TGF-β in T Cell Biology: Implications for Cancer Immunotherapy


{kind=link}
{kind=link}
Abstract
1. Introduction
2. TGF-β Secretion, Activation and Signaling
3. TGF-β: Suppressor of T-Cell Proliferation and Effector Functions
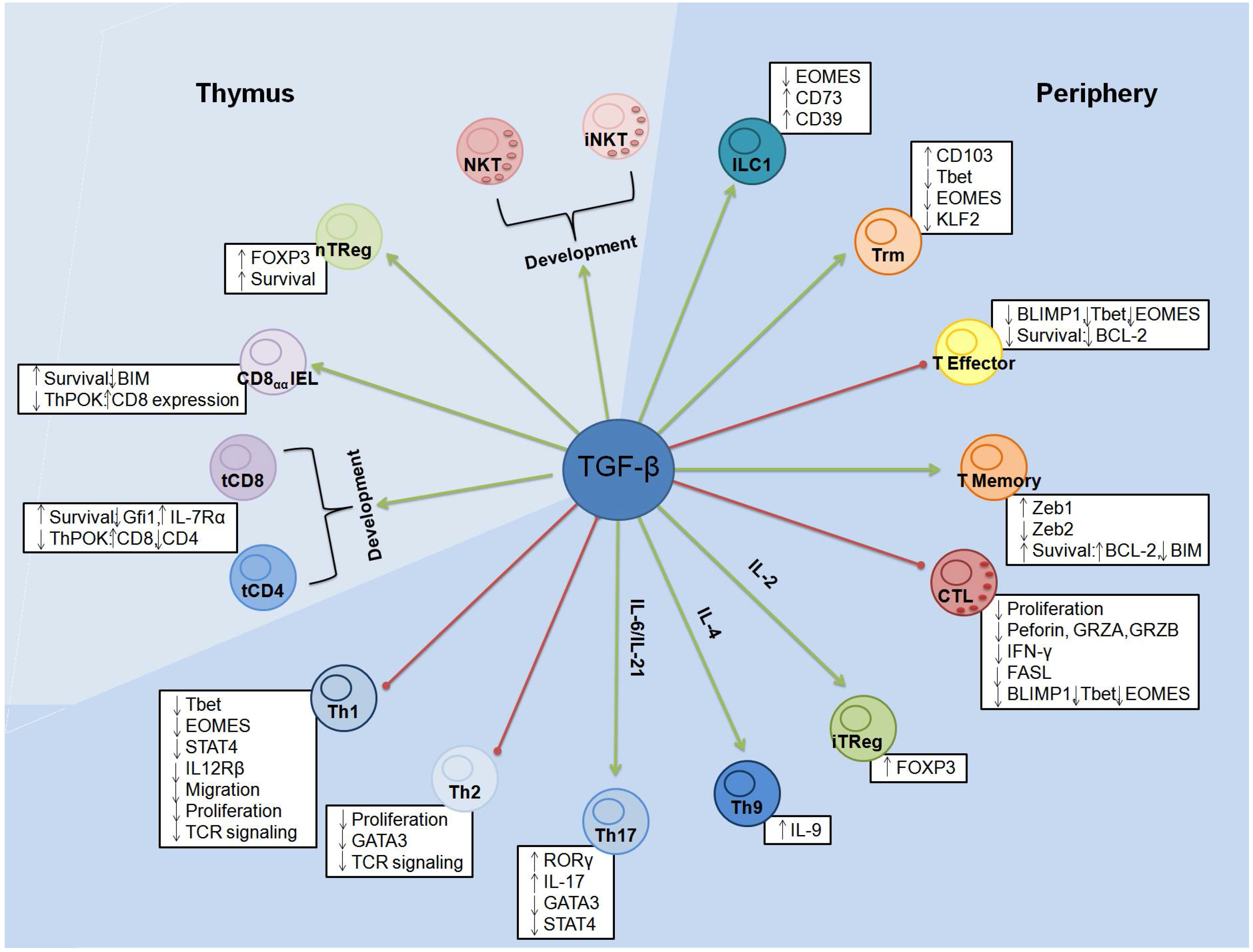
4. TGF-β: Master Regulator of T-cell Homeostasis and Differentiation
5. TGF-β: Architect of the Immune Tumor Microenvironment and Therapeutic Opportunities
6. Conclusions and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- David, C.J.; Massague, J. Contextual determinants of TGFβ action in development, immunity and cancer. Nat. Rev. Mol. Cell Biol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Gaarenstroom, T.; Hill, C.S. TGF-β signaling to chromatin: How Smads regulate transcription during self-renewal and differentiation. Semin. Cell Dev. Biol. 2014, 32, 107–118. [Google Scholar] [CrossRef] [PubMed]
- Savage-Dunn, C. TGF-β signaling. WormBook 2005, 9, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Itoh, S.; ten Dijke, P. Negative regulation of TGF-β receptor/Smad signal transduction. Curr. Opin. Cell Biol. 2007, 19, 176–184. [Google Scholar] [CrossRef] [PubMed]
- Clarke, D.C.; Liu, X. Decoding the quantitative nature of TGF-β/Smad signaling. Trends Cell Biol. 2008, 18, 430–442. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.G. Endocytic regulation of TGF-β signaling. Cell Res. 2009, 19, 58–70. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.E. Non-Smad pathways in TGF-β signaling. Cell Res. 2009, 19, 128–139. [Google Scholar] [CrossRef] [PubMed]
- Mu, Y.; Gudey, S.K.; Landstrom, M. Non-Smad signaling pathways. Cell Tissue Res. 2011. [Google Scholar] [CrossRef] [PubMed]
- Ayyaz, A.; Attisano, L.; Wrana, J.L. Recent advances in understanding contextual TGFβ signaling. F1000Research 2017, 6, 749. [Google Scholar] [CrossRef] [PubMed]
- Tian, M.; Neil, J.R.; Schiemann, W.P. Transforming growth factor-β and the hallmarks of cancer. Cell. Signal. 2011, 23, 951–962. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Pang, Y.; Moses, H.L. TGF-β and immune cells: An important regulatory axis in the tumor microenvironment and progression. Trends Immunol. 2010, 31, 220–227. [Google Scholar] [CrossRef] [PubMed]
- Zitvogel, L.; Tesniere, A.; Kroemer, G. Cancer despite immunosurveillance: Immunoselection and immunosubversion. Nat. Rev. Immunol. 2006, 6, 715–727. [Google Scholar] [CrossRef] [PubMed]
- Dunn, G.P.; Old, L.J.; Schreiber, R.D. The immunobiology of cancer immunosurveillance and immunoediting. Immunity 2004, 21, 137–148. [Google Scholar] [CrossRef] [PubMed]
- Flavell, R.A.; Sanjabi, S.; Wrzesinski, S.H.; Licona-Limon, P. The polarization of immune cells in the tumour environment by TGFβ. Nat. Rev. Immunol. 2010, 10, 554–567. [Google Scholar] [CrossRef] [PubMed]
- Neuzillet, C.; Tijeras-Raballand, A.; Cohen, R.; Cros, J.; Faivre, S.; Raymond, E.; de Gramont, A. Targeting the TGFβ pathway for cancer therapy. Pharmacol. Ther. 2015, 147, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Pickup, M.W.; Owens, P.; Moses, H.L. TGF-β, Bone Morphogenetic Protein, and Activin Signaling and the Tumor Microenvironment. Cold Spring Harb. Perspect. Biol. 2017, 9, a022285. [Google Scholar] [CrossRef] [PubMed]
- Topalian, S.L.; Weiner, G.J.; Pardoll, D.M. Cancer immunotherapy comes of age. J. Clin. Oncol. 2011, 29, 4828–4836. [Google Scholar] [CrossRef] [PubMed]
- Sanjabi, S.; Oh, S.A.; Li, M.O. Regulation of the Immune Response by TGF-β: From Conception to Autoimmunity and Infection. Cold Spring Harb. Perspect. Biol. 2017, 9, a022236. [Google Scholar] [CrossRef] [PubMed]
- Travis, M.A.; Sheppard, D. TGF-β activation and function in immunity. Annu. Rev. Immunol. 2014, 32, 51–82. [Google Scholar] [CrossRef] [PubMed]
- Shull, M.M.; Ormsby, I.; Kier, A.B.; Pawlowski, S.; Diebold, R.J.; Yin, M.; Allen, R.; Sidman, C.; Proetzel, G.; Calvin, D.; et al. Targeted disruption of the mouse transforming growth factor-β1 gene results in multifocal inflammatory disease. Nature 1992, 359, 693–699. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, A.B.; Huh, C.G.; Becker, D.; Geiser, A.; Lyght, M.; Flanders, K.C.; Roberts, A.B.; Sporn, M.B.; Ward, J.M.; Karlsson, S. Transforming growth factor β1 null mutation in mice causes excessive inflammatory response and early death. Proc. Natl. Acad. Sci. USA 1993, 90, 770–774. [Google Scholar] [CrossRef] [PubMed]
- Yaswen, L.; Kulkarni, A.B.; Fredrickson, T.; Mittleman, B.; Schiffman, R.; Payne, S.; Longenecker, G.; Mozes, E.; Karlsson, S. Autoimmune manifestations in the transforming growth factor-β1 knockout mouse. Blood 1996, 87, 1439–1445. [Google Scholar] [PubMed]
- Li, M.O.; Wan, Y.Y.; Sanjabi, S.; Robertson, A.K.; Flavell, R.A. Transforming growth factor-β regulation of immune responses. Annu. Rev. Immunol. 2006, 24, 99–146. [Google Scholar] [CrossRef] [PubMed]
- Gorelik, L.; Flavell, R.A. Abrogation of TGFβ signaling in T cells leads to spontaneous T cell differentiation and autoimmune disease. Immunity 2000, 12, 171–181. [Google Scholar] [CrossRef]
- Li, M.O.; Sanjabi, S.; Flavell, R.A. Transforming growth factor-β controls development, homeostasis, and tolerance of T cells by regulatory T cell-dependent and -independent mechanisms. Immunity 2006, 25, 455–471. [Google Scholar] [CrossRef] [PubMed]
- Marie, J.C.; Liggitt, D.; Rudensky, A.Y. Cellular mechanisms of fatal early-onset autoimmunity in mice with the T cell-specific targeting of transforming growth factor-β receptor. Immunity 2006, 25, 441–454. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhang, P.; Li, J.; Kulkarni, A.B.; Perruche, S.; Chen, W. A critical function for TGF-β signaling in the development of natural CD4+CD25+Foxp3+ regulatory T cells. Nat. Immunol. 2008, 9, 632–640. [Google Scholar] [CrossRef] [PubMed]
- Diebold, R.J.; Eis, M.J.; Yin, M.; Ormsby, I.; Boivin, G.P.; Darrow, B.J.; Saffitz, J.E.; Doetschman, T. Early-onset multifocal inflammation in the transforming growth factor β1-null mouse is lymphocyte mediated. Proc. Natl. Acad. Sci. USA 1995, 92, 12215–12219. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, S.; Sakaguchi, N.; Asano, M.; Itoh, M.; Toda, M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor α-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J. Immunol. 1995, 155, 1151–1164. [Google Scholar] [PubMed]
