Endolysosomal Ca2+ Signalling and Cancer Hallmarks: Two-Pore Channels on the Move, TRPML1 Lags Behind!

 ,
, 


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. The Endolysosomal Ca2+ Store: Functions and Mechanisms of Ca2+ Refilling and Release
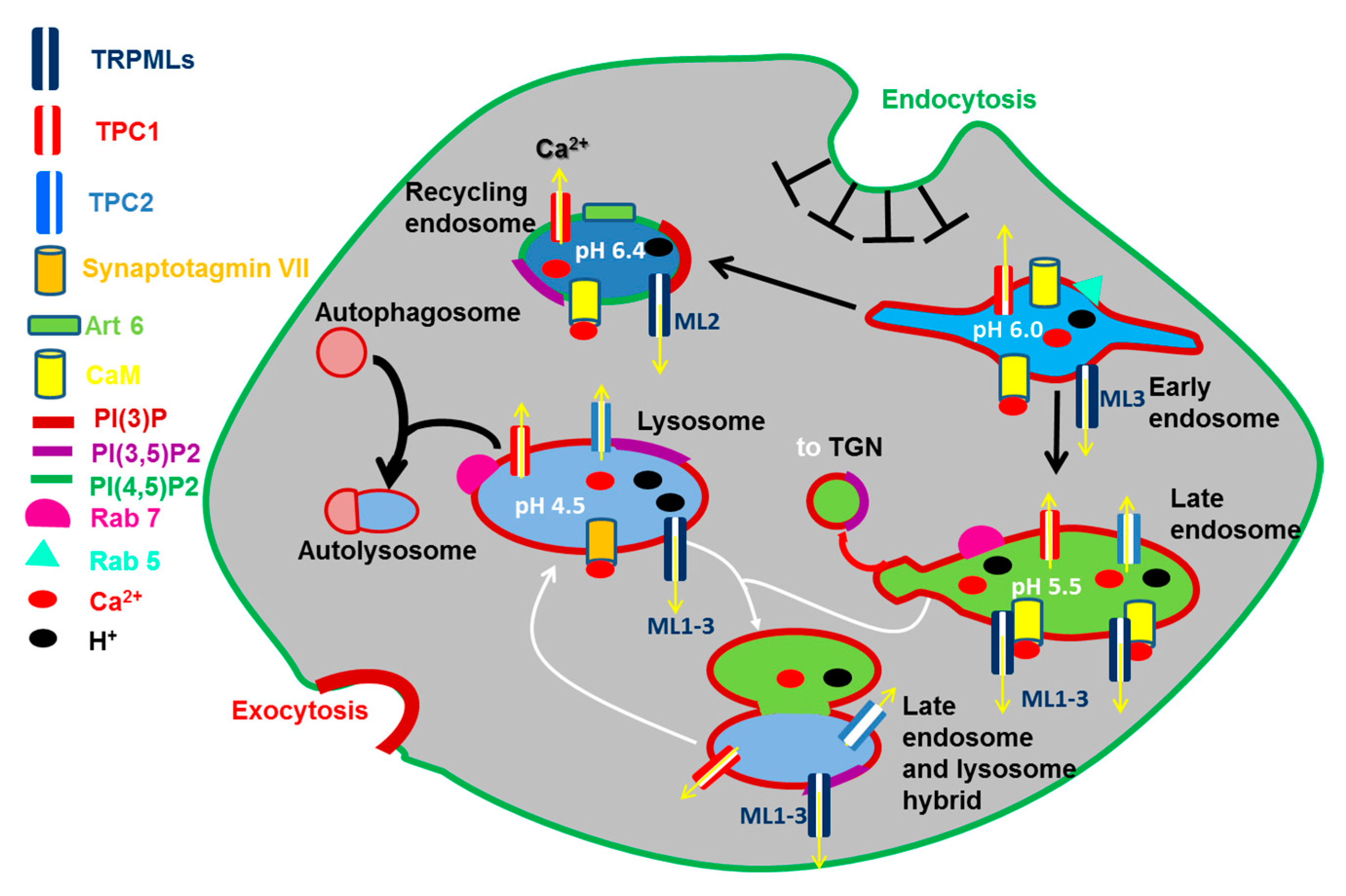
2.1. The Function of Endosomes and Lysosomes: Their Emerging Role in Ca2+ Signaling
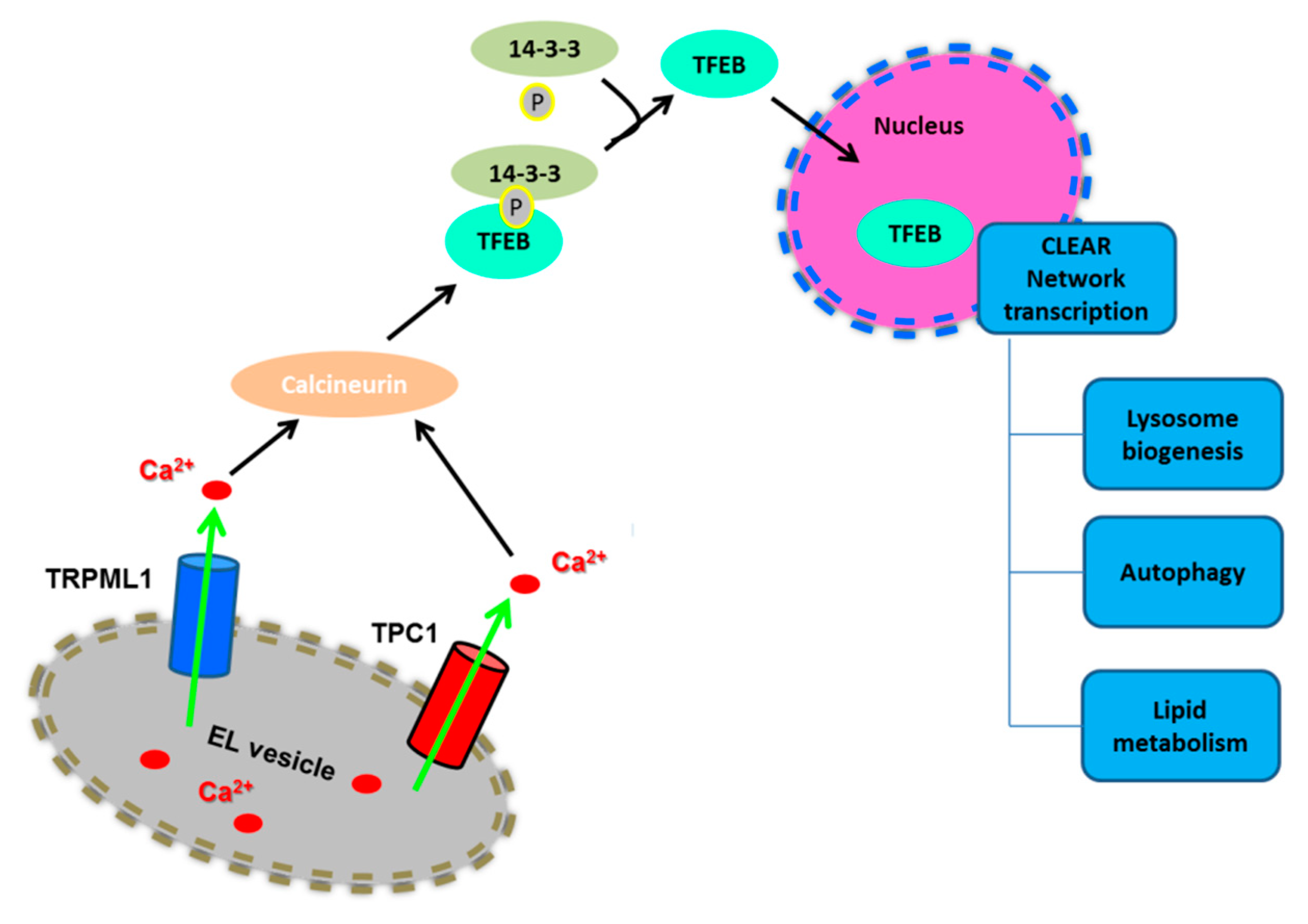
2.2. Lysosomal Ca2+ Signaling Controls Lysosome Biogenesis and Autophagy
2.3. Ca2+ Uptake Mechanisms in the Lysosomes
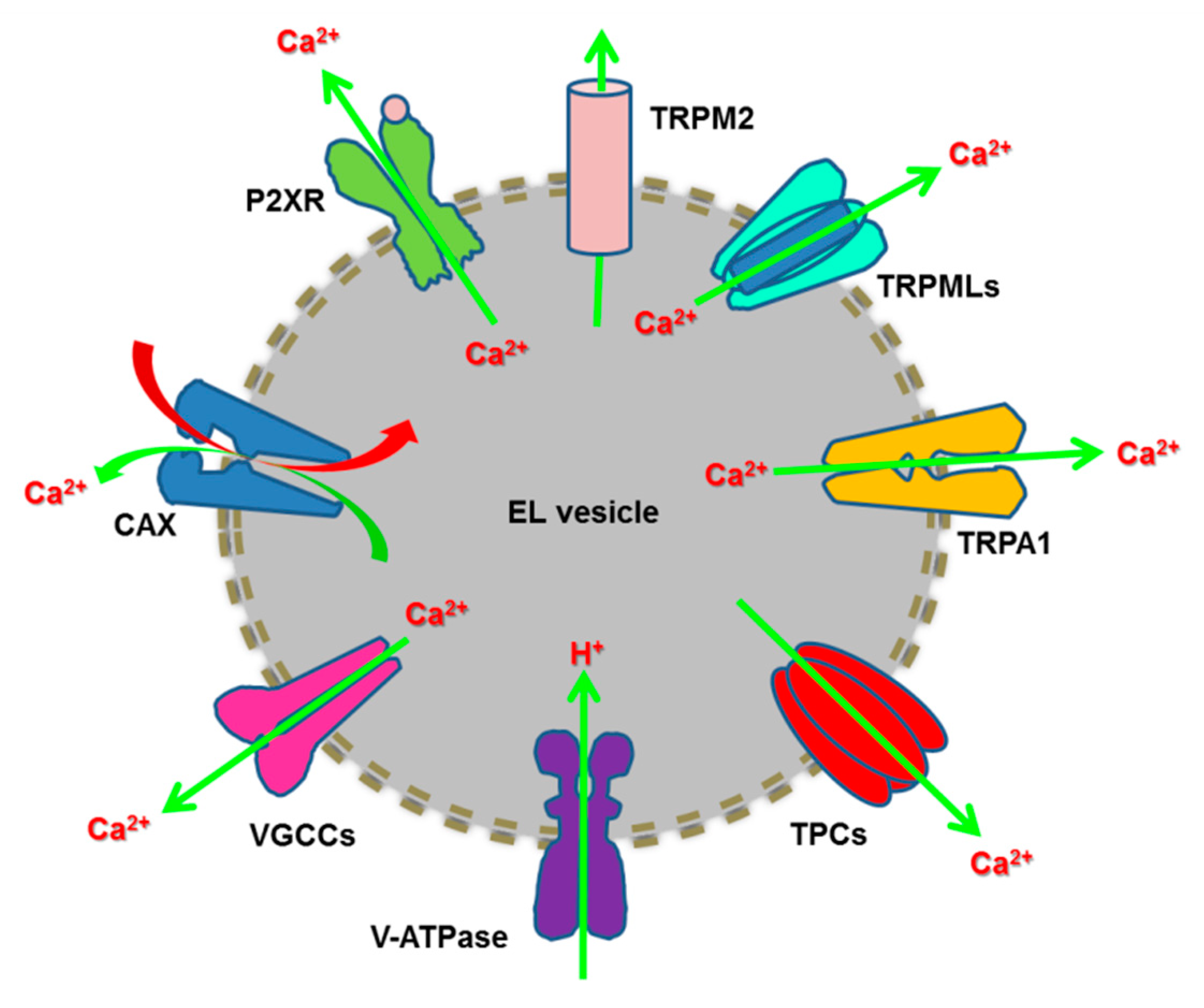
2.4. Endoysosomal Ca2+ Release Channels
2.4.1. TRPML1-3 Channels
2.4.2. TPC1-2 Channels: Structure, Gating Mechanisms and Controversies
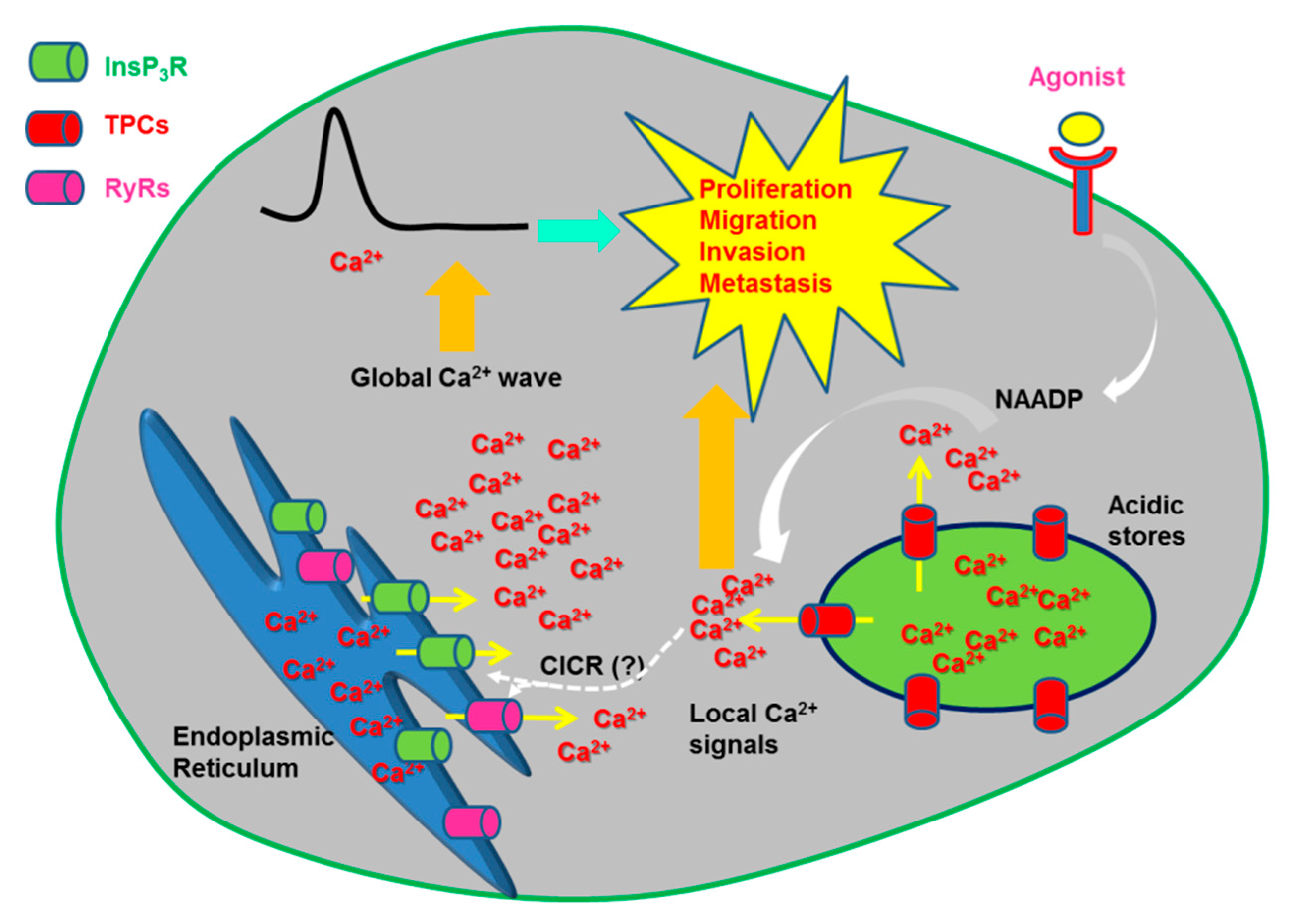
2.4.2.1. The Physiological Role of TPCs: Local vs. Global Ca2+ Signals
2.4.3. Other endolysosomal Ca2+ releasing channels
3. The Role of Lysosomal Ca2+ Signals in Cancer: Two-Pore Channels Drive Metastasis and Angiogenesis
3.1. Lysosomes Contribute to Cancer Hallmarks
3.2. The Role of Endolysosomal Ca2+ Signaling in Cancer: Are Two-Pore Channels the Guilty Ones?
3.3. Two-Pore Channels 1 and 2 Support Cancer Cell Migration and Promote Tumor Vascularization
3.4. TPCs as Therapeutic Target in Cancer
4. Conclusions
Funding
Conflicts of Interest
References
- Berridge, M.J.; Bootman, M.D.; Roderick, H.L. Calcium signalling: Dynamics, homeostasis and remodelling. Nat. Rev. Mol. Cell Biol. 2003, 4, 517–529. [Google Scholar] [CrossRef] [PubMed]
- Clapham, D.E. Calcium signaling. Cell 2007, 131, 1047–1058. [Google Scholar] [CrossRef] [PubMed]
- De Stefani, D.; Rizzuto, R.; Pozzan, T. Enjoy the Trip: Calcium in Mitochondria Back and Forth. Annu. Rev. Biochem. 2016, 85, 161–192. [Google Scholar] [CrossRef] [PubMed]
- Missiaen, L.; Dode, L.; Vanoevelen, J.; Raeymaekers, L.; Wuytack, F. Calcium in the Golgi apparatus. Cell Calcium 2007, 41, 405–416. [Google Scholar] [CrossRef] [PubMed]
- Thillaiappan, N.B.; Chavda, A.P.; Tovey, S.C.; Prole, D.L.; Taylor, C.W. Ca2+ signals initiate at immobile IP3 receptors adjacent to ER-plasma membrane junctions. Nat. Commun. 2017, 8, 1505. [Google Scholar] [CrossRef] [PubMed]
- Keebler, M.V.; Taylor, C.W. Endogenous signalling pathways and caged IP3 evoke Ca2+ puffs at the same abundant immobile intracellular sites. J. Cell Sci. 2017, 130, 3728–3739. [Google Scholar] [CrossRef] [PubMed]
- Berridge, M.J. The endoplasmic reticulum: A multifunctional signaling organelle. Cell Calcium 2002, 32, 235–249. [Google Scholar] [CrossRef]
- Prakriya, M.; Lewis, R.S. Store-Operated Calcium Channels. Physiol. Rev. 2015, 95, 1383–1436. [Google Scholar] [CrossRef]
- Moccia, F.; Dragoni, S.; Lodola, F.; Bonetti, E.; Bottino, C.; Guerra, G.; Laforenza, U.; Rosti, V.; Tanzi, F. Store-dependent Ca2+ entry in endothelial progenitor cells as a perspective tool to enhance cell-based therapy and adverse tumour vascularization. Curr. Med. Chem. 2012, 19, 5802–5818. [Google Scholar] [CrossRef]
- Berridge, M.J. Inositol trisphosphate and calcium signalling mechanisms. Biochim. Biophys. Acta 2009, 1793, 933–940. [Google Scholar] [CrossRef]
- Laude, A.J.; Simpson, A.W. Compartmentalized signalling: Ca2+ compartments, microdomains and the many facets of Ca2+ signalling. FEBS J. 2009, 276, 1800–1816. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.; Docampo, R. Acidic calcium stores open for business: Expanding the potential for intracellular Ca2+ signaling. Trends Cell Biol. 2010, 20, 277–286. [Google Scholar] [CrossRef] [PubMed]
- Galione, A. A primer of NAADP-mediated Ca2+ signalling: From sea urchin eggs to mammalian cells. Cell Calcium 2015, 58, 27–47. [Google Scholar] [CrossRef] [PubMed]
- Morgan, A.J.; Platt, F.M.; Lloyd-Evans, E.; Galione, A. Molecular mechanisms of endolysosomal Ca2+ signalling in health and disease. Biochem. J. 2011, 439, 349–374. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.; Cai, X. Evolution of acidic Ca2+ stores and their resident Ca2+-permeable channels. Cell Calcium 2015, 57, 222–230. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Zhao, Z.; Gu, M.; Feng, X.; Xu, H. Release and uptake mechanisms of vesicular Ca2+ stores. Protein Cell 2018. [Google Scholar] [CrossRef] [PubMed]
- Lam, A.K.; Galione, A. The endoplasmic reticulum and junctional membrane communication during calcium signaling. Biochim. Biophys. Acta 2013, 1833, 2542–2559. [Google Scholar] [CrossRef]
- Morgan, A.J.; Davis, L.C.; Wagner, S.K.; Lewis, A.M.; Parrington, J.; Churchill, G.C.; Galione, A. Bidirectional Ca2+ signaling occurs between the endoplasmic reticulum and acidic organelles. J. Cell Biol. 2013, 200, 789–805. [Google Scholar] [CrossRef]
- Grimm, C.; Bartel, K.; Vollmar, A.M.; Biel, M. Endolysosomal Cation Channels and Cancer-A Link with Great Potential. Pharmaceuticals (Basel) 2018, 11, 4. [Google Scholar] [CrossRef]
- Patel, S.; Kilpatrick, B.S. Two-pore channels and disease. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 1678–1686. [Google Scholar] [CrossRef]
- Sterea, A.M.; Almasi, S.; El Hiani, Y. The hidden potential of lysosomal ion channels: A new era of oncogenes. Cell Calcium 2018, 72, 91–103. [Google Scholar] [CrossRef] [PubMed]
- Pizzo, P.; Lissandron, V.; Capitanio, P.; Pozzan, T. Ca2+ signalling in the Golgi apparatus. Cell Calcium 2011, 50, 184–192. [Google Scholar] [CrossRef] [PubMed]
- Wong, A.K.; Capitanio, P.; Lissandron, V.; Bortolozzi, M.; Pozzan, T.; Pizzo, P. Heterogeneity of Ca2+ handling among and within Golgi compartments. J. Mol. Cell Biol. 2013, 5, 266–276. [Google Scholar] [CrossRef] [PubMed]
- Huotari, J.; Helenius, A. Endosome maturation. EMBO J. 2011, 30, 3481–3500. [Google Scholar] [CrossRef] [PubMed]
- Gerasimenko, J.V.; Tepikin, A.V.; Petersen, O.H.; Gerasimenko, O.V. Calcium uptake via endocytosis with rapid release from acidifying endosomes. Curr. Biol. 1998, 8, 1335–1338. [Google Scholar] [CrossRef]
- Albrecht, T.; Zhao, Y.; Nguyen, T.H.; Campbell, R.E.; Johnson, J.D. Fluorescent biosensors illuminate calcium levels within defined beta-cell endosome subpopulations. Cell Calcium 2015, 57, 263–274. [Google Scholar] [CrossRef] [PubMed]
- Scott, C.C.; Gruenberg, J. Ion flux and the function of endosomes and lysosomes: pH is just the start: The flux of ions across endosomal membranes influences endosome function not only through regulation of the luminal pH. Bioessays 2011, 33, 103–110. [Google Scholar] [CrossRef]
- Prekeris, R.; Klumperman, J.; Chen, Y.A.; Scheller, R.H. Syntaxin 13 mediates cycling of plasma membrane proteins via tubulovesicular recycling endosomes. J. Cell Biol. 1998, 143, 957–971. [Google Scholar] [CrossRef]
- Xu, H.; Ren, D. Lysosomal physiology. Annu. Rev. Physiol. 2015, 77, 57–80. [Google Scholar] [CrossRef]
- Appelqvist, H.; Waster, P.; Kagedal, K.; Ollinger, K. The lysosome: From waste bag to potential therapeutic target. J. Mol. Cell Biol. 2013, 5, 214–226. [Google Scholar] [CrossRef]
- Maxson, M.E.; Grinstein, S. The vacuolar-type H(+)-ATPase at a glance—More than a proton pump. J. Cell Sci. 2014, 127 Pt 23, 4987–4993. [Google Scholar] [CrossRef]
