1 |
| Threonine | Phosphatases | PPgamma | |
| Tyrosine | Phosphatases | EYA1/3 | |
| Readers | Serine | 14-3-3 proteins BRCT domain | 14-3-3β/γ/η/ε/μ |
| Threonine | BIR domain | XRCC1, NBS1, BARD1 | |
| Tyrosine | PTB domain | ||
| Ubiquitynation | |||
| Writers | Lysine | Ubiquitin-ligases | BRCA1-BARD1, RING1A/RING1B/BMI1 |
| Erasers | Lysine | Isopeptidases | OTUB1/2, BRCC36, USP3/16/26/44 |
| Readers | Lysine | 53BP1 | |
| ADP-ribosylation | |||
| Writers | Glutamate Arginine Glutamate | ADP-ribosyltransferases | PARP1 |
| Erasers | Glutamate Arginine Glutamate | ADP-ribosylhydrolases | PARG, MDO1/2, TARG |
| Readers | Glutamate Arginine Glutamate | Macrodomains PBZ WWE domain | RNF146 APLF, CHFR |
| Target | Intervention | Status | Condition | Study Type | Phase | NCT Number |
|---|---|---|---|---|---|---|
| EZH2 | SHR2554 | RECRUITING | AML and myelodysplastic syndromes | INTERVENTIONAL | I | NCT03603951 |
| EPZ-6438 | RECRUITING | Advanced Solid Tumors and Hematologic Malignancies | INTERVENTIONAL | I/II | NCT01897571 | |
| CPI-1205 | RECRUITING | Advanced Solid Tumors | INTERVENTIONAL | I/II | NCT03525795 | |
| DOT1L | EPZ-5676 | COMPLETED | AML and myelodysplastic syndromes | INTERVENTIONAL | I | NCT02141828 |
| PRMT5 | JNJ-64619178 | RECTUITING | Advanced solid tumors | INTERVENTIONAL | I | NCT03573310 |
| GSK3326595 | RECTUITING | Advanced solid tumors | INTERVENTIONAL | I | NCT02783300 | |
| LSD1 | IMG-7289 | ACTIVE | AML and myelodysplastic syndromes | INTERVENTIONAL | I | NCT02842827 |
| INCB059872 | RECRUITING | Advanced Solid Tumors and Hematologic Malignancies | INTERVENTIONAL | I/II | NCT02712905 | |
| HDAC | Panobinostat (LBH589) | COMPLETED | HL and MM | INTERVENTIONAL | III | NCT01034163 |
| ACTIVE | Hematologic Malignancies | INTERVENTIONAL | II | NCT01802879 | ||
| COMPLETED | Advanced Solid Tumors and Hematologic Malignancies | INTERVENTIONAL | I | NCT00472368 | ||
| COMPLETED | CTCL | INTERVENTIONAL | II/III | NCT00425555 | ||
| Belinostat (PXD101) | COMPLETED | AML and myelodysplastic syndromes | INTERVENTIONAL | II | NCT00357032 | |
| COMPLETED | OC | INTERVENTIONAL | II | NCT00301756 | ||
| Vorinostat | COMPLETED | Advanced BC | I | NCT00719875 | ||
| COMPLETED | Advanced CTCL | INTERVENTIONAL | II | NCT00091559 | ||
| ACTIVE | Advanced NSCLC | I | NCT01059552 | |||
| CHR-3996 | COMPLETED | Advanced Solid Tumors | INTERVENTIONAL | I/II | NCT00697879 | |
| Givinostat | RECRUITING | Chronic myeloproliferative neoplasms | INTERVENTIONAL | II | NCT01761968 | |
| Romidepsin | COMPLETED | T cell lymphoma | INTERVENTIONAL | II | NCT00007345 | |
| KA2507 | RECRUITING | Advanced solid tumors | I | NCT03008018 | ||
| DNMT | SGI-110 | COMPLETED | AML and myelodysplastic syndromes | INTERVENTIONAL | I/II | NCT01261312 |
| Deoxycytidine (Aza TdC) | RECRUITING | Advanced solid tumors | INTERVENTIONAL | I | NCT03366116 | |
| Decitabine | COMPLETED | Metastatic PTC or FTC | INTERVENTIONAL | II | NCT00085293 | |
| COMPLETED | AML and myelodysplastic syndromes | INTERVENTIONAL | II | NCT00492401 | ||
| Disulfiram | COMPLETED | PC | INTERVENTIONAL | II | NCT01118741 | |
| RECRUITING | Metastatic BC | INTERVENTIONAL | II | NCT03323346 |
| BET Inhibitors | Intervention | Status | Condition | Study Type | Phase | NCT Number |
|---|---|---|---|---|---|---|
| I-BET762 (GSK525762) | GSK525762 + FULVESTRANT vs. GSK525762 + PLACEBO | RECRUITING | ER and/or PR-positive/HER2-Negative Advanced or Metastatic Breast Cancer | INTERVENTIONAL | II | NCT02964507 |