- Annes, J.P.; Munger, J.S.; Rifkin, D.B. Making sense of latent TGFβ activation. J. Cell Sci. 2003, 116, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Mu, Z.; Dabovic, B.; Jurukovski, V.; Yu, D.; Sung, J.; Xiong, X.; Munger, J.S. Absence of integrin-mediated TGFβ1 activation in vivo recapitulates the phenotype of TGFβ1-null mice. J. Cell Biol. 2007, 176, 787–793. [Google Scholar] [CrossRef] [PubMed]
- Travis, M.A.; Reizis, B.; Melton, A.C.; Masteller, E.; Tang, Q.; Proctor, J.M.; Wang, Y.; Bernstein, X.; Huang, X.; Reichardt, L.F.; et al. Loss of integrin α(v)β8 on dendritic cells causes autoimmunity and colitis in mice. Nature 2007, 449, 361–365. [Google Scholar] [CrossRef] [PubMed]
- Worthington, J.J.; Czajkowska, B.I.; Melton, A.C.; Travis, M.A. Intestinal dendritic cells specialize to activate transforming growth factor-β and induce Foxp3+ regulatory T cells via integrin αvβ8. Gastroenterology 2011, 141, 1802–1812. [Google Scholar] [CrossRef] [PubMed]
- Melton, A.C.; Bailey-Bucktrout, S.L.; Travis, M.A.; Fife, B.T.; Bluestone, J.A.; Sheppard, D. Expression of αvβ8 integrin on dendritic cells regulates Th17 cell development and experimental autoimmune encephalomyelitis in mice. J. Clin. Investig. 2010, 120, 4436–4444. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Wan, Q.; Kozhaya, L.; Fujii, H.; Unutmaz, D. Identification of a regulatory T cell specific cell surface molecule that mediates suppressive signals and induces Foxp3 expression. PLoS ONE 2008, 3, e2705. [Google Scholar] [CrossRef] [PubMed]
- Stockis, J.; Colau, D.; Coulie, P.G.; Lucas, S. Membrane protein GARP is a receptor for latent TGF-β on the surface of activated human Treg. Eur. J. Immunol. 2009, 39, 3315–3322. [Google Scholar] [CrossRef] [PubMed]
- Tran, D.Q.; Andersson, J.; Wang, R.; Ramsey, H.; Unutmaz, D.; Shevach, E.M. GARP (LRRC32) is essential for the surface expression of latent TGF-β on platelets and activated FOXP3+ regulatory T cells. Proc. Natl. Acad. Sci. USA 2009, 106, 13445–13450. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Kozhaya, L.; Mercer, F.; Khaitan, A.; Fujii, H.; Unutmaz, D. Expression of GARP selectively identifies activated human FOXP3+ regulatory T cells. Proc. Natl. Acad. Sci. USA 2009, 106, 13439–13444. [Google Scholar] [CrossRef] [PubMed]
- Edwards, J.P.; Thornton, A.M.; Shevach, E.M. Release of active TGF-β1 from the latent TGF-β1/GARP complex on T regulatory cells is mediated by integrin β8. J. Immunol. 2014, 193, 2843–2849. [Google Scholar] [CrossRef] [PubMed]
- Worthington, J.J.; Kelly, A.; Smedley, C.; Bauche, D.; Campbell, S.; Marie, J.C.; Travis, M.A. Integrin αvβ8-Mediated TGF-β Activation by Effector Regulatory T Cells Is Essential for Suppression of T-Cell-Mediated Inflammation. Immunity 2015, 42, 903–915. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.H.; Derynck, R. Specificity and versatility in TGF-β signaling through Smads. Annu. Rev. Cell Dev. Biol. 2005, 21, 659–693. [Google Scholar] [CrossRef] [PubMed]
- Massague, J.; Seoane, J.; Wotton, D. Smad transcription factors. Genes Dev. 2005, 19, 2783–2810. [Google Scholar] [CrossRef] [PubMed]
- Gorelik, L.; Constant, S.; Flavell, R.A. Mechanism of transforming growth factor β-induced inhibition of T helper type 1 differentiation. J. Exp. Med. 2002, 195, 1499–1505. [Google Scholar] [CrossRef] [PubMed]
- Lucas, P.J.; Kim, S.J.; Melby, S.J.; Gress, R.E. Disruption of T cell homeostasis in mice expressing a T cell-specific dominant negative transforming growth factor β II receptor. J. Exp. Med. 2000, 191, 1187–1196. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Bevan, M.J. TGF-β signaling to T cells inhibits autoimmunity during lymphopenia-driven proliferation. Nat. Immunol. 2012, 13, 667–673. [Google Scholar] [CrossRef] [PubMed]
- Lucas, P.J.; McNeil, N.; Hilgenfeld, E.; Choudhury, B.; Kim, S.J.; Eckhaus, M.A.; Ried, T.; Gress, R.E. Transforming growth factor-β pathway serves as a primary tumor suppressor in CD8+ T cell tumorigenesis. Cancer Res. 2004, 64, 6524–6529. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Richardson, J.A.; Parada, L.F.; Graff, J.M. Smad3 mutant mice develop metastatic colorectal cancer. Cell 1998, 94, 703–714. [Google Scholar] [CrossRef]
- Hahn, J.N.; Falck, V.G.; Jirik, F.R. Smad4 deficiency in T cells leads to the Th17-associated development of premalignant gastroduodenal lesions in mice. J. Clin. Investig. 2011, 121, 4030–4042. [Google Scholar] [CrossRef] [PubMed]
- Blank, U.; Karlsson, S. TGF-β signaling in the control of hematopoietic stem cells. Blood 2015, 125, 3542–3550. [Google Scholar] [CrossRef] [PubMed]
- Chabanon, A.; Desterke, C.; Rodenburger, E.; Clay, D.; Guerton, B.; Boutin, L.; Bennaceur-Griscelli, A.; Pierre-Louis, O.; Uzan, G.; Abecassis, L.; et al. A cross-talk between stromal cell-derived factor-1 and transforming growth factor-β controls the quiescence/cycling switch of CD34(+) progenitors through FoxO3 and mammalian target of rapamycin. Stem Cells 2008, 26, 3150–3161. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Han, X.; Wang, J.; Wang, C.; Sun, X.; Xie, J.; Wu, G.; Phan, H.; Liu, Z.; Zhang, C.; et al. SHP-1 regulates hematopoietic stem cell quiescence by coordinating TGF-β signaling. J. Exp. Med. 2018, 215, 1337–1347. [Google Scholar] [CrossRef] [PubMed]
- Kehrl, J.H.; Wakefield, L.M.; Roberts, A.B.; Jakowlew, S.; Alvarez-Mon, M.; Derynck, R.; Sporn, M.B.; Fauci, A.S. Production of transforming growth factor β by human T lymphocytes and its potential role in the regulation of T cell growth. J. Exp. Med. 1986, 163, 1037–1050. [Google Scholar] [CrossRef] [PubMed]
- McKarns, S.C.; Schwartz, R.H.; Kaminski, N.E. Smad3 is essential for TGF-β1 to suppress IL-2 production and TCR-induced proliferation, but not IL-2-induced proliferation. J. Immunol. 2004, 172, 4275–4284. [Google Scholar] [CrossRef] [PubMed]
- Brabletz, T.; Pfeuffer, I.; Schorr, E.; Siebelt, F.; Wirth, T.; Serfling, E. Transforming growth factor β and cyclosporin A inhibit the inducible activity of the interleukin-2 gene in T cells through a noncanonical octamer-binding site. Mol. Cell. Biol. 1993, 13, 1155–1162. [Google Scholar] [CrossRef] [PubMed]