- Hesketh, G.G.; Wartosch, L.; Davis, L.J.; Bright, N.A.; Luzio, J.P. The Lysosome and Intracellular Signalling. Prog. Mol. Subcell. Biol. 2018, 57, 151–180. [Google Scholar] [PubMed]
- Perera, R.M.; Zoncu, R. The Lysosome as a Regulatory Hub. Annu. Rev. Cell Dev. Biol. 2016, 32, 223–253. [Google Scholar] [CrossRef] [PubMed]
- Carroll, B.; Dunlop, E.A. The lysosome: A crucial hub for AMPK and mTORC1 signalling. Biochem. J. 2017, 474, 1453–1466. [Google Scholar] [CrossRef] [PubMed]
- Macgregor, A.; Yamasaki, M.; Rakovic, S.; Sanders, L.; Parkesh, R.; Churchill, G.C.; Galione, A.; Terrar, D.A. NAADP controls cross-talk between distinct Ca2+ stores in the heart. J. Biol. Chem. 2007, 282, 15302–15311. [Google Scholar] [CrossRef] [PubMed]
- Atakpa, P.; Thillaiappan, N.B.; Mataragka, S.; Prole, D.L.; Taylor, C.W. IP3 Receptors Preferentially Associate with ER-Lysosome Contact Sites and Selectively Deliver Ca2+ to Lysosomes. Cell Rep. 2018, 25, 3180–3193. [Google Scholar] [CrossRef] [PubMed]
- Christensen, K.A.; Myers, J.T.; Swanson, J.A. pH-dependent regulation of lysosomal calcium in macrophages. J. Cell Sci. 2002, 115 Pt 3, 599–607. [Google Scholar]
- Lloyd-Evans, E.; Morgan, A.J.; He, X.; Smith, D.A.; Elliot-Smith, E.; Sillence, D.J.; Churchill, G.C.; Schuchman, E.H.; Galione, A.; Platt, F.M. Niemann-Pick disease type C1 is a sphingosine storage disease that causes deregulation of lysosomal calcium. Nat. Med. 2008, 14, 1247–1255. [Google Scholar] [CrossRef]
- Kilpatrick, B.S.; Eden, E.R.; Schapira, A.H.; Futter, C.E.; Patel, S. Direct mobilisation of lysosomal Ca2+ triggers complex Ca2+ signals. J. Cell Sci. 2013, 126 Pt 1, 60–66. [Google Scholar] [CrossRef]
- Ronco, V.; Potenza, D.M.; Denti, F.; Vullo, S.; Gagliano, G.; Tognolina, M.; Guerra, G.; Pinton, P.; Genazzani, A.A.; Mapelli, L.; et al. A novel Ca2+-mediated cross-talk between endoplasmic reticulum and acidic organelles: Implications for NAADP-dependent Ca2+ signalling. Cell Calcium 2015, 57, 89–100. [Google Scholar] [CrossRef]
- Zuccolo, E.; Dragoni, S.; Poletto, V.; Catarsi, P.; Guido, D.; Rappa, A.; Reforgiato, M.; Lodola, F.; Lim, D.; Rosti, V.; et al. Arachidonic acid-evoked Ca2+ signals promote nitric oxide release and proliferation in human endothelial colony forming cells. Vasc. Pharmacol. 2016, 87, 159–171. [Google Scholar] [CrossRef] [PubMed]
- Zhong, X.Z.; Yang, Y.; Sun, X.; Dong, X.P. Methods for monitoring Ca2+ and ion channels in the lysosome. Cell Calcium 2017, 64, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Medina, D.L.; Di Paola, S.; Peluso, I.; Armani, A.; De Stefani, D.; Venditti, R.; Montefusco, S.; Scotto-Rosato, A.; Prezioso, C.; Forrester, A.; et al. Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB. Nat. Cell Biol. 2015, 17, 288–299. [Google Scholar] [CrossRef] [PubMed]
- Korolchuk, V.I.; Saiki, S.; Lichtenberg, M.; Siddiqi, F.H.; Roberts, E.A.; Imarisio, S.; Jahreiss, L.; Sarkar, S.; Futter, M.; Menzies, F.M.; et al. Lysosomal positioning coordinates cellular nutrient responses. Nat. Cell Biol. 2011, 13, 453–460. [Google Scholar] [CrossRef] [PubMed]
- Efeyan, A.; Zoncu, R.; Sabatini, D.M. Amino acids and mTORC1: From lysosomes to disease. Trends Mol. Med. 2012, 18, 524–533. [Google Scholar] [CrossRef] [PubMed]
- Rabanal-Ruiz, Y.; Korolchuk, V.I. mTORC1 and Nutrient Homeostasis: The Central Role of the Lysosome. Int. J. Mol. Sci. 2018, 19, 818. [Google Scholar] [CrossRef] [PubMed]
- Sardiello, M.; Palmieri, M.; di Ronza, A.; Medina, D.L.; Valenza, M.; Gennarino, V.A.; Di Malta, C.; Donaudy, F.; Embrione, V.; Polishchuk, R.S.; et al. A gene network regulating lysosomal biogenesis and function. Science 2009, 325, 473–477. [Google Scholar] [CrossRef]
- Palmieri, M.; Impey, S.; Kang, H.; di Ronza, A.; Pelz, C.; Sardiello, M.; Ballabio, A. Characterization of the CLEAR network reveals an integrated control of cellular clearance pathways. Hum. Mol. Genet. 2011, 20, 3852–3866. [Google Scholar] [CrossRef]
- Settembre, C.; Fraldi, A.; Medina, D.L.; Ballabio, A. Signals from the lysosome: A control centre for cellular clearance and energy metabolism. Nat. Rev. Mol. Cell Biol. 2013, 14, 283–296. [Google Scholar] [CrossRef]
- Settembre, C.; Di Malta, C.; Polito, V.A.; Garcia Arencibia, M.; Vetrini, F.; Erdin, S.; Erdin, S.U.; Huynh, T.; Medina, D.; Colella, P.; et al. TFEB links autophagy to lysosomal biogenesis. Science 2011, 332, 1429–1433. [Google Scholar] [CrossRef]
- Morgan, A.J. Ca2+ dialogue between acidic vesicles and ER. Biochem. Soc. Trans. 2016, 44, 546–553. [Google Scholar] [CrossRef] [PubMed]
- Churchill, G.C.; Okada, Y.; Thomas, J.M.; Genazzani, A.A.; Patel, S.; Galione, A. NAADP mobilizes Ca2+ from reserve granules, lysosome-related organelles, in sea urchin eggs. Cell 2002, 111, 703–708. [Google Scholar] [CrossRef]
- Srinivas, S.P.; Ong, A.; Goon, L.; Goon, L.; Bonanno, J.A. Lysosomal Ca2+ stores in bovine corneal endothelium. Investig. Ophthalmol. Vis. Sci. 2002, 43, 2341–2350. [Google Scholar]
- Melchionda, M.; Pittman, J.K.; Mayor, R.; Patel, S. Ca2+/H+ exchange by acidic organelles regulates cell migration in vivo. J. Cell Biol. 2016, 212, 803–813. [Google Scholar] [CrossRef] [PubMed]
- Inesi, G.; Tadini-Buoninsegni, F. Ca2+/H+ exchange, lumenal Ca2+ release and Ca (2+)/ATP coupling ratios in the sarcoplasmic reticulum ATPase. J. Cell Commun. Signal. 2014, 8, 5–11. [Google Scholar] [CrossRef] [PubMed]
- Lopez, J.J.; Camello-Almaraz, C.; Pariente, J.A.; Salido, G.M.; Rosado, J.A. Ca2+ accumulation into acidic organelles mediated by Ca2+- and vacuolar H+-ATPases in human platelets. Biochem. J. 2005, 390 Pt 1, 243–252. [Google Scholar] [CrossRef]
- Lopez Sanjurjo, C.I.; Tovey, S.C.; Taylor, C.W. Rapid recycling of Ca2+ between IP3-sensitive stores and lysosomes. PLoS ONE 2014, 9, e111275. [Google Scholar] [CrossRef]
- Lopez-Sanjurjo, C.I.; Tovey, S.C.; Prole, D.L.; Taylor, C.W. Lysosomes shape Ins(1,4,5)P3-evoked Ca2+ signals by selectively sequestering Ca2+ released from the endoplasmic reticulum. J. Cell Sci. 2013, 126 Pt 1, 289–300. [Google Scholar] [CrossRef]
- Garrity, A.G.; Wang, W.; Collier, C.M.; Levey, S.A.; Gao, Q.; Xu, H. The endoplasmic reticulum, not the pH gradient, drives calcium refilling of lysosomes. eLife 2016, 5, e15887. [Google Scholar] [CrossRef]
- Sbano, L.; Bonora, M.; Marchi, S.; Baldassari, F.; Medina, D.L.; Ballabio, A.; Giorgi, C.; Pinton, P. TFEB-mediated increase in peripheral lysosomes regulates store-operated calcium entry. Sci. Rep. 2017, 7, 40797. [Google Scholar] [CrossRef]
- Xiong, J.; Zhu, M.X. Regulation of lysosomal ion homeostasis by channels and transporters. Sci. China Life Sci. 2016, 59, 777–791. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Zhang, X.; Gao, Q.; Lawas, M.; Yu, L.; Cheng, X.; Gu, M.; Sahoo, N.; Li, X.; Li, P.; et al. A voltage-dependent K(+) channel in the lysosome is required for refilling lysosomal Ca2+ stores. J. Cell Biol. 2017, 216, 1715–1730. [Google Scholar] [CrossRef] [PubMed]