| GSK525762 + ABIRATERONE/ENZALUTAMIDE +PREDNISONE | RECRUITING | Castration-resistant Prostate Cancer | INTERVENTIONAL | I | NCT03150056 | |
| GSK525762 monotherapy | RECRUITING | Relapsed Refractory Hematologic Malignancies | INTERVENTIONAL | I | NCT01943851 | |
| GSK525762 monotherapy | ACTIVE | NUT Midline Carcinoma | INTERVENTIONAL | I | NCT03702036 | |
| MK-8628 monotherapy | COMPLETED | Advanced Solid Tumor | INTERVENTIONAL | I | NCT02259114 | |
| MK-8628 monotherapy | COMPLETED | Hematologic Malignancies | INTERVENTIONAL | I | NCT01713582 | |
| MK-8628 monotherapy | ACTIVE | Hematologic Malignancies | INTERVENTIONAL | I | NCT02698189 | |
| FT-1101 | FT-1101 + AZACITIDINE vs. FT-1101 + PLACEBO | RECRUITING | Hematologic Malignancies | INTERVENTIONAL | I | NCT02543879 |
| CPI-0610 | CPI-0610 + Ruxolitinib vs. CPI-0610 + PLACEBO | RECRUITING | Hematologic Malignancies | INTERVENTIONAL | I-II | NCT02158858 |
| CPI-0610 monotherapy | COMPLETED | Multiple Myeloma | INTERVENTIONAL | I | NCT02157636 | |
| CPI-0610 monotherapy | ACTIVE | Lymphoma | INTERVENTIONAL | I | NCT01949883 | |
| INCB054329 | INCB054329 monotherapy | COMPLETED | Advanced Solid Tumors and Hematologic Malignancies | INTERVENTIONAL | I-II | NCT02431260 |
| RO6870810 | RO6870810 + Atezolizumab vs. RO6870810 + PLACEBO | RECRUITING | Advanced Ovarian Cancer and TNBC | INTERVENTIONAL | I | NCT03292172 |
| RO6870810 and VENETOCLAX + RITUXIMAB vs. RO6870810 and VENETOCLAX + PLACEBO | RECRUITING | DLBCL | INTERVENTIONAL | I | NCT03255096 | |
| GSK2820151 | GSK2820151 monotherapy | ACTIVE | Advanced or Recurrent Solid Tumors | INTERVENTIONAL | I | NCT02630251 |
| ZEN003694 | ZEN003694 monotherapy | COMPLETED | Metastatic Castration-resistant Prostate Cancer | INTERVENTIONAL | I | NCT02705469 |
| ZEN003694 + ENZALUTAMIDE vs. ZEN003694 + PLACEBO | RECRUITING | Metastatic Castration-resistant Prostate Cancer | INTERVENTIONAL | I-II | NCT02711956 | |
| BMS-986158 | BMS-986158 and NIVOLUMAB | RECRUITING | Advanced Tumors | INTERVENTIONAL | I-II | NCT02419417 |
| ABBV-075 | ABBV-075 and VENETOCLAX | RECRUITING | Solid Tumors | INTERVENTIONAL | I | NCT02391480 |
| GS-5829 | GS-5829 + ENZALUTAMIDE vs. GS-5829 + PLACEBO | ACTIVE | Metastatic Castration-resistant Prostate Cancer | INTERVENTIONAL | I-II | NCT02607228 |
| GS-5829 + FULVESTRANT vs. GS-5829 + EXEMESTANE | COMPLETED | Advanced Solid Tumors and Lymphomas | INTERVENTIONAL | I | NCT02392611 | |
| PLX51107 | PLX51107 monotherapy | RECRUITING | Advanced Solid Tumors and Hematologic Malignancies | INTERVENTIONAL | I | NCT02683395 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mio, C.; Bulotta, S.; Russo, D.; Damante, G. Reading Cancer: Chromatin Readers as Druggable Targets for Cancer Treatment. Cancers 2019, 11, 61. https://doi.org/10.3390/cancers11010061
Mio C, Bulotta S, Russo D, Damante G. Reading Cancer: Chromatin Readers as Druggable Targets for Cancer Treatment. Cancers. 2019; 11(1):61. https://doi.org/10.3390/cancers11010061
Chicago/Turabian StyleMio, Catia, Stefania Bulotta, Diego Russo, and Giuseppe Damante. 2019. "Reading Cancer: Chromatin Readers as Druggable Targets for Cancer Treatment" Cancers 11, no. 1: 61. https://doi.org/10.3390/cancers11010061
APA StyleMio, C., Bulotta, S., Russo, D., & Damante, G. (2019). Reading Cancer: Chromatin Readers as Druggable Targets for Cancer Treatment. Cancers, 11(1), 61. https://doi.org/10.3390/cancers11010061