- Wolfraim, L.A.; Walz, T.M.; James, Z.; Fernandez, T.; Letterio, J.J. p21Cip1 and p27Kip1 act in synergy to alter the sensitivity of naive T cells to TGF-β-mediated G1 arrest through modulation of IL-2 responsiveness. J. Immunol. 2004, 173, 3093–3102. [Google Scholar] [CrossRef] [PubMed]
- Ruegemer, J.J.; Ho, S.N.; Augustine, J.A.; Schlager, J.W.; Bell, M.P.; McKean, D.J.; Abraham, R.T. Regulatory effects of transforming growth factor-β on IL-2- and IL-4-dependent T cell-cycle progression. J. Immunol. 1990, 144, 1767–1776. [Google Scholar] [PubMed]
- Li, L.; Iwamoto, Y.; Berezovskaya, A.; Boussiotis, V.A. A pathway regulated by cell cycle inhibitor p27Kip1 and checkpoint inhibitor Smad3 is involved in the induction of T cell tolerance. Nat. Immunol. 2006, 7, 1157–1165. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Letterio, J.J.; Lechleider, R.J.; Chen, L.; Hayman, R.; Gu, H.; Roberts, A.B.; Deng, C. Targeted disruption of SMAD3 results in impaired mucosal immunity and diminished T cell responsiveness to TGF-β. EMBO J. 1999, 18, 1280–1291. [Google Scholar] [CrossRef] [PubMed]
- Giroux, M.; Delisle, J.S.; Gauthier, S.D.; Heinonen, K.M.; Hinsinger, J.; Houde, B.; Gaboury, L.; Brochu, S.; Wu, J.; Hebert, M.J.; et al. SMAD3 prevents graft-versus-host disease by restraining Th1 differentiation and granulocyte-mediated tissue damage. Blood 2011, 117, 1734–1744. [Google Scholar] [CrossRef] [PubMed]
- Datto, M.B.; Frederick, J.P.; Pan, L.; Borton, A.J.; Zhuang, Y.; Wang, X.F. Targeted disruption of Smad3 reveals an essential role in transforming growth factor β-mediated signal transduction. Mol. Cell. Biol. 1999, 19, 2495–2504. [Google Scholar] [CrossRef] [PubMed]
- McKarns, S.C.; Schwartz, R.H. Distinct effects of TGF-β 1 on CD4+ and CD8+ T cell survival, division, and IL-2 production: A role for T cell intrinsic Smad3. J. Immunol. 2005, 174, 2071–2083. [Google Scholar] [CrossRef] [PubMed]
- Ishigame, H.; Mosaheb, M.M.; Sanjabi, S.; Flavell, R.A. Truncated form of TGF-βRII, but not its absence, induces memory CD8+ T cell expansion and lymphoproliferative disorder in mice. J. Immunol. 2013, 190, 6340–6350. [Google Scholar] [CrossRef] [PubMed]
- Delisle, J.S.; Giroux, M.; Boucher, G.; Landry, J.R.; Hardy, M.P.; Lemieux, S.; Jones, R.G.; Wilhelm, B.T.; Perreault, C. The TGF-β-Smad3 pathway inhibits CD28-dependent cell growth and proliferation of CD4 T cells. Genes Immun. 2013, 14, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Viel, S.; Marcais, A.; Guimaraes, F.S.; Loftus, R.; Rabilloud, J.; Grau, M.; Degouve, S.; Djebali, S.; Sanlaville, A.; Charrier, E.; et al. TGF-β inhibits the activation and functions of NK cells by repressing the mTOR pathway. Sci. Signal. 2016, 9, ra19. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.H.; Seguin-Devaux, C.; Burke, N.A.; Oriss, T.B.; Watkins, S.C.; Clipstone, N.; Ray, A. Transforming growth factor β blocks Tec kinase phosphorylation, Ca2+ influx, and NFATc translocation causing inhibition of T cell differentiation. J. Exp. Med. 2003, 197, 1689–1699. [Google Scholar] [CrossRef] [PubMed]
- Boussiotis, V.A.; Chen, Z.M.; Zeller, J.C.; Murphy, W.J.; Berezovskaya, A.; Narula, S.; Roncarolo, M.G.; Blazar, B.R. Altered T-cell receptor + CD28-mediated signaling and blocked cell cycle progression in interleukin 10 and transforming growth factor-β-treated alloreactive T cells that do not induce graft-versus-host disease. Blood 2001, 97, 565–571. [Google Scholar] [CrossRef] [PubMed]
- Choudhry, M.A.; Sir, O.; Sayeed, M.M. TGF-β abrogates TCR-mediated signaling by upregulating tyrosine phosphatases in T cells. Shock 2001, 15, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Riese, M.J.; Wang, L.C.; Moon, E.K.; Joshi, R.P.; Ranganathan, A.; June, C.H.; Koretzky, G.A.; Albelda, S.M. Enhanced effector responses in activated CD8+ T cells deficient in diacylglycerol kinases. Cancer Res. 2013, 73, 3566–3577. [Google Scholar] [CrossRef] [PubMed]
- Giroux, M.; Delisle, J.S.; O’Brien, A.; Hebert, M.J.; Perreault, C. T cell activation leads to protein kinase C theta-dependent inhibition of TGF-β signaling. J. Immunol. 2010, 185, 1568–1576. [Google Scholar] [CrossRef] [PubMed]
- Cottrez, F.; Groux, H. Regulation of TGF-β response during T cell activation is modulated by IL-10. J. Immunol. 2001, 167, 773–778. [Google Scholar] [CrossRef] [PubMed]
- Thomas, D.A.; Massague, J. TGF-β directly targets cytotoxic T cell functions during tumor evasion of immune surveillance. Cancer Cell 2005, 8, 369–380. [Google Scholar] [CrossRef] [PubMed]
- Gorelik, L.; Flavell, R.A. Immune-mediated eradication of tumors through the blockade of transforming growth factor-β signaling in T cells. Nat. Med. 2001, 7, 1118–1122. [Google Scholar] [CrossRef] [PubMed]
- Donkor, M.K.; Sarkar, A.; Savage, P.A.; Franklin, R.A.; Johnson, L.K.; Jungbluth, A.A.; Allison, J.P.; Li, M.O. T cell surveillance of oncogene-induced prostate cancer is impeded by T cell-derived TGF-β1 cytokine. Immunity 2011, 35, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Bollard, C.M.; Rossig, C.; Calonge, M.J.; Huls, M.H.; Wagner, H.J.; Massague, J.; Brenner, M.K.; Heslop, H.E.; Rooney, C.M. Adapting a transforming growth factor β-related tumor protection strategy to enhance antitumor immunity. Blood 2002, 99, 3179–3187. [Google Scholar] [CrossRef] [PubMed]
- Chou, C.K.; Schietinger, A.; Liggitt, H.D.; Tan, X.; Funk, S.; Freeman, G.J.; Ratliff, T.L.; Greenberg, N.M.; Greenberg, P.D. Cell-intrinsic abrogation of TGF-β signaling delays but does not prevent dysfunction of self/tumor-specific CD8 T cells in a murine model of autochthonous prostate cancer. J. Immunol. 2012, 189, 3936–3946. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, B.M.; Juedes, A.; Szabo, S.J.; von Herrath, M.; Glimcher, L.H. Antigen-driven effector CD8 T cell function regulated by T-bet. Proc. Natl. Acad. Sci. USA 2003, 100, 15818–15823. [Google Scholar] [CrossRef] [PubMed]
- Pearce, E.L.; Mullen, A.C.; Martins, G.A.; Krawczyk, C.M.; Hutchins, A.S.; Zediak, V.P.; Banica, M.; DiCioccio, C.B.; Gross, D.A.; Mao, C.A.; et al. Control of effector CD8+ T cell function by the transcription factor Eomesodermin. Science 2003, 302, 1041–1043. [Google Scholar] [CrossRef] [PubMed]