- Cao, Q.; Zhong, X.Z.; Zou, Y.; Zhang, Z.; Toro, L.; Dong, X.P. BK Channels Alleviate Lysosomal Storage Diseases by Providing Positive Feedback Regulation of Lysosomal Ca2+ Release. Dev. Cell 2015, 33, 427–441. [Google Scholar] [CrossRef] [PubMed]
- Di Paola, S.; Scotto-Rosato, A.; Medina, D.L. TRPML1: The Ca(2+)retaker of the lysosome. Cell Calcium 2018, 69, 112–121. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Shen, D.; Samie, M.; Xu, H. Mucolipins: Intracellular TRPML1-3 channels. FEBS Lett. 2010, 584, 2013–2021. [Google Scholar] [CrossRef] [PubMed]
- Grimm, C.; Hassan, S.; Wahl-Schott, C.; Biel, M. Role of TRPML and two-pore channels in endolysosomal cation homeostasis. J. Pharmacol. Exp. Ther. 2012, 342, 236–244. [Google Scholar] [CrossRef] [PubMed]
- Gees, M.; Colsoul, B.; Nilius, B. The role of transient receptor potential cation channels in Ca2+ signaling. Cold Spring Harb. Perspect. Biol. 2010, 2, a003962. [Google Scholar] [CrossRef] [PubMed]
- Venkatachalam, K.; Hofmann, T.; Montell, C. Lysosomal localization of TRPML3 depends on TRPML2 and the mucolipidosis-associated protein TRPML1. J. Biol. Chem. 2006, 281, 17517–17527. [Google Scholar] [CrossRef]
- Pryor, P.R.; Reimann, F.; Gribble, F.M.; Luzio, J.P. Mucolipin-1 is a lysosomal membrane protein required for intracellular lactosylceramide traffic. Traffic 2006, 7, 1388–1398. [Google Scholar] [CrossRef]
- Medina, D.L.; Fraldi, A.; Bouche, V.; Annunziata, F.; Mansueto, G.; Spampanato, C.; Puri, C.; Pignata, A.; Martina, J.A.; Sardiello, M.; et al. Transcriptional activation of lysosomal exocytosis promotes cellular clearance. Dev. Cell 2011, 21, 421–430. [Google Scholar] [CrossRef]
- Dong, X.P.; Cheng, X.; Mills, E.; Delling, M.; Wang, F.; Kurz, T.; Xu, H. The type IV mucolipidosis-associated protein TRPML1 is an endolysosomal iron release channel. Nature 2008, 455, 992–996. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Delling, M.; Li, L.; Dong, X.; Clapham, D.E. Activating mutation in a mucolipin transient receptor potential channel leads to melanocyte loss in varitint-waddler mice. Proc. Natl. Acad. Sci. USA 2007, 104, 18321–18326. [Google Scholar] [CrossRef] [PubMed]
- Miedel, M.T.; Rbaibi, Y.; Guerriero, C.J.; Colletti, G.; Weixel, K.M.; Weisz, O.A.; Kiselyov, K. Membrane traffic and turnover in TRP-ML1-deficient cells: A revised model for mucolipidosis type IV pathogenesis. J. Exp. Med. 2008, 205, 1477–1490. [Google Scholar] [CrossRef] [PubMed]
- Raychowdhury, M.K.; Gonzalez-Perrett, S.; Montalbetti, N.; Timpanaro, G.A.; Chasan, B.; Goldmann, W.H.; Stahl, S.; Cooney, A.; Goldin, E.; Cantiello, H.F. Molecular pathophysiology of mucolipidosis type IV: pH dysregulation of the mucolipin-1 cation channel. Hum. Mol. Genet. 2004, 13, 617–627. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Zhang, W.K.; Benvin, N.M.; Zhou, X.; Su, D.; Li, H.; Wang, S.; Michailidis, I.E.; Tong, L.; Li, X.; et al. Structural basis of dual Ca2+/pH regulation of the endolysosomal TRPML1 channel. Nat. Struct. Mol. Biol. 2017, 24, 205–213. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Li, N.; Zeng, W.; Gao, N.; Yang, M. Cryo-EM structures of the mammalian endo-lysosomal TRPML1 channel elucidate the combined regulation mechanism. Protein Cell 2017, 8, 834–847. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.P.; Shen, D.; Wang, X.; Dawson, T.; Li, X.; Zhang, Q.; Cheng, X.; Zhang, Y.; Weisman, L.S.; Delling, M.; et al. PI(3,5)P(2) controls membrane trafficking by direct activation of mucolipin Ca2+ release channels in the endolysosome. Nat. Commun. 2010, 1, 38. [Google Scholar] [CrossRef]
- Zhang, X.; Li, X.; Xu, H. Phosphoinositide isoforms determine compartment-specific ion channel activity. Proc. Natl. Acad. Sci. USA 2012, 109, 11384–11389. [Google Scholar] [CrossRef]
- Zhang, X.; Cheng, X.; Yu, L.; Yang, J.; Calvo, R.; Patnaik, S.; Hu, X.; Gao, Q.; Yang, M.; Lawas, M.; et al. MCOLN1 is a ROS sensor in lysosomes that regulates autophagy. Nat. Commun. 2016, 7, 12109. [Google Scholar] [CrossRef]
- Grimm, C.; Butz, E.; Chen, C.C.; Wahl-Schott, C.; Biel, M. From mucolipidosis type IV to Ebola: TRPML and two-pore channels at the crossroads of endo-lysosomal trafficking and disease. Cell Calcium 2017, 67, 148–155. [Google Scholar] [CrossRef]
- Zhang, X.; Hu, M.; Yang, Y.; Xu, H. Organellar TRP channels. Nat. Struct. Mol. Biol. 2018, 25, 1009–1018. [Google Scholar] [CrossRef] [PubMed]
- Li, R.J.; Xu, J.; Fu, C.; Zhang, J.; Zheng, Y.G.; Jia, H.; Liu, J.O. Regulation of mTORC1 by lysosomal calcium and calmodulin. eLife 2016, 5, e19360. [Google Scholar] [CrossRef] [PubMed]
- Settembre, C.; Zoncu, R.; Medina, D.L.; Vetrini, F.; Erdin, S.; Erdin, S.; Huynh, T.; Ferron, M.; Karsenty, G.; Vellard, M.C.; et al. A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J. 2012, 31, 1095–1108. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Gao, Q.; Yang, M.; Zhang, X.; Yu, L.; Lawas, M.; Li, X.; Bryant-Genevier, M.; Southall, N.T.; Marugan, J.; et al. Up-regulation of lysosomal TRPML1 channels is essential for lysosomal adaptation to nutrient starvation. Proc. Natl. Acad. Sci. USA 2015, 112, E1373–E1381. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Yang, Y.; Zhong, X.Z.; Cao, Q.; Zhu, X.H.; Zhu, X.; Dong, X.P. A negative feedback regulation of MTORC1 activity by the lysosomal Ca2+ channel MCOLN1 (mucolipin 1) using a CALM (calmodulin)-dependent mechanism. Autophagy 2018, 14, 38–52. [Google Scholar] [CrossRef] [PubMed]
- De Leo, M.G.; Staiano, L.; Vicinanza, M.; Luciani, A.; Carissimo, A.; Mutarelli, M.; Di Campli, A.; Polishchuk, E.; Di Tullio, G.; Morra, V.; et al. Autophagosome-lysosome fusion triggers a lysosomal response mediated by TLR9 and controlled by OCRL. Nat. Cell Biol. 2016, 18, 839–850. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Anoveros, J.; Wiwatpanit, T. TRPML2 and mucolipin evolution. Handb. Exp. Pharmacol. 2014, 222, 647–658. [Google Scholar] [PubMed]
- Li, X.; Rydzewski, N.; Hider, A.; Zhang, X.; Yang, J.; Wang, W.; Gao, Q.; Cheng, X.; Xu, H. A molecular mechanism to regulate lysosome motility for lysosome positioning and tubulation. Nat. Cell Biol. 2016, 18, 404–417. [Google Scholar] [CrossRef]
- Lev, S.; Zeevi, D.A.; Frumkin, A.; Offen-Glasner, V.; Bach, G.; Minke, B. Constitutive activity of the human TRPML2 channel induces cell degeneration. J. Biol. Chem. 2010, 285, 2771–2782. [Google Scholar] [CrossRef]
- Karacsonyi, C.; Miguel, A.S.; Puertollano, R. Mucolipin-2 localizes to the Arf6-associated pathway and regulates recycling of GPI-APs. Traffic 2007, 8, 1404–1414. [Google Scholar] [CrossRef]
- Curcio-Morelli, C.; Zhang, P.; Venugopal, B.; Charles, F.A.; Browning, M.F.; Cantiello, H.F.; Slaugenhaupt, S.A. Functional multimerization of mucolipin channel proteins. J. Cell. Physiol. 2010, 222, 328–335. [Google Scholar] [CrossRef] [PubMed]
- Zeevi, D.A.; Lev, S.; Frumkin, A.; Minke, B.; Bach, G. Heteromultimeric TRPML channel assemblies play a crucial role in the regulation of cell viability models and starvation-induced autophagy. J. Cell Sci. 2010, 123 Pt 18, 3112–3124. [Google Scholar] [CrossRef]
- Zeevi, D.A.; Frumkin, A.; Offen-Glasner, V.; Kogot-Levin, A.; Bach, G. A potentially dynamic lysosomal role for the endogenous TRPML proteins. J. Pathol. 2009, 219, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Li, Q.; Tjon-Kon-Sang, S.; So, I.; Kiselyov, K.; Muallem, S. Gain-of-function mutation in TRPML3 causes the mouse Varitint-Waddler phenotype. J. Biol. Chem. 2007, 282, 36138–36142. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Yamaguchi, S.; Li, Q.; So, I.; Muallem, S. Properties of the TRPML3 channel pore and its stable expansion by the Varitint-Waddler-causing mutation. J. Biol. Chem. 2010, 285, 16513–16520. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Li, Q.; Tjon-Kon-Sang, S.; So, I.; Kiselyov, K.; Soyombo, A.A.; Muallem, S. A novel mode of TRPML3 regulation by extracytosolic pH absent in the varitint-waddler phenotype. EMBO J. 2008, 27, 1197–1205. [Google Scholar] [CrossRef] [PubMed]
- Grimm, C.; Cuajungco, M.P.; van Aken, A.F.; Schnee, M.; Jors, S.; Kros, C.J.; Ricci, A.J.; Heller, S. A helix-breaking mutation in TRPML3 leads to constitutive activity underlying deafness in the varitint-waddler mouse. Proc. Natl. Acad. Sci. USA 2007, 104, 19583–19588. [Google Scholar] [CrossRef]
- Feng, X.; Huang, Y.; Lu, Y.; Xiong, J.; Wong, C.O.; Yang, P.; Xia, J.; Chen, D.; Du, G.; Venkatachalam, K.; et al. Drosophila TRPML forms PI(3,5)P2-activated cation channels in both endolysosomes and plasma membrane. J. Biol. Chem. 2014, 289, 4262–4272. [Google Scholar] [CrossRef]
- Grimm, C.; Barthmes, M.; Wahl-Schott, C. Trpml3. Handb. Exp. Pharmacol. 2014, 222, 659–674. [Google Scholar]
- Miao, Y.; Li, G.; Zhang, X.; Xu, H.; Abraham, S.N. A TRP Channel Senses Lysosome Neutralization by Pathogens to Trigger Their Expulsion. Cell 2015, 161, 1306–1319. [Google Scholar] [CrossRef]
- Kim, H.J.; Soyombo, A.A.; Tjon-Kon-Sang, S.; So, I.; Muallem, S. The Ca2+ channel TRPML3 regulates membrane trafficking and autophagy. Traffic 2009, 10, 1157–1167. [Google Scholar] [CrossRef]
- Choi, S.; Kim, H.J. The Ca2+ channel TRPML3 specifically interacts with the mammalian ATG8 homologue GATE16 to regulate autophagy. Biochem. Biophys. Res. Commun. 2014, 443, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Patel, S. Function and dysfunction of two-pore channels. Sci. Signal. 2015, 8, re7. [Google Scholar] [CrossRef]
- Yu, F.H.; Catterall, W.A. The VGL-chanome: A protein superfamily specialized for electrical signaling and ionic homeostasis. Sci. STKE 2004, 2004, re15. [Google Scholar] [CrossRef] [PubMed]
- Ishibashi, K.; Suzuki, M.; Imai, M. Molecular cloning of a novel form (two-repeat) protein related to voltage-gated sodium and calcium channels. Biochem. Biophys. Res. Commun. 2000, 270, 370–376. [Google Scholar] [CrossRef]
- Furuichi, T.; Cunningham, K.W.; Muto, S. A putative two pore channel AtTPC1 mediates Ca2+ flux in Arabidopsis leaf cells. Plant Cell Physiol. 2001, 42, 900–905. [Google Scholar] [CrossRef] [PubMed]
- Churamani, D.; Hooper, R.; Brailoiu, E.; Patel, S. Domain assembly of NAADP-gated two-pore channels. Biochem. J. 2012, 441, 317–323. [Google Scholar] [CrossRef]
- Rietdorf, K.; Funnell, T.M.; Ruas, M.; Heinemann, J.; Parrington, J.; Galione, A. Two-pore channels form homo- and heterodimers. J. Biol. Chem. 2011, 286, 37058–37062. [Google Scholar] [CrossRef]
- Hooper, R.; Churamani, D.; Brailoiu, E.; Taylor, C.W.; Patel, S. Membrane topology of NAADP-sensitive two-pore channels and their regulation by N-linked glycosylation. J. Biol. Chem. 2011, 286, 9141–9149. [Google Scholar] [CrossRef]
- Rahman, T.; Cai, X.; Brailoiu, G.C.; Abood, M.E.; Brailoiu, E.; Patel, S. Two-pore channels provide insight into the evolution of voltage-gated Ca2+ and Na+ channels. Sci. Signal. 2014, 7, ra109. [Google Scholar] [CrossRef]
- Larisch, N.; Kirsch, S.A.; Schambony, A.; Studtrucker, T.; Bockmann, R.A.; Dietrich, P. The function of the two-pore channel TPC1 depends on dimerization of its carboxy-terminal helix. Cell. Mol. Life Sci. 2016, 73, 2565–2581. [Google Scholar] [CrossRef] [PubMed]
- Brailoiu, E.; Hooper, R.; Cai, X.; Brailoiu, G.C.; Keebler, M.V.; Dun, N.J.; Marchant, J.S.; Patel, S. An ancestral deuterostome family of two-pore channels mediates nicotinic acid adenine dinucleotide phosphate-dependent calcium release from acidic organelles. J. Biol. Chem. 2010, 285, 2897–2901. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Patel, S. Degeneration of an intracellular ion channel in the primate lineage by relaxation of selective constraints. Mol. Biol. Evol. 2010, 27, 2352–2359. [Google Scholar] [CrossRef] [PubMed]
- Kintzer, A.F.; Stroud, R.M. Structure, inhibition and regulation of two-pore channel TPC1 from Arabidopsis thaliana. Nature 2016, 531, 258–262. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Zeng, W.; Chen, Q.; Lee, C.; Chen, L.; Yang, Y.; Cang, C.; Ren, D.; Jiang, Y. Structure of the voltage-gated two-pore channel TPC1 from Arabidopsis thaliana. Nature 2016, 531, 196–201. [Google Scholar] [CrossRef] [PubMed]
- She, J.; Guo, J.; Chen, Q.; Zeng, W.; Jiang, Y.; Bai, X.C. Structural insights into the voltage and phospholipid activation of the mammalian TPC1 channel. Nature 2018, 556, 130–134. [Google Scholar] [CrossRef] [PubMed]
- Calcraft, P.J.; Ruas, M.; Pan, Z.; Cheng, X.; Arredouani, A.; Hao, X.; Tang, J.; Rietdorf, K.; Teboul, L.; Chuang, K.T.; et al. NAADP mobilizes calcium from acidic organelles through two-pore channels. Nature 2009, 459, 596–600. [Google Scholar] [CrossRef]
- Zong, X.; Schieder, M.; Cuny, H.; Fenske, S.; Gruner, C.; Rotzer, K.; Griesbeck, O.; Harz, H.; Biel, M.; Wahl-Schott, C. The two-pore channel TPCN2 mediates NAADP-dependent Ca2+-release from lysosomal stores. Pflugers Arch. 2009, 458, 891–899. [Google Scholar] [CrossRef]
- Ruas, M.; Rietdorf, K.; Arredouani, A.; Davis, L.C.; Lloyd-Evans, E.; Koegel, H.; Funnell, T.M.; Morgan, A.J.; Ward, J.A.; Watanabe, K.; et al. Purified TPC isoforms form NAADP receptors with distinct roles for Ca2+ signaling and endolysosomal trafficking. Curr. Biol. 2010, 20, 703–709. [Google Scholar] [CrossRef]
- Brailoiu, E.; Rahman, T.; Churamani, D.; Prole, D.L.; Brailoiu, G.C.; Hooper, R.; Taylor, C.W.; Patel, S. An NAADP-gated two-pore channel targeted to the plasma membrane uncouples triggering from amplifying Ca2+ signals. J. Biol. Chem. 2010, 285, 38511–38516. [Google Scholar] [CrossRef]
- Fameli, N.; Ogunbayo, O.A.; van Breemen, C.; Evans, A.M. Cytoplasmic nanojunctions between lysosomes and sarcoplasmic reticulum are required for specific calcium signaling. F1000Research 2014, 3, 93. [Google Scholar] [CrossRef] [PubMed]
- Kinnear, N.P.; Wyatt, C.N.; Clark, J.H.; Calcraft, P.J.; Fleischer, S.; Jeyakumar, L.H.; Nixon, G.F.; Evans, A.M. Lysosomes co-localize with ryanodine receptor subtype 3 to form a trigger zone for calcium signalling by NAADP in rat pulmonary arterial smooth muscle. Cell Calcium 2008, 44, 190–201. [Google Scholar] [CrossRef] [PubMed]