- Intlekofer, A.M.; Banerjee, A.; Takemoto, N.; Gordon, S.M.; Dejong, C.S.; Shin, H.; Hunter, C.A.; Wherry, E.J.; Lindsten, T.; Reiner, S.L. Anomalous type 17 response to viral infection by CD8+ T cells lacking T-bet and eomesodermin. Science 2008, 321, 408–411. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Guilloty, F.; Pipkin, M.E.; Djuretic, I.M.; Levanon, D.; Lotem, J.; Lichtenheld, M.G.; Groner, Y.; Rao, A. Runx3 and T-box proteins cooperate to establish the transcriptional program of effector CTLs. J. Exp. Med. 2009, 206, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Ju, S.; Chen, E.; Dai, S.; Li, C.; Morel, P.; Liu, L.; Zhang, X.; Lu, B. T-bet and eomesodermin are required for T cell-mediated antitumor immune responses. J. Immunol. 2010, 185, 3174–3183. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.H.; Jung, S.M.; Park, S.H.; Kato, M.; Yamashita, T.; Lee, I.K.; Sudo, K.; Nakae, S.; Han, J.S.; Kim, O.H.; et al. Activin receptor-like kinase5 inhibition suppresses mouse melanoma by ubiquitin degradation of Smad4, thereby derepressing eomesodermin in cytotoxic T lymphocytes. EMBO Mol. Med. 2013, 5, 1720–1739. [Google Scholar] [CrossRef] [PubMed]
- Ahmadzadeh, M.; Rosenberg, S.A. TGF-β1 attenuates the acquisition and expression of effector function by tumor antigen-specific human memory CD8 T cells. J. Immunol. 2005, 174, 5215–5223. [Google Scholar] [CrossRef] [PubMed]
- Rutishauser, R.L.; Martins, G.A.; Kalachikov, S.; Chandele, A.; Parish, I.A.; Meffre, E.; Jacob, J.; Calame, K.; Kaech, S.M. Transcriptional repressor Blimp-1 promotes CD8+ T cell terminal differentiation and represses the acquisition of central memory T cell properties. Immunity 2009, 31, 296–308. [Google Scholar] [CrossRef] [PubMed]
- Kallies, A.; Xin, A.; Belz, G.T.; Nutt, S.L. Blimp-1 transcription factor is required for the differentiation of effector CD8(+) T cells and memory responses. Immunity 2009, 31, 283–295. [Google Scholar] [CrossRef] [PubMed]
- Lin, R.; Chen, L.; Chen, G.; Hu, C.; Jiang, S.; Sevilla, J.; Wan, Y.; Sampson, J.H.; Zhu, B.; Li, Q.J. Targeting miR-23a in CD8+ cytotoxic T lymphocytes prevents tumor-dependent immunosuppression. J. Clin. Investig. 2014, 124, 5352–5367. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, W.; Beckett, O.; Ma, Q.; Li, M.O. Transforming growth factor-β signaling curbs thymic negative selection promoting regulatory T cell development. Immunity 2010, 32, 642–653. [Google Scholar] [CrossRef] [PubMed]
- Stritesky, G.L.; Jameson, S.C.; Hogquist, K.A. Selection of self-reactive T cells in the thymus. Annu. Rev. Immunol. 2012, 30, 95–114. [Google Scholar] [CrossRef] [PubMed]
- Robinson, R.T.; Gorham, J.D. TGF-β1 regulates antigen-specific CD4+ T cell responses in the periphery. J. Immunol. 2007, 179, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Filippi, C.M.; Juedes, A.E.; Oldham, J.E.; Ling, E.; Togher, L.; Peng, Y.; Flavell, R.A.; von Herrath, M.G. Transforming growth factor-β suppresses the activation of CD8+ T-cells when naive but promotes their survival and function once antigen experienced: A two-faced impact on autoimmunity. Diabetes 2008, 57, 2684–2692. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Zhang, N. Transforming growth factor-β signaling is constantly shaping memory T-cell population. Proc. Natl. Acad. Sci. USA 2015, 112, 11013–11017. [Google Scholar] [CrossRef] [PubMed]
- Lewis, G.M.; Wehrens, E.J.; Labarta-Bajo, L.; Streeck, H.; Zuniga, E.I. TGF-β receptor maintains CD4 T helper cell identity during chronic viral infections. J. Clin. Investig. 2016, 126, 3799–3813. [Google Scholar] [CrossRef] [PubMed]
- Sanjabi, S.; Mosaheb, M.M.; Flavell, R.A. Opposing effects of TGF-β and IL-15 cytokines control the number of short-lived effector CD8+ T cells. Immunity 2009, 31, 131–144. [Google Scholar] [CrossRef] [PubMed]
- Guan, T.; Dominguez, C.X.; Amezquita, R.A.; Laidlaw, B.J.; Cheng, J.; Henao-Mejia, J.; Williams, A.; Flavell, R.A.; Lu, J.; Kaech, S.M. ZEB1, ZEB2, and the miR-200 family form a counterregulatory network to regulate CD8(+) T cell fates. J. Exp. Med. 2018, 215, 1153–1168. [Google Scholar] [CrossRef] [PubMed]
- Gregory, P.A.; Bracken, C.P.; Smith, E.; Bert, A.G.; Wright, J.A.; Roslan, S.; Morris, M.; Wyatt, L.; Farshid, G.; Lim, Y.Y.; et al. An autocrine TGF-β/ZEB/miR-200 signaling network regulates establishment and maintenance of epithelial-mesenchymal transition. Mol. Biol. Cell 2011, 22, 1686–1698. [Google Scholar] [CrossRef] [PubMed]
- Larsen, J.E.; Nathan, V.; Osborne, J.K.; Farrow, R.K.; Deb, D.; Sullivan, J.P.; Dospoy, P.D.; Augustyn, A.; Hight, S.K.; Sato, M.; et al. ZEB1 drives epithelial-to-mesenchymal transition in lung cancer. J. Clin. Investig. 2016, 126, 3219–3235. [Google Scholar] [CrossRef] [PubMed]
- Rateitschak, K.; Kaderali, L.; Wolkenhauer, O.; Jaster, R. Autocrine TGF-β/ZEB/microRNA-200 signal transduction drives epithelial-mesenchymal transition: Kinetic models predict minimal drug dose to inhibit metastasis. Cell. Signal. 2016, 28, 861–870. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Yamane, H.; Paul, W.E. Differentiation of effector CD4 T cell populations. Annu. Rev. Immunol. 2010, 28, 445–489. [Google Scholar] [CrossRef] [PubMed]
- Gorelik, L.; Fields, P.E.; Flavell, R.A. Cutting edge: TGF-β inhibits Th type 2 development through inhibition of GATA-3 expression. J. Immunol. 2000, 165, 4773–4777. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.T.; Martin, S.L.; Xia, L.; Gorham, J.D. TGF-β 1 uses distinct mechanisms to inhibit IFN-gamma expression in CD4+ T cells at priming and at recall: Differential involvement of Stat4 and T-bet. J. Immunol. 2005, 174, 5950–5958. [Google Scholar] [CrossRef] [PubMed]
- Fantini, M.C.; Becker, C.; Monteleone, G.; Pallone, F.; Galle, P.R.; Neurath, M.F. Cutting edge: TGF-β induces a regulatory phenotype in CD4+CD25− T cells through Foxp3 induction and down-regulation of Smad7. J. Immunol. 2004, 172, 5149–5153. [Google Scholar] [CrossRef] [PubMed]