- Grimm, C.; Chen, C.C.; Wahl-Schott, C.; Biel, M. Two-Pore Channels: Catalyzers of Endolysosomal Transport and Function. Front. Pharmacol. 2017, 8, 45. [Google Scholar] [CrossRef] [PubMed]
- Pitt, S.J.; Reilly-O’Donnell, B.; Sitsapesan, R. Exploring the biophysical evidence that mammalian two-pore channels are NAADP-activated calcium-permeable channels. J. Physiol. 2016, 594, 4171–4179. [Google Scholar] [CrossRef] [PubMed]
- Morgan, A.J.; Davis, L.C.; Ruas, M.; Galione, A. TPC: The NAADP discovery channel? Biochem. Soc. Trans. 2015, 43, 384–389. [Google Scholar] [CrossRef] [PubMed]
- Morgan, A.J.; Galione, A. Two-pore channels (TPCs): Current controversies. Bioessays 2014, 36, 173–183. [Google Scholar] [CrossRef] [PubMed]
- Guse, A.H.; Diercks, B.P. Integration of nicotinic acid adenine dinucleotide phosphate (NAADP)-dependent calcium signalling. J. Physiol. 2018, 596, 2735–2743. [Google Scholar] [CrossRef] [PubMed]
- Brailoiu, E.; Churamani, D.; Cai, X.; Schrlau, M.G.; Brailoiu, G.C.; Gao, X.; Hooper, R.; Boulware, M.J.; Dun, N.J.; Marchant, J.S.; et al. Essential requirement for two-pore channel 1 in NAADP-mediated calcium signaling. J. Cell Biol. 2009, 186, 201–209. [Google Scholar] [CrossRef]
- Ruas, M.; Davis, L.C.; Chen, C.C.; Morgan, A.J.; Chuang, K.T.; Walseth, T.F.; Grimm, C.; Garnham, C.; Powell, T.; Platt, N.; et al. Expression of Ca2+-permeable two-pore channels rescues NAADP signalling in TPC-deficient cells. EMBO J. 2015, 34, 1743–1758. [Google Scholar] [CrossRef]
- Davis, L.C.; Morgan, A.J.; Chen, J.L.; Snead, C.M.; Bloor-Young, D.; Shenderov, E.; Stanton-Humphreys, M.N.; Conway, S.J.; Churchill, G.C.; Parrington, J.; et al. NAADP activates two-pore channels on T cell cytolytic granules to stimulate exocytosis and killing. Curr. Biol. 2012, 22, 2331–2337. [Google Scholar] [CrossRef]
- Tugba Durlu-Kandilci, N.; Ruas, M.; Chuang, K.T.; Brading, A.; Parrington, J.; Galione, A. TPC2 proteins mediate nicotinic acid adenine dinucleotide phosphate (NAADP)- and agonist-evoked contractions of smooth muscle. J. Biol. Chem. 2010, 285, 24925–24932. [Google Scholar] [CrossRef] [PubMed]
- Pitt, S.J.; Funnell, T.M.; Sitsapesan, M.; Venturi, E.; Rietdorf, K.; Ruas, M.; Ganesan, A.; Gosain, R.; Churchill, G.C.; Zhu, M.X.; et al. TPC2 is a novel NAADP-sensitive Ca2+ release channel, operating as a dual sensor of luminal pH and Ca2+. J. Biol. Chem. 2010, 285, 35039–35046. [Google Scholar] [CrossRef] [PubMed]
- Pitt, S.J.; Lam, A.K.; Rietdorf, K.; Galione, A.; Sitsapesan, R. Reconstituted human TPC1 is a proton-permeable ion channel and is activated by NAADP or Ca2+. Sci. Signal. 2014, 7, ra46. [Google Scholar] [CrossRef] [PubMed]
- Rybalchenko, V.; Ahuja, M.; Coblentz, J.; Churamani, D.; Patel, S.; Kiselyov, K.; Muallem, S. Membrane potential regulates nicotinic acid adenine dinucleotide phosphate (NAADP) dependence of the pH- and Ca2+-sensitive organellar two-pore channel TPC1. J. Biol. Chem. 2012, 287, 20407–20416. [Google Scholar] [CrossRef] [PubMed]
- Jha, A.; Ahuja, M.; Patel, S.; Brailoiu, E.; Muallem, S. Convergent regulation of the lysosomal two-pore channel-2 by Mg(2)(+), NAADP, PI(3,5)P(2) and multiple protein kinases. EMBO J. 2014, 33, 501–511. [Google Scholar] [CrossRef] [PubMed]
- Schieder, M.; Rotzer, K.; Bruggemann, A.; Biel, M.; Wahl-Schott, C.A. Characterization of two-pore channel 2 (TPCN2)-mediated Ca2+ currents in isolated lysosomes. J. Biol. Chem. 2010, 285, 21219–21222. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhang, X.; Dong, X.P.; Samie, M.; Li, X.; Cheng, X.; Goschka, A.; Shen, D.; Zhou, Y.; Harlow, J.; et al. TPC proteins are phosphoinositide- activated sodium-selective ion channels in endosomes and lysosomes. Cell 2012, 151, 372–383. [Google Scholar] [CrossRef] [PubMed]
- Cang, C.; Zhou, Y.; Navarro, B.; Seo, Y.J.; Aranda, K.; Shi, L.; Battaglia-Hsu, S.; Nissim, I.; Clapham, D.E.; Ren, D. mTOR regulates lysosomal ATP-sensitive two-pore Na(+) channels to adapt to metabolic state. Cell 2013, 152, 778–790. [Google Scholar] [CrossRef] [PubMed]
- Ruas, M.; Galione, A.; Parrington, J. Two-Pore Channels: Lessons from Mutant Mouse Models. Messenger 2015, 4, 4–22. [Google Scholar] [CrossRef]
- Walseth, T.F.; Lin-Moshier, Y.; Jain, P.; Ruas, M.; Parrington, J.; Galione, A.; Marchant, J.S.; Slama, J.T. Photoaffinity labeling of high affinity nicotinic acid adenine dinucleotide phosphate (NAADP)-binding proteins in sea urchin egg. J. Biol. Chem. 2012, 287, 2308–2315. [Google Scholar] [CrossRef]
- Lin-Moshier, Y.; Walseth, T.F.; Churamani, D.; Davidson, S.M.; Slama, J.T.; Hooper, R.; Brailoiu, E.; Patel, S.; Marchant, J.S. Photoaffinity labeling of nicotinic acid adenine dinucleotide phosphate (NAADP) targets in mammalian cells. J. Biol. Chem. 2012, 287, 2296–2307. [Google Scholar] [CrossRef] [PubMed]
- Cang, C.; Bekele, B.; Ren, D. The voltage-gated sodium channel TPC1 confers endolysosomal excitability. Nat. Chem. Biol. 2014, 10, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Suaga, P.; Luzon-Toro, B.; Churamani, D.; Zhang, L.; Bloor-Young, D.; Patel, S.; Woodman, P.G.; Churchill, G.C.; Hilfiker, S. Leucine-rich repeat kinase 2 regulates autophagy through a calcium-dependent pathway involving NAADP. Hum. Mol. Genet. 2012, 21, 511–525. [Google Scholar] [CrossRef] [PubMed]
- Pryor, P.R.; Mullock, B.M.; Bright, N.A.; Gray, S.R.; Luzio, J.P. The role of intraorganellar Ca2+ in late endosome-lysosome heterotypic fusion and in the reformation of lysosomes from hybrid organelles. J. Cell Biol. 2000, 149, 1053–1062. [Google Scholar] [CrossRef] [PubMed]
- Marchant, J.S.; Patel, S. Two-pore channels at the intersection of endolysosomal membrane traffic. Biochem. Soc. Trans. 2015, 43, 434–441. [Google Scholar] [CrossRef] [PubMed]
- Hockey, L.N.; Kilpatrick, B.S.; Eden, E.R.; Lin-Moshier, Y.; Brailoiu, G.C.; Brailoiu, E.; Futter, C.E.; Schapira, A.H.; Marchant, J.S.; Patel, S. Dysregulation of lysosomal morphology by pathogenic LRRK2 is corrected by TPC2 inhibition. J. Cell Sci. 2015, 128, 232–238. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, Y.; Kolokoltsov, A.A.; Chen, C.C.; Tidwell, M.W.; Bauta, W.E.; Klugbauer, N.; Grimm, C.; Wahl-Schott, C.; Biel, M.; Davey, R.A. Ebola virus. Two-pore channels control Ebola virus host cell entry and are drug targets for disease treatment. Science 2015, 347, 995–998. [Google Scholar] [CrossRef] [PubMed]
- Grimm, C.; Holdt, L.M.; Chen, C.C.; Hassan, S.; Muller, C.; Jors, S.; Cuny, H.; Kissing, S.; Schroder, B.; Butz, E.; et al. High susceptibility to fatty liver disease in two-pore channel 2-deficient mice. Nat. Commun. 2014, 5, 4699. [Google Scholar] [CrossRef]
- Kilpatrick, B.S.; Eden, E.R.; Hockey, L.N.; Yates, E.; Futter, C.E.; Patel, S. An Endosomal NAADP-Sensitive Two-Pore Ca2+ Channel Regulates ER-Endosome Membrane Contact Sites to Control Growth Factor Signaling. Cell Rep. 2017, 18, 1636–1645. [Google Scholar] [CrossRef]
- Ruas, M.; Chuang, K.T.; Davis, L.C.; Al-Douri, A.; Tynan, P.W.; Tunn, R.; Teboul, L.; Galione, A.; Parrington, J. TPC1 has two variant isoforms, and their removal has different effects on endo-lysosomal functions compared to loss of TPC2. Mol. Cell. Biol. 2014, 34, 3981–3992. [Google Scholar] [CrossRef]
- Lin-Moshier, Y.; Keebler, M.V.; Hooper, R.; Boulware, M.J.; Liu, X.; Churamani, D.; Abood, M.E.; Walseth, T.F.; Brailoiu, E.; Patel, S.; et al. The Two-pore channel (TPC) interactome unmasks isoform-specific roles for TPCs in endolysosomal morphology and cell pigmentation. Proc. Natl. Acad. Sci. USA 2014, 111, 13087–13092. [Google Scholar] [CrossRef] [PubMed]
- Atlas, D. Voltage-gated calcium channels function as Ca2+-activated signaling receptors. Trends Biochem. Sci. 2014, 39, 45–52. [Google Scholar] [CrossRef]
- Ogunbayo, O.A.; Duan, J.; Xiong, J.; Wang, Q.; Feng, X.; Ma, J.; Zhu, M.X.; Evans, A.M. mTORC1 controls lysosomal Ca2+ release through the two-pore channel TPC2. Sci. Signal. 2018, 11, eaao5775. [Google Scholar] [CrossRef] [PubMed]
- Hoglinger, D.; Haberkant, P.; Aguilera-Romero, A.; Riezman, H.; Porter, F.D.; Platt, F.M.; Galione, A.; Schultz, C. Intracellular sphingosine releases calcium from lysosomes. eLife 2015, 4, e10616. [Google Scholar] [CrossRef] [PubMed]
- Rah, S.Y.; Lee, Y.H.; Kim, U.H. NAADP-mediated Ca2+ signaling promotes autophagy and protects against LPS-induced liver injury. FASEB J. 2017, 31, 3126–3137. [Google Scholar] [CrossRef]
- Pereira, G.J.; Hirata, H.; Fimia, G.M.; do Carmo, L.G.; Bincoletto, C.; Han, S.W.; Stilhano, R.S.; Ureshino, R.P.; Bloor-Young, D.; Churchill, G.; et al. Nicotinic acid adenine dinucleotide phosphate (NAADP) regulates autophagy in cultured astrocytes. J. Biol. Chem. 2011, 286, 27875–27881. [Google Scholar] [CrossRef]
- Pereira, G.J.; Antonioli, M.; Hirata, H.; Ureshino, R.P.; Nascimento, A.R.; Bincoletto, C.; Vescovo, T.; Piacentini, M.; Fimia, G.M.; Smaili, S.S. Glutamate induces autophagy via the two-pore channels in neural cells. Oncotarget 2017, 8, 12730–12740. [Google Scholar] [CrossRef]
- Garcia-Rua, V.; Feijoo-Bandin, S.; Rodriguez-Penas, D.; Mosquera-Leal, A.; Abu-Assi, E.; Beiras, A.; Maria Seoane, L.; Lear, P.; Parrington, J.; Portoles, M.; et al. Endolysosomal two-pore channels regulate autophagy in cardiomyocytes. J. Physiol. 2016, 594, 3061–3077. [Google Scholar] [CrossRef]
- Lin, P.H.; Duann, P.; Komazaki, S.; Park, K.H.; Li, H.; Sun, M.; Sermersheim, M.; Gumpper, K.; Parrington, J.; Galione, A.; et al. Lysosomal two-pore channel subtype 2 (TPC2) regulates skeletal muscle autophagic signaling. J. Biol. Chem. 2015, 290, 3377–3389. [Google Scholar] [CrossRef]
- Cancela, J.M.; Van Coppenolle, F.; Galione, A.; Tepikin, A.V.; Petersen, O.H. Transformation of local Ca2+ spikes to global Ca2+ transients: The combinatorial roles of multiple Ca2+ releasing messengers. EMBO J. 2002, 21, 909–919. [Google Scholar] [CrossRef]
- Churchill, G.C.; Galione, A. NAADP induces Ca2+ oscillations via a two-pool mechanism by priming IP3- and cADPR-sensitive Ca2+ stores. EMBO J. 2001, 20, 2666–2671. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.L.; Lin, A.H.; Xia, Y.; Lee, S.; Paudel, O.; Sun, H.; Yang, X.R.; Ran, P.; Sham, J.S. Nicotinic acid adenine dinucleotide phosphate (NAADP) activates global and heterogeneous local Ca2+ signals from NAADP- and ryanodine receptor-gated Ca2+ stores in pulmonary arterial myocytes. J. Biol. Chem. 2013, 288, 10381–10394. [Google Scholar] [CrossRef] [PubMed]
- Penny, C.J.; Kilpatrick, B.S.; Eden, E.R.; Patel, S. Coupling acidic organelles with the ER through Ca2+ microdomains at membrane contact sites. Cell Calcium 2015, 58, 387–396. [Google Scholar] [CrossRef]
- Henne, W.M. Discovery and Roles of ER-Endolysosomal Contact Sites in Disease. Adv. Exp. Med. Biol. 2017, 997, 135–147. [Google Scholar]
- Wu, H.; Carvalho, P.; Voeltz, G.K. Here, there, and everywhere: The importance of ER membrane contact sites. Science 2018, 361, eaan5835. [Google Scholar] [CrossRef]
- Eden, E.R.; White, I.J.; Tsapara, A.; Futter, C.E. Membrane contacts between endosomes and ER provide sites for PTP1B-epidermal growth factor receptor interaction. Nat. Cell Biol. 2010, 12, 267–272. [Google Scholar] [CrossRef] [PubMed]
- Cabukusta, B.; Neefjes, J. Mechanisms of lysosomal positioning and movement. Traffic 2018, 19, 761–769. [Google Scholar] [CrossRef] [PubMed]
- Brailoiu, E.; Miyamoto, M.D.; Dun, N.J. Nicotinic acid adenine dinucleotide phosphate enhances quantal neurosecretion at the frog neuromuscular junction: Possible action on synaptic vesicles in the releasable pool. Mol. Pharmacol. 2001, 60, 718–724. [Google Scholar]
- Brailoiu, E.; Hoard, J.L.; Filipeanu, C.M.; Brailoiu, G.C.; Dun, S.L.; Patel, S.; Dun, N.J. Nicotinic acid adenine dinucleotide phosphate potentiates neurite outgrowth. J. Biol. Chem. 2005, 280, 5646–5650. [Google Scholar] [CrossRef]
- Favia, A.; Desideri, M.; Gambara, G.; D’Alessio, A.; Ruas, M.; Esposito, B.; Del Bufalo, D.; Parrington, J.; Ziparo, E.; Palombi, F.; et al. VEGF-induced neoangiogenesis is mediated by NAADP and two-pore channel-2-dependent Ca2+ signaling. Proc. Natl. Acad. Sci. USA 2014, 111, E4706–E4715. [Google Scholar] [CrossRef]
- Di Nezza, F.; Zuccolo, E.; Poletto, V.; Rosti, V.; De Luca, A.; Moccia, F.; Guerra, G.; Ambrosone, L. Liposomes as a Putative Tool to Investigate NAADP Signaling in Vasculogenesis. J. Cell. Biochem. 2017, 118, 3722–3729. [Google Scholar] [CrossRef] [PubMed]
- Zuccolo, E.; Laforenza, U.; Negri, S.; Botta, L.; Berra-Romani, R.; Faris, P.; Scarpellino, G.; Forcaia, G.; Pellavio, G.; Sancini, G.; et al. Muscarinic M5 receptors trigger acetylcholine-induced Ca2+ signals and nitric oxide release in human brain microvascular endothelial cells. J. Cell. Physiol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Zuccolo, E.; Negri, S.; Pellavio, G.; Scarpellino, G.; Laforenza, U.; Sancini, G.; Guerra, G.; Moccia, F. Acetylcholine induces Ca2+ signals and nitric oxide release from human brain microvascular endothelial cells. Vasc. Pharmacol. 2018, 103, 65. [Google Scholar] [CrossRef]
- Faouzi, M.; Penner, R. Trpm2. Handb. Exp. Pharmacol. 2014, 222, 403–426. [Google Scholar] [PubMed]
- Lange, I.; Yamamoto, S.; Partida-Sanchez, S.; Mori, Y.; Fleig, A.; Penner, R. TRPM2 functions as a lysosomal Ca2+-release channel in beta cells. Sci. Signal. 2009, 2, ra23. [Google Scholar] [CrossRef] [PubMed]
- Sumoza-Toledo, A.; Lange, I.; Cortado, H.; Bhagat, H.; Mori, Y.; Fleig, A.; Penner, R.; Partida-Sanchez, S. Dendritic cell maturation and chemotaxis is regulated by TRPM2-mediated lysosomal Ca2+ release. FASEB J. 2011, 25, 3529–3542. [Google Scholar] [CrossRef] [PubMed]
- Zygmunt, P.M.; Hogestatt, E.D. Trpa1. Handb. Exp. Pharmacol. 2014, 222, 583–630. [Google Scholar]
- Shang, S.; Zhu, F.; Liu, B.; Chai, Z.; Wu, Q.; Hu, M.; Wang, Y.; Huang, R.; Zhang, X.; Wu, X.; et al. Intracellular TRPA1 mediates Ca2+ release from lysosomes in dorsal root ganglion neurons. J. Cell Biol. 2016, 215, 369–381. [Google Scholar] [CrossRef]