- Selvaraj, R.K.; Geiger, T.L. A kinetic and dynamic analysis of Foxp3 induced in T cells by TGF-β. J. Immunol. 2007, 178, 7667–7677. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Jin, W.; Hardegen, N.; Lei, K.J.; Li, L.; Marinos, N.; McGrady, G.; Wahl, S.M. Conversion of peripheral CD4+CD25− naive T cells to CD4+CD25+ regulatory T cells by TGF-β induction of transcription factor Foxp3. J. Exp. Med. 2003, 198, 1875–1886. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Laouar, Y.; Li, M.O.; Green, E.A.; Flavell, R.A. TGF-β regulates in vivo expansion of Foxp3-expressing CD4+CD25+ regulatory T cells responsible for protection against diabetes. Proc. Natl. Acad. Sci. USA 2004, 101, 4572–4577. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, A.; Oberle, N.; Krammer, P.H. Molecular mechanisms of treg-mediated T cell suppression. Front. Immunol. 2012, 3, 51. [Google Scholar] [CrossRef] [PubMed]
- Tone, Y.; Furuuchi, K.; Kojima, Y.; Tykocinski, M.L.; Greene, M.I.; Tone, M. Smad3 and NFAT cooperate to induce Foxp3 expression through its enhancer. Nat. Immunol. 2008, 9, 194–202. [Google Scholar] [CrossRef] [PubMed]
- Ghiringhelli, F.; Puig, P.E.; Roux, S.; Parcellier, A.; Schmitt, E.; Solary, E.; Kroemer, G.; Martin, F.; Chauffert, B.; Zitvogel, L. Tumor cells convert immature myeloid dendritic cells into TGF-β-secreting cells inducing CD4+CD25+ regulatory T cell proliferation. J. Exp. Med. 2005, 202, 919–929. [Google Scholar] [CrossRef] [PubMed]
- Valzasina, B.; Piconese, S.; Guiducci, C.; Colombo, M.P. Tumor-induced expansion of regulatory T cells by conversion of CD4+CD25− lymphocytes is thymus and proliferation independent. Cancer Res. 2006, 66, 4488–4495. [Google Scholar] [CrossRef] [PubMed]
- Liu, V.C.; Wong, L.Y.; Jang, T.; Shah, A.H.; Park, I.; Yang, X.; Zhang, Q.; Lonning, S.; Teicher, B.A.; Lee, C. Tumor evasion of the immune system by converting CD4+CD25− T cells into CD4+CD25+ T regulatory cells: Role of tumor-derived TGF-β. J. Immunol. 2007, 178, 2883–2892. [Google Scholar] [CrossRef] [PubMed]
- Bailey, S.R.; Nelson, M.H.; Himes, R.A.; Li, Z.; Mehrotra, S.; Paulos, C.M. Th17 cells in cancer: The ultimate identity crisis. Front. Immunol. 2014, 5, 276. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, I.I.; McKenzie, B.S.; Zhou, L.; Tadokoro, C.E.; Lepelley, A.; Lafaille, J.J.; Cua, D.J.; Littman, D.R. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell 2006, 126, 1121–1133. [Google Scholar] [CrossRef] [PubMed]
- Salehi, S.; Bankoti, R.; Benevides, L.; Willen, J.; Couse, M.; Silva, J.S.; Dhall, D.; Meffre, E.; Targan, S.; Martins, G.A. B lymphocyte-induced maturation protein-1 contributes to intestinal mucosa homeostasis by limiting the number of IL-17-producing CD4+ T cells. J. Immunol. 2012, 189, 5682–5693. [Google Scholar] [CrossRef] [PubMed]
- Das, J.; Ren, G.; Zhang, L.; Roberts, A.I.; Zhao, X.; Bothwell, A.L.; Van Kaer, L.; Shi, Y.; Das, G. Transforming growth factor β is dispensable for the molecular orchestration of Th17 cell differentiation. J. Exp. Med. 2009, 206, 2407–2416. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Davidson, T.S.; Wei, G.; Jankovic, D.; Cui, K.; Schones, D.E.; Guo, L.; Zhao, K.; Shevach, E.M.; Paul, W.E. Down-regulation of Gfi-1 expression by TGF-β is important for differentiation of Th17 and CD103+ inducible regulatory T cells. J. Exp. Med. 2009, 206, 329–341. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Lopes, J.E.; Chong, M.M.; Ivanov, I.I.; Min, R.; Victora, G.D.; Shen, Y.; Du, J.; Rubtsov, Y.P.; Rudensky, A.Y.; et al. TGF-β-induced Foxp3 inhibits T(H)17 cell differentiation by antagonizing RORgammat function. Nature 2008, 453, 236–240. [Google Scholar] [CrossRef] [PubMed]
- Hasan, M.; Neumann, B.; Haupeltshofer, S.; Stahlke, S.; Fantini, M.C.; Angstwurm, K.; Bogdahn, U.; Kleiter, I. Activation of TGF-β-induced non-Smad signaling pathways during Th17 differentiation. Immunol. Cell Biol. 2015, 93, 662–672. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Takaku, M.; Zou, L.; Gu, A.D.; Chou, W.C.; Zhang, G.; Wu, B.; Kong, Q.; Thomas, S.Y.; Serody, J.S.; et al. Reversing SKI-SMAD4-mediated suppression is essential for Th17 cell differentiation. Nature 2017, 551, 105–109. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.O.; Nurieva, R.; Martinez, G.J.; Kang, H.S.; Chung, Y.; Pappu, B.P.; Shah, B.; Chang, S.H.; Schluns, K.S.; Watowich, S.S.; et al. Molecular antagonism and plasticity of regulatory and inflammatory T cell programs. Immunity 2008, 29, 44–56. [Google Scholar] [CrossRef] [PubMed]
- Allard, B.; Longhi, M.S.; Robson, S.C.; Stagg, J. The ectonucleotidases CD39 and CD73: Novel checkpoint inhibitor targets. Immunol. Rev. 2017, 276, 121–144. [Google Scholar] [CrossRef] [PubMed]
- Chalmin, F.; Mignot, G.; Bruchard, M.; Chevriaux, A.; Vegran, F.; Hichami, A.; Ladoire, S.; Derangere, V.; Vincent, J.; Masson, D.; et al. STAT3 and Gfi-1 transcription factors control Th17 cell immunosuppressive activity via the regulation of ectonucleotidase expression. Immunity 2012, 36, 362–373. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.; Schwartz, J.A.; Sandrock, C.; Bellemore, S.M.; Nikoopour, E. Modulation of autoimmune diseases by interleukin (IL)-17 producing regulatory T helper (Th17) cells. Indian J. Med. Res. 2013, 138, 591–594. [Google Scholar] [PubMed]
- Bellemore, S.M.; Nikoopour, E.; Schwartz, J.A.; Krougly, O.; Lee-Chan, E.; Singh, B. Preventative role of interleukin-17 producing regulatory T helper type 17 (Treg 17) cells in type 1 diabetes in non-obese diabetic mice. Clin. Exp. Immunol. 2015, 182, 261–269. [Google Scholar] [CrossRef] [PubMed]
- Ghoreschi, K.; Laurence, A.; Yang, X.P.; Tato, C.M.; McGeachy, M.J.; Konkel, J.E.; Ramos, H.L.; Wei, L.; Davidson, T.S.; Bouladoux, N.; et al. Generation of pathogenic T(H)17 cells in the absence of TGF-β signalling. Nature 2010, 467, 967–971. [Google Scholar] [CrossRef] [PubMed]