- North, R.A. P2X receptors. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2016, 371. [Google Scholar] [CrossRef]
- Qureshi, O.S.; Paramasivam, A.; Yu, J.C.; Murrell-Lagnado, R.D. Regulation of P2X4 receptors by lysosomal targeting, glycan protection and exocytosis. J. Cell Sci. 2007, 120 Pt 21, 3838–3849. [Google Scholar] [CrossRef]
- Huang, P.; Zou, Y.; Zhong, X.Z.; Cao, Q.; Zhao, K.; Zhu, M.X.; Murrell-Lagnado, R.; Dong, X.P. P2X4 forms functional ATP-activated cation channels on lysosomal membranes regulated by luminal pH. J. Biol. Chem. 2014, 289, 17658–17667. [Google Scholar] [CrossRef] [PubMed]
- Cao, Q.; Zhong, X.Z.; Zou, Y.; Murrell-Lagnado, R.; Zhu, M.X.; Dong, X.P. Calcium release through P2X4 activates calmodulin to promote endolysosomal membrane fusion. J. Cell Biol. 2015, 209, 879–894. [Google Scholar] [CrossRef] [PubMed]
- Catterall, W.A. Voltage-gated calcium channels. Cold Spring Harb. Perspect. Biol. 2011, 3, a003947. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.; Gala, U.; Zhang, Y.; Shang, W.; Nagarkar Jaiswal, S.; di Ronza, A.; Jaiswal, M.; Yamamoto, S.; Sandoval, H.; Duraine, L.; et al. A voltage-gated calcium channel regulates lysosomal fusion with endosomes and autophagosomes and is required for neuronal homeostasis. PLoS Biol. 2015, 13, e1002103. [Google Scholar] [CrossRef] [PubMed]
- Hamalisto, S.; Jaattela, M. Lysosomes in cancer-living on the edge (of the cell). Curr. Opin. Cell Biol. 2016, 39, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Bargal, R.; Avidan, N.; Ben-Asher, E.; Olender, Z.; Zeigler, M.; Frumkin, A.; Raas-Rothschild, A.; Glusman, G.; Lancet, D.; Bach, G. Identification of the gene causing mucolipidosis type IV. Nat. Genet. 2000, 26, 118–123. [Google Scholar] [CrossRef] [PubMed]
- Piao, S.; Amaravadi, R.K. Targeting the lysosome in cancer. Ann. N. Y. Acad. Sci. 2016, 1371, 45–54. [Google Scholar] [CrossRef]
- Davidson, S.M.; Vander Heiden, M.G. Critical Functions of the Lysosome in Cancer Biology. Annu. Rev. Pharmacol. Toxicol. 2017, 57, 481–507. [Google Scholar] [CrossRef]
- Goveia, J.; Stapor, P.; Carmeliet, P. Principles of targeting endothelial cell metabolism to treat angiogenesis and endothelial cell dysfunction in disease. EMBO Mol. Med. 2014, 6, 1105–1120. [Google Scholar] [CrossRef]
- Kirkegaard, T.; Jaattela, M. Lysosomal involvement in cell death and cancer. Biochim. Biophys. Acta 2009, 1793, 746–754. [Google Scholar] [CrossRef]
- Carmeliet, P.; Jain, R.K. Molecular mechanisms and clinical applications of angiogenesis. Nature 2011, 473, 298–307. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.K.; Carmeliet, P. SnapShot: Tumor angiogenesis. Cell 2012, 149, 1408. [Google Scholar] [CrossRef] [PubMed]
- Stransky, L.; Cotter, K.; Forgac, M. The Function of V-ATPases in Cancer. Physiol. Rev. 2016, 96, 1071–1091. [Google Scholar] [CrossRef] [PubMed]
- Zhitomirsky, B.; Assaraf, Y.G. Lysosomes as mediators of drug resistance in cancer. Drug Resist. Updates 2016, 24, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Pietrocola, F.; Bravo-San Pedro, J.M.; Amaravadi, R.K.; Baehrecke, E.H.; Cecconi, F.; Codogno, P.; Debnath, J.; Gewirtz, D.A.; Karantza, V.; et al. Autophagy in malignant transformation and cancer progression. EMBO J. 2015, 34, 856–880. [Google Scholar] [CrossRef] [PubMed]
- Pierro, C.; Cook, S.J.; Foets, T.C.; Bootman, M.D.; Roderick, H.L. Oncogenic K-Ras suppresses IP(3)-dependent Ca2+ release through remodelling of the isoform composition of IP(3)Rs and ER luminal Ca2+ levels in colorectal cancer cell lines. J. Cell Sci. 2014, 127 Pt 7, 1607–1619. [Google Scholar] [CrossRef]
- Poletto, V.; Dragoni, S.; Lim, D.; Biggiogera, M.; Aronica, A.; Cinelli, M.; De Luca, A.; Rosti, V.; Porta, C.; Guerra, G.; et al. Endoplasmic Reticulum Ca2+ Handling and Apoptotic Resistance in Tumor-Derived Endothelial Colony Forming Cells. J. Cell. Biochem. 2016, 117, 2260–2271. [Google Scholar] [CrossRef]
- Moccia, F.; Poletto, V. May the remodeling of the Ca2+ toolkit in endothelial progenitor cells derived from cancer patients suggest alternative targets for anti-angiogenic treatment? Biochim. Biophys. Acta 2015, 1853, 1958–1973. [Google Scholar] [CrossRef]
- Moccia, F. Endothelial Ca2+ Signaling and the Resistance to Anticancer Treatments: Partners in Crime. Int. J. Mol. Sci. 2018, 19, 217. [Google Scholar] [CrossRef]
- Dubois, C.; Vanden Abeele, F.; Lehen’kyi, V.; Gkika, D.; Guarmit, B.; Lepage, G.; Slomianny, C.; Borowiec, A.S.; Bidaux, G.; Benahmed, M.; et al. Remodeling of channel-forming ORAI proteins determines an oncogenic switch in prostate cancer. Cancer Cell 2014, 26, 19–32. [Google Scholar] [CrossRef]
- Prevarskaya, N.; Skryma, R.; Shuba, Y. Ion Channels in Cancer: Are Cancer Hallmarks Oncochannelopathies? Physiol. Rev. 2018, 98, 559–621. [Google Scholar] [CrossRef] [PubMed]
- Fiorio Pla, A.; Gkika, D. Emerging role of TRP channels in cell migration: From tumor vascularization to metastasis. Front. Physiol. 2013, 4, 311. [Google Scholar] [CrossRef] [PubMed]
- Moccia, F.; Zuccolo, E.; Poletto, V.; Turin, I.; Guerra, G.; Pedrazzoli, P.; Rosti, V.; Porta, C.; Montagna, D. Targeting Stim and Orai Proteins as an Alternative Approach in Anticancer Therapy. Curr. Med. Chem. 2016, 23, 3450–3480. [Google Scholar] [CrossRef] [PubMed]
- Lodola, F.; Laforenza, U.; Bonetti, E.; Lim, D.; Dragoni, S.; Bottino, C.; Ong, H.L.; Guerra, G.; Ganini, C.; Massa, M.; et al. Store-operated Ca2+ entry is remodelled and controls in vitro angiogenesis in endothelial progenitor cells isolated from tumoral patients. PLoS ONE 2012, 7, e42541. [Google Scholar] [CrossRef] [PubMed]
- Iamshanova, O.; Fiorio Pla, A.; Prevarskaya, N. Molecular mechanisms of tumour invasion: Regulation by calcium signals. J. Physiol. 2017, 595, 3063–3075. [Google Scholar] [CrossRef] [PubMed]
- Perera, R.M.; Stoykova, S.; Nicolay, B.N.; Ross, K.N.; Fitamant, J.; Boukhali, M.; Lengrand, J.; Deshpande, V.; Selig, M.K.; Ferrone, C.R.; et al. Transcriptional control of autophagy-lysosome function drives pancreatic cancer metabolism. Nature 2015, 524, 361–365. [Google Scholar] [CrossRef] [PubMed]
- Giatromanolaki, A.; Kalamida, D.; Sivridis, E.; Karagounis, I.V.; Gatter, K.C.; Harris, A.L.; Koukourakis, M.I. Increased expression of transcription factor EB (TFEB) is associated with autophagy, migratory phenotype and poor prognosis in non-small cell lung cancer. Lung Cancer 2015, 90, 98–105. [Google Scholar] [CrossRef]
- Calcagni, A.; Kors, L.; Verschuren, E.; De Cegli, R.; Zampelli, N.; Nusco, E.; Confalonieri, S.; Bertalot, G.; Pece, S.; Settembre, C.; et al. Modelling TFE renal cell carcinoma in mice reveals a critical role of WNT signaling. eLife 2016, 5, e17047. [Google Scholar] [CrossRef]
- Morelli, M.B.; Nabissi, M.; Amantini, C.; Tomassoni, D.; Rossi, F.; Cardinali, C.; Santoni, M.; Arcella, A.; Oliva, M.A.; Santoni, A.; et al. Overexpression of transient receptor potential mucolipin-2 ion channels in gliomas: Role in tumor growth and progression. Oncotarget 2016, 7, 43654–43668. [Google Scholar] [CrossRef]
- Jahidin, A.H.; Stewart, T.A.; Thompson, E.W.; Roberts-Thomson, S.J.; Monteith, G.R. Differential effects of two-pore channel protein 1 and 2 silencing in MDA-MB-468 breast cancer cells. Biochem. Biophys. Res. Commun. 2016, 477, 731–736. [Google Scholar] [CrossRef]
- Lopez, J.; Dionisio, N.; Berna-Erro, A.; Galan, C.; Salido, G.M.; Rosado, J.A. Two-pore channel 2 (TPC2) modulates store-operated Ca2+ entry. Biochim. Biophys. Acta 2012, 1823, 1976–1983. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, O.N.; Grimm, C.; Schneider, L.S.; Chao, Y.K.; Atzberger, C.; Bartel, K.; Watermann, A.; Ulrich, M.; Mayr, D.; Wahl-Schott, C.; et al. Two-Pore Channel Function Is Crucial for the Migration of Invasive Cancer Cells. Cancer Res. 2017, 77, 1427–1438. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Yue, J. TPC2 mediates autophagy progression and extracellular vesicle secretion in cancer cells. Exp. Cell Res. 2018, 370, 478–489. [Google Scholar] [CrossRef] [PubMed]
- Favia, A.; Pafumi, I.; Desideri, M.; Padula, F.; Montesano, C.; Passeri, D.; Nicoletti, C.; Orlandi, A.; Del Bufalo, D.; Sergi, M.; et al. NAADP-Dependent Ca2+ Signaling Controls Melanoma Progression, Metastatic Dissemination and Neoangiogenesis. Sci. Rep. 2016, 6, 18925. [Google Scholar] [CrossRef] [PubMed]
- Pafumi, I.; Festa, M.; Papacci, F.; Lagostena, L.; Giunta, C.; Gutla, V.; Cornara, L.; Favia, A.; Palombi, F.; Gambale, F.; et al. Naringenin Impairs Two-Pore Channel 2 Activity And Inhibits VEGF-Induced Angiogenesis. Sci. Rep. 2017, 7, 5121. [Google Scholar] [CrossRef] [PubMed]
- Moccia, F.; Zuccolo, E.; Poletto, V.; Cinelli, M.; Bonetti, E.; Guerra, G.; Rosti, V. Endothelial progenitor cells support tumour growth and metastatisation: Implications for the resistance to anti-angiogenic therapy. Tumour Biol. 2015, 36, 6603–6614. [Google Scholar] [CrossRef] [PubMed]
- Dragoni, S.; Reforgiato, M.; Zuccolo, E.; Poletto, V.; Lodola, F.; Ruffinatti, F.A.; Bonetti, E.; Guerra, G.; Barosi, G.; Rosti, V.; et al. Dysregulation of VEGF-induced proangiogenic Ca2+ oscillations in primary myelofibrosis-derived endothelial colony-forming cells. Exp. Hematol. 2015, 43, 1019–1030. [Google Scholar] [CrossRef]
- Lodola, F.; Laforenza, U.; Cattaneo, F.; Ruffinatti, F.A.; Poletto, V.; Massa, M.; Tancredi, R.; Zuccolo, E.; Khdar, A.D.; Riccardi, A.; et al. VEGF-induced intracellular Ca2+ oscillations are down-regulated and do not stimulate angiogenesis in breast cancer-derived endothelial colony forming cells. Oncotarget 2017, 8, 95223–95246. [Google Scholar] [CrossRef]
- Poletto, V.; Rosti, V.; Biggiogera, M.; Guerra, G.; Moccia, F.; Porta, C. The role of endothelial colony forming cells in kidney cancer’s pathogenesis, and in resistance to anti-VEGFR agents and mTOR inhibitors: A speculative review. Crit. Rev. Oncol. Hematol. 2018, 132, 89–99. [Google Scholar] [CrossRef]
- Naylor, E.; Arredouani, A.; Vasudevan, S.R.; Lewis, A.M.; Parkesh, R.; Mizote, A.; Rosen, D.; Thomas, J.M.; Izumi, M.; Ganesan, A.; et al. Identification of a chemical probe for NAADP by virtual screening. Nat. Chem. Biol. 2009, 5, 220–226. [Google Scholar] [CrossRef]
- Gunaratne, G.S.; Yang, Y.; Li, F.; Walseth, T.F.; Marchant, J.S. NAADP-dependent Ca2+ signaling regulates Middle East respiratory syndrome-coronavirus pseudovirus translocation through the endolysosomal system. Cell Calcium 2018, 75, 30–41. [Google Scholar] [CrossRef] [PubMed]
- Billington, R.A.; Genazzani, A.A. PPADS is a reversible competitive antagonist of the NAADP receptor. Cell Calcium 2007, 41, 505–511. [Google Scholar] [CrossRef] [PubMed]
- Genazzani, A.A.; Mezna, M.; Dickey, D.M.; Michelangeli, F.; Walseth, T.F.; Galione, A. Pharmacological properties of the Ca2+-release mechanism sensitive to NAADP in the sea urchin egg. Br. J. Pharmacol. 1997, 121, 1489–1495. [Google Scholar] [CrossRef]
- Moccia, F.; Lim, D.; Nusco, G.A.; Ercolano, E.; Santella, L. NAADP activates a Ca2+ current that is dependent on F-actin cytoskeleton. FASEB J. 2003, 17, 1907–1909. [Google Scholar] [CrossRef] [PubMed]
- Monteith, G.R.; McAndrew, D.; Faddy, H.M.; Roberts-Thomson, S.J. Calcium and cancer: Targeting Ca2+ transport. Nat. Rev. Cancer 2007, 7, 519–530. [Google Scholar] [CrossRef] [PubMed]
- Moccia, F.; Lim, D.; Kyozuka, K.; Santella, L. NAADP triggers the fertilization potential in starfish oocytes. Cell Calcium 2004, 36, 515–524. [Google Scholar] [CrossRef] [PubMed]
- Gunaratne, G.S.; Johns, M.E.; Hintz, H.M.; Walseth, T.F.; Marchant, J.S. A screening campaign in sea urchin egg homogenate as a platform for discovering modulators of NAADP-dependent Ca2+ signaling in human cells. Cell Calcium 2018, 75, 42–52. [Google Scholar] [CrossRef]
- Penny, C.J.; Vassileva, K.; Jha, A.; Yuan, Y.; Chee, X.; Yates, E.; Mazzon, M.; Kilpatrick, B.S.; Muallem, S.; Marsh, M.; et al. Mining of Ebola virus entry inhibitors identifies approved drugs as two-pore channel pore blockers. Biochim. Biophys. Acta Mol. Cell Res. 2018. [Google Scholar] [CrossRef]
- Zhang, B.; Watt, J.M.; Cordiglieri, C.; Dammermann, W.; Mahon, M.F.; Flugel, A.; Guse, A.H.; Potter, B.V.L. Small Molecule Antagonists of NAADP-Induced Ca2+ Release in T-Lymphocytes Suggest Potential Therapeutic Agents for Autoimmune Disease. Sci. Rep. 2018, 8, 16775. [Google Scholar] [CrossRef]
- Cordiglieri, C.; Odoardi, F.; Zhang, B.; Nebel, M.; Kawakami, N.; Klinkert, W.E.; Lodygin, D.; Luhder, F.; Breunig, E.; Schild, D.; et al. Nicotinic acid adenine dinucleotide phosphate-mediated calcium signalling in effector T cells regulates autoimmunity of the central nervous system. Brain 2010, 133 Pt 7, 1930–1943. [Google Scholar] [CrossRef]
- Nebel, M.; Schwoerer, A.P.; Warszta, D.; Siebrands, C.C.; Limbrock, A.C.; Swarbrick, J.M.; Fliegert, R.; Weber, K.; Bruhn, S.; Hohenegger, M.; et al. Nicotinic acid adenine dinucleotide phosphate (NAADP)-mediated calcium signaling and arrhythmias in the heart evoked by beta-adrenergic stimulation. J. Biol. Chem. 2013, 288, 16017–16030. [Google Scholar] [CrossRef] [PubMed]




© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Faris, P.; Shekha, M.; Montagna, D.; Guerra, G.; Moccia, F. Endolysosomal Ca2+ Signalling and Cancer Hallmarks: Two-Pore Channels on the Move, TRPML1 Lags Behind! Cancers 2019, 11, 27. https://doi.org/10.3390/cancers11010027
Faris P, Shekha M, Montagna D, Guerra G, Moccia F. Endolysosomal Ca2+ Signalling and Cancer Hallmarks: Two-Pore Channels on the Move, TRPML1 Lags Behind! Cancers. 2019; 11(1):27. https://doi.org/10.3390/cancers11010027
Chicago/Turabian StyleFaris, Pawan, Mudhir Shekha, Daniela Montagna, Germano Guerra, and Francesco Moccia. 2019. "Endolysosomal Ca2+ Signalling and Cancer Hallmarks: Two-Pore Channels on the Move, TRPML1 Lags Behind!" Cancers 11, no. 1: 27. https://doi.org/10.3390/cancers11010027
APA StyleFaris, P., Shekha, M., Montagna, D., Guerra, G., & Moccia, F. (2019). Endolysosomal Ca2+ Signalling and Cancer Hallmarks: Two-Pore Channels on the Move, TRPML1 Lags Behind! Cancers, 11(1), 27. https://doi.org/10.3390/cancers11010027