- Rivera Vargas, T.; Humblin, E.; Vegran, F.; Ghiringhelli, F.; Apetoh, L. TH9 cells in anti-tumor immunity. Semin. Immunopathol. 2017, 39, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Fischer, M.; Bijman, M.; Molin, D.; Cormont, F.; Uyttenhove, C.; van Snick, J.; Sundstrom, C.; Enblad, G.; Nilsson, G. Increased serum levels of interleukin-9 correlate to negative prognostic factors in Hodgkin’s lymphoma. Leukemia 2003, 17, 2513–2516. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Chen, X.; Bai, Q.; Qin, C.; Mohamud, A.O.; Zhu, Z.; Ball, T.W.; Ruth, C.M.; Newcomer, D.R.; Herrick, E.J.; et al. IL-9 inhibits HTB-72 melanoma cell growth through upregulation of p21 and TRAIL. J. Surg. Oncol. 2015, 111, 969–974. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Hong, S.; Li, H.; Park, J.; Hong, B.; Wang, L.; Zheng, Y.; Liu, Z.; Xu, J.; He, J.; et al. Th9 cells promote antitumor immune responses in vivo. J. Clin. Investig. 2012, 122, 4160–4171. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Chu, X.; Chen, J.; Wang, Y.; Gao, S.; Jiang, Y.; Zhu, X.; Tan, G.; Zhao, W.; Yi, H.; et al. Dectin-1-activated dendritic cells trigger potent antitumour immunity through the induction of Th9 cells. Nat. Commun. 2016, 7, 12368. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.K.; Kim, B.S.; Koh, C.H.; Seok, J.W.; Park, J.S.; Shin, K.S.; Bae, E.A.; Lee, G.E.; Jeon, H.; Cho, J.; et al. Glucocorticoid-induced tumor necrosis factor receptor-related protein co-stimulation facilitates tumor regression by inducing IL-9-producing helper T cells. Nat. Med. 2015, 21, 1010–1017. [Google Scholar] [CrossRef] [PubMed]
- Xiao, X.; Shi, X.; Fan, Y.; Zhang, X.; Wu, M.; Lan, P.; Minze, L.; Fu, Y.X.; Ghobrial, R.M.; Liu, W.; et al. GITR subverts Foxp3(+) Tregs to boost Th9 immunity through regulation of histone acetylation. Nat. Commun. 2015, 6, 8266. [Google Scholar] [CrossRef] [PubMed]
- Nonomura, Y.; Otsuka, A.; Nakashima, C.; Seidel, J.A.; Kitoh, A.; Dainichi, T.; Nakajima, S.; Sawada, Y.; Matsushita, S.; Aoki, M.; et al. Peripheral blood Th9 cells are a possible pharmacodynamic biomarker of nivolumab treatment efficacy in metastatic melanoma patients. Oncoimmunology 2016, 5, e1248327. [Google Scholar] [CrossRef] [PubMed]
- Chaput, N.; Louafi, S.; Bardier, A.; Charlotte, F.; Vaillant, J.C.; Menegaux, F.; Rosenzwajg, M.; Lemoine, F.; Klatzmann, D.; Taieb, J. Identification of CD8+CD25+Foxp3+ suppressive T cells in colorectal cancer tissue. Gut 2009, 58, 520–529. [Google Scholar] [CrossRef] [PubMed]
- Dwivedi, V.P.; Tousif, S.; Bhattacharya, D.; Prasad, D.V.; Van Kaer, L.; Das, J.; Das, G. Transforming growth factor-β protein inversely regulates in vivo differentiation of interleukin-17 (IL-17)-producing CD4+ and CD8+ T cells. J. Biol. Chem. 2012, 287, 2943–2947. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Hong, B.; Li, H.; Zheng, Y.; Zhang, M.; Wang, S.; Qian, J.; Yi, Q. Tumor-specific IL-9-producing CD8+ Tc9 cells are superior effector than type-I cytotoxic Tc1 cells for adoptive immunotherapy of cancers. Proc. Natl. Acad. Sci. USA 2014, 111, 2265–2270. [Google Scholar] [CrossRef] [PubMed]
- Schenkel, J.M.; Masopust, D. Tissue-resident memory T cells. Immunity 2014, 41, 886–897. [Google Scholar] [CrossRef] [PubMed]
- Mackay, L.K.; Rahimpour, A.; Ma, J.Z.; Collins, N.; Stock, A.T.; Hafon, M.L.; Vega-Ramos, J.; Lauzurica, P.; Mueller, S.N.; Stefanovic, T.; et al. The developmental pathway for CD103(+)CD8+ tissue-resident memory T cells of skin. Nat. Immunol. 2013, 14, 1294–1301. [Google Scholar] [CrossRef] [PubMed]
- Skon, C.N.; Lee, J.Y.; Anderson, K.G.; Masopust, D.; Hogquist, K.A.; Jameson, S.C. Transcriptional downregulation of S1pr1 is required for the establishment of resident memory CD8+ T cells. Nat. Immunol. 2013, 14, 1285–1293. [Google Scholar] [CrossRef] [PubMed]
- Mackay, L.K.; Wynne-Jones, E.; Freestone, D.; Pellicci, D.G.; Mielke, L.A.; Newman, D.M.; Braun, A.; Masson, F.; Kallies, A.; Belz, G.T.; et al. T-box Transcription Factors Combine with the Cytokines TGF-β and IL-15 to Control Tissue-Resident Memory T Cell Fate. Immunity 2015, 43, 1101–1111. [Google Scholar] [CrossRef] [PubMed]
- Webb, J.R.; Milne, K.; Watson, P.; Deleeuw, R.J.; Nelson, B.H. Tumor-infiltrating lymphocytes expressing the tissue resident memory marker CD103 are associated with increased survival in high-grade serous ovarian cancer. Clin. Cancer Res. 2014, 20, 434–444. [Google Scholar] [CrossRef] [PubMed]
- Djenidi, F.; Adam, J.; Goubar, A.; Durgeau, A.; Meurice, G.; de Montpreville, V.; Validire, P.; Besse, B.; Mami-Chouaib, F. CD8+CD103+ tumor-infiltrating lymphocytes are tumor-specific tissue-resident memory T cells and a prognostic factor for survival in lung cancer patients. J. Immunol. 2015, 194, 3475–3486. [Google Scholar] [CrossRef] [PubMed]
- Webb, J.R.; Milne, K.; Nelson, B.H. PD-1 and CD103 Are Widely Coexpressed on Prognostically Favorable Intraepithelial CD8 T Cells in Human Ovarian Cancer. Cancer Immunol. Res. 2015, 3, 926–935. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.Q.; Milne, K.; Derocher, H.; Webb, J.R.; Nelson, B.H.; Watson, P.H. CD103 and Intratumoral Immune Response in Breast Cancer. Clin. Cancer Res. 2016. [Google Scholar] [CrossRef] [PubMed]
- Ansell, S.M.; Lesokhin, A.M.; Borrello, I.; Halwani, A.; Scott, E.C.; Gutierrez, M.; Schuster, S.J.; Millenson, M.M.; Cattry, D.; Freeman, G.J.; et al. PD-1 blockade with nivolumab in relapsed or refractory Hodgkin’s lymphoma. N. Engl. J. Med. 2015, 372, 311–319. [Google Scholar] [CrossRef] [PubMed]
- Borghaei, H.; Paz-Ares, L.; Horn, L.; Spigel, D.R.; Steins, M.; Ready, N.E.; Chow, L.Q.; Vokes, E.E.; Felip, E.; Holgado, E.; et al. Nivolumab versus Docetaxel in Advanced Nonsquamous Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 373, 1627–1639. [Google Scholar] [CrossRef] [PubMed]
- Brahmer, J.; Reckamp, K.L.; Baas, P.; Crino, L.; Eberhardt, W.E.; Poddubskaya, E.; Antonia, S.; Pluzanski, A.; Vokes, E.E.; Holgado, E.; et al. Nivolumab versus Docetaxel in Advanced Squamous-Cell Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 373, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, S.A.; Restifo, N.P.; Yang, J.C.; Morgan, R.A.; Dudley, M.E. Adoptive cell transfer: A clinical path to effective cancer immunotherapy. Nat. Rev. Cancer 2008, 8, 299–308. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, S.A. Raising the bar: The curative potential of human cancer immunotherapy. Sci. Transl. Med. 2012, 4, 127–128. [Google Scholar] [CrossRef] [PubMed]
- Rook, A.H.; Kehrl, J.H.; Wakefield, L.M.; Roberts, A.B.; Sporn, M.B.; Burlington, D.B.; Lane, H.C.; Fauci, A.S. Effects of transforming growth factor β on the functions of natural killer cells: Depressed cytolytic activity and blunting of interferon responsiveness. J. Immunol. 1986, 136, 3916–3920. [Google Scholar] [PubMed]
- Yu, J.; Wei, M.; Becknell, B.; Trotta, R.; Liu, S.; Boyd, Z.; Jaung, M.S.; Blaser, B.W.; Sun, J.; Benson, D.M., Jr.; et al. Pro- and antiinflammatory cytokine signaling: Reciprocal antagonism regulates interferon-gamma production by human natural killer cells. Immunity 2006, 24, 575–590. [Google Scholar] [CrossRef] [PubMed]
- Trotta, R.; Col, J.D.; Yu, J.; Ciarlariello, D.; Thomas, B.; Zhang, X.; Allard, J., 2nd; Wei, M.; Mao, H.; Byrd, J.C.; et al. TGF-β utilizes SMAD3 to inhibit CD16-mediated IFN-gamma production and antibody-dependent cellular cytotoxicity in human NK cells. J. Immunol. 2008, 181, 3784–3792. [Google Scholar] [CrossRef] [PubMed]
- Castriconi, R.; Cantoni, C.; Della Chiesa, M.; Vitale, M.; Marcenaro, E.; Conte, R.; Biassoni, R.; Bottino, C.; Moretta, L.; Moretta, A. Transforming growth factor β1 inhibits expression of NKp30 and NKG2D receptors: Consequences for the NK-mediated killing of dendritic cells. Proc. Natl. Acad. Sci. USA 2003, 100, 4120–4125. [Google Scholar] [CrossRef] [PubMed]
- Casetti, R.; Agrati, C.; Wallace, M.; Sacchi, A.; Martini, F.; Martino, A.; Rinaldi, A.; Malkovsky, M. Cutting edge: TGF-β1 and IL-15 Induce FOXP3+ gammadelta regulatory T cells in the presence of antigen stimulation. J. Immunol. 2009, 183, 3574–3577. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Kang, N.; Zhang, X.; Dong, X.; Wei, W.; Cui, L.; Ba, D.; He, W. Generation of human regulatory gammadelta T cells by TCRgammadelta stimulation in the presence of TGF-β and their involvement in the pathogenesis of systemic lupus erythematosus. J. Immunol. 2011, 186, 6693–6700. [Google Scholar] [CrossRef] [PubMed]
- Cortez, V.S.; Cervantes-Barragan, L.; Robinette, M.L.; Bando, J.K.; Wang, Y.; Geiger, T.L.; Gilfillan, S.; Fuchs, A.; Vivier, E.; Sun, J.C.; et al. Transforming Growth Factor-β Signaling Guides the Differentiation of Innate Lymphoid Cells in Salivary Glands. Immunity 2016, 44, 1127–1139. [Google Scholar] [CrossRef] [PubMed]
- Dadi, S.; Chhangawala, S.; Whitlock, B.M.; Franklin, R.A.; Luo, C.T.; Oh, S.A.; Toure, A.; Pritykin, Y.; Huse, M.; Leslie, C.S.; et al. Cancer Immunosurveillance by Tissue-Resident Innate Lymphoid Cells and Innate-like T Cells. Cell 2016, 164, 365–377. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Souza-Fonseca-Guimaraes, F.; Bald, T.; Ng, S.S.; Young, A.; Ngiow, S.F.; Rautela, J.; Straube, J.; Waddell, N.; Blake, S.J.; et al. Tumor immunoevasion by the conversion of effector NK cells into type 1 innate lymphoid cells. Nat. Immunol. 2017, 18, 1004–1015. [Google Scholar] [CrossRef] [PubMed]
- Carli, C.; Giroux, M.; Delisle, J.S. Roles of Transforming Growth Factor-β in Graft-versus-Host and Graft-versus-Tumor Effects. Biol. Blood Marrow Transplant. 2012, 18, 1329–1340. [Google Scholar] [CrossRef] [PubMed]
- Fuxe, J.; Vincent, T.; Garcia de Herreros, A. Transcriptional crosstalk between TGF-β and stem cell pathways in tumor cell invasion: Role of EMT promoting Smad complexes. Cell Cycle 2010, 9, 2363–2374. [Google Scholar] [CrossRef] [PubMed]
- Wendt, M.K.; Allington, T.M.; Schiemann, W.P. Mechanisms of the epithelial-mesenchymal transition by TGF-β. Future Oncol. 2009, 5, 1145–1168. [Google Scholar] [CrossRef] [PubMed]
- Pardali, E.; ten Dijke, P. Transforming growth factor-β signaling and tumor angiogenesis. Front. Biosci. 2009, 14, 4848–4861. [Google Scholar] [CrossRef]
- Massague, J. TGFβ in Cancer. Cell 2008, 134, 215–230. [Google Scholar] [CrossRef] [PubMed]
- Galon, J.; Costes, A.; Sanchez-Cabo, F.; Kirilovsky, A.; Mlecnik, B.; Lagorce-Pages, C.; Tosolini, M.; Camus, M.; Berger, A.; Wind, P.; et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science 2006, 313, 1960–1964. [Google Scholar] [CrossRef] [PubMed]
- Galon, J.; Mlecnik, B.; Bindea, G.; Angell, H.K.; Berger, A.; Lagorce, C.; Lugli, A.; Zlobec, I.; Hartmann, A.; Bifulco, C.; et al. Towards the introduction of the ‘Immunoscore’ in the classification of malignant tumours. J. Pathol. 2014, 232, 199–209. [Google Scholar] [CrossRef] [PubMed]
- Thorsson, V.; Gibbs, D.L.; Brown, S.D.; Wolf, D.; Bortone, D.S.; Ou Yang, T.H.; Porta-Pardo, E.; Gao, G.F.; Plaisier, C.L.; Eddy, J.A.; et al. The Immune Landscape of Cancer. Immunity 2018, 48, 812–830.e14. [Google Scholar] [CrossRef] [PubMed]
- Spranger, S.; Sivan, A.; Corrales, L.; Gajewski, T.F. Tumor and Host Factors Controlling Antitumor Immunity and Efficacy of Cancer Immunotherapy. Adv. Immunol. 2016, 130, 75–93. [Google Scholar] [PubMed]
- Colak, S.; Ten Dijke, P. Targeting TGF-β Signaling in Cancer. Trends Cancer 2017, 3, 56–71. [Google Scholar] [CrossRef] [PubMed]
- Guinney, J.; Dienstmann, R.; Wang, X.; de Reynies, A.; Schlicker, A.; Soneson, C.; Marisa, L.; Roepman, P.; Nyamundanda, G.; Angelino, P.; et al. The consensus molecular subtypes of colorectal cancer. Nat. Med. 2015, 21, 1350–1356. [Google Scholar] [CrossRef] [PubMed]
- Calon, A.; Espinet, E.; Palomo-Ponce, S.; Tauriello, D.V.; Iglesias, M.; Cespedes, M.V.; Sevillano, M.; Nadal, C.; Jung, P.; Zhang, X.H.; et al. Dependency of colorectal cancer on a TGF-β-driven program in stromal cells for metastasis initiation. Cancer Cell 2012, 22, 571–584. [Google Scholar] [CrossRef] [PubMed]
- Calon, A.; Lonardo, E.; Berenguer-Llergo, A.; Espinet, E.; Hernando-Momblona, X.; Iglesias, M.; Sevillano, M.; Palomo-Ponce, S.; Tauriello, D.V.; Byrom, D.; et al. Stromal gene expression defines poor-prognosis subtypes in colorectal cancer. Nat. Genet. 2015, 47, 320–329. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Kim, T.J.; Peng, D.H.; Duan, D.; Gibbons, D.L.; Yamauchi, M.; Jackson, J.R.; Le Saux, C.J.; Calhoun, C.; Peters, J.; et al. Fibroblast-specific inhibition of TGF-β1 signaling attenuates lung and tumor fibrosis. J. Clin. Investig. 2017, 127, 3675–3688. [Google Scholar] [CrossRef] [PubMed]
- Principe, D.R.; DeCant, B.; Mascarinas, E.; Wayne, E.A.; Diaz, A.M.; Akagi, N.; Hwang, R.; Pasche, B.; Dawson, D.W.; Fang, D.; et al. TGFβ Signaling in the Pancreatic Tumor Microenvironment Promotes Fibrosis and Immune Evasion to Facilitate Tumorigenesis. Cancer Res. 2016, 76, 2525–2539. [Google Scholar] [CrossRef] [PubMed]
- Tauriello, D.V.F.; Palomo-Ponce, S.; Stork, D.; Berenguer-Llergo, A.; Badia-Ramentol, J.; Iglesias, M.; Sevillano, M.; Ibiza, S.; Canellas, A.; Hernando-Momblona, X.; et al. TGFβ drives immune evasion in genetically reconstituted colon cancer metastasis. Nature 2018, 554, 538–543. [Google Scholar] [CrossRef] [PubMed]
- Mariathasan, S.; Turley, S.J.; Nickles, D.; Castiglioni, A.; Yuen, K.; Wang, Y.; Kadel Iii, E.E.; Koeppen, H.; Astarita, J.L.; Cubas, R.; et al. TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 2018, 554, 544–548. [Google Scholar] [CrossRef] [PubMed]
- Bhowmick, N.A.; Chytil, A.; Plieth, D.; Gorska, A.E.; Dumont, N.; Shappell, S.; Washington, M.K.; Neilson, E.G.; Moses, H.L. TGF-β signaling in fibroblasts modulates the oncogenic potential of adjacent epithelia. Science 2004, 303, 848–851. [Google Scholar] [CrossRef] [PubMed]
- Donkor, M.K.; Sarkar, A.; Li, M.O. TGF-β1 produced by activated CD4(+) T Cells Antagonizes T Cell Surveillance of Tumor Development. Oncoimmunology 2012, 1, 162–171. [Google Scholar] [CrossRef] [PubMed]
- Fridlender, Z.G.; Sun, J.; Kim, S.; Kapoor, V.; Cheng, G.; Ling, L.; Worthen, G.S.; Albelda, S.M. Polarization of tumor-associated neutrophil phenotype by TGF-β: “N1” versus “N2” TAN. Cancer Cell 2009, 16, 183–194. [Google Scholar] [CrossRef] [PubMed]
- Novitskiy, S.V.; Pickup, M.W.; Chytil, A.; Polosukhina, D.; Owens, P.; Moses, H.L. Deletion of TGF-β signaling in myeloid cells enhances their anti-tumorigenic properties. J. Leukoc. Biol. 2012, 92, 641–651. [Google Scholar] [CrossRef] [PubMed]
- Park, B.V.; Freeman, Z.T.; Ghasemzadeh, A.; Chattergoon, M.A.; Rutebemberwa, A.; Steigner, J.; Winter, M.E.; Huynh, T.V.; Sebald, S.M.; Lee, S.J.; et al. TGFβ1-Mediated SMAD3 Enhances PD-1 Expression on Antigen-Specific T Cells in Cancer. Cancer Discov. 2016, 6, 1366–1381. [Google Scholar] [CrossRef] [PubMed]
- Stephen, T.L.; Payne, K.K.; Chaurio, R.A.; Allegrezza, M.J.; Zhu, H.; Perez-Sanz, J.; Perales-Puchalt, A.; Nguyen, J.M.; Vara-Ailor, A.E.; Eruslanov, E.B.; et al. SATB1 Expression Governs Epigenetic Repression of PD-1 in Tumor-Reactive T Cells. Immunity 2017, 46, 51–64. [Google Scholar] [CrossRef] [PubMed]
- Garrison, K.; Hahn, T.; Lee, W.C.; Ling, L.E.; Weinberg, A.D.; Akporiaye, E.T. The small molecule TGF-β signaling inhibitor SM16 synergizes with agonistic OX40 antibody to suppress established mammary tumors and reduce spontaneous metastasis. Cancer Immunol. Immunother. 2012, 61, 511–521. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Wrzesinski, S.H.; Stern, E.; Look, M.; Criscione, J.; Ragheb, R.; Jay, S.M.; Demento, S.L.; Agawu, A.; Licona Limon, P.; et al. Combination delivery of TGF-β inhibitor and IL-2 by nanoscale liposomal polymeric gels enhances tumour immunotherapy. Nat. Mater. 2012, 11, 895–905. [Google Scholar] [CrossRef] [PubMed]
- Vanpouille-Box, C.; Diamond, J.M.; Pilones, K.A.; Zavadil, J.; Babb, J.S.; Formenti, S.C.; Barcellos-Hoff, M.H.; Demaria, S. TGFβ Is a Master Regulator of Radiation Therapy-Induced Antitumor Immunity. Cancer Res. 2015, 75, 2232–2242. [Google Scholar] [CrossRef] [PubMed]
- Courau, T.; Nehar-Belaid, D.; Florez, L.; Levacher, B.; Vazquez, T.; Brimaud, F.; Bellier, B.; Klatzmann, D. TGF-β and VEGF cooperatively control the immunotolerant tumor environment and the efficacy of cancer immunotherapies. JCI Insight 2016, 1, e85974. [Google Scholar] [CrossRef] [PubMed]
- Hutzen, B.; Chen, C.Y.; Wang, P.Y.; Sprague, L.; Swain, H.M.; Love, J.; Conner, J.; Boon, L.; Cripe, T.P. TGF-β Inhibition Improves Oncolytic Herpes Viroimmunotherapy in Murine Models of Rhabdomyosarcoma. Mol. Ther. Oncolytics 2017, 7, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Terabe, M.; Ambrosino, E.; Takaku, S.; O’Konek, J.J.; Venzon, D.; Lonning, S.; McPherson, J.M.; Berzofsky, J.A. Synergistic enhancement of CD8+ T cell-mediated tumor vaccine efficacy by an anti-transforming growth factor-β monoclonal antibody. Clin. Cancer Res. 2009, 15, 6560–6569. [Google Scholar] [CrossRef] [PubMed]
- Ueda, R.; Fujita, M.; Zhu, X.; Sasaki, K.; Kastenhuber, E.R.; Kohanbash, G.; McDonald, H.A.; Harper, J.; Lonning, S.; Okada, H. Systemic inhibition of transforming growth factor-β in glioma-bearing mice improves the therapeutic efficacy of glioma-associated antigen peptide vaccines. Clin. Cancer Res. 2009, 15, 6551–6559. [Google Scholar] [CrossRef] [PubMed]
- Takaku, S.; Terabe, M.; Ambrosino, E.; Peng, J.; Lonning, S.; McPherson, J.M.; Berzofsky, J.A. Blockade of TGF-β enhances tumor vaccine efficacy mediated by CD8(+) T cells. Int. J. Cancer 2010, 126, 1666–1674. [Google Scholar] [CrossRef] [PubMed]
- Terabe, M.; Robertson, F.C.; Clark, K.; De Ravin, E.; Bloom, A.; Venzon, D.J.; Kato, S.; Mirza, A.; Berzofsky, J.A. Blockade of only TGF-β 1 and 2 is sufficient to enhance the efficacy of vaccine and PD-1 checkpoint blockade immunotherapy. Oncoimmunology 2017, 6, e1308616. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Wang, Y.; Zhang, L.; Huang, L. Nanoparticle-delivered transforming growth factor-β siRNA enhances vaccination against advanced melanoma by modifying tumor microenvironment. ACS Nano 2014, 8, 3636–3645. [Google Scholar] [CrossRef] [PubMed]
- Rooney, C.M.; Leen, A.M.; Vera, J.F.; Heslop, H.E. T lymphocytes targeting native receptors. Immunol. Rev. 2014, 257, 39–55. [Google Scholar] [CrossRef] [PubMed]
- Riviere, I.; Sadelain, M. Chimeric antigen receptors: A cell and gene therapy perspective. Mol. Ther. 2017, 25, 1117–1124. [Google Scholar] [CrossRef] [PubMed]
- Barrett, D.M.; Singh, N.; Porter, D.L.; Grupp, S.A.; June, C.H. Chimeric antigen receptor therapy for cancer. Annu. Rev. Med. 2014, 65, 333–347. [Google Scholar] [CrossRef] [PubMed]
- Kloss, C.C.; Lee, J.; Zhang, A.; Chen, F.; Melenhorst, J.J.; Lacey, S.F.; Maus, M.V.; Fraietta, J.A.; Zhao, Y.; June, C.H. Dominant-Negative TGF-β Receptor Enhances PSMA-Targeted Human CAR T Cell Proliferation And Augments Prostate Cancer Eradication. Mol. Ther. 2018. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wen, W.; Yuan, J.; Helfand, B.; Li, Y.; Shi, C.; Tian, F.; Zheng, J.; Wang, F.; Chen, L.; et al. Immunotherapy for human renal cell carcinoma by adoptive transfer of autologous transforming growth factor β-insensitive CD8+ T cells. Clin. Cancer Res. 2010, 16, 164–173. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Yang, X.; Pins, M.; Javonovic, B.; Kuzel, T.; Kim, S.J.; Parijs, L.V.; Greenberg, N.M.; Liu, V.; Guo, Y.; et al. Adoptive transfer of tumor-reactive transforming growth factor-β-insensitive CD8+ T cells: Eradication of autologous mouse prostate cancer. Cancer Res. 2005, 65, 1761–1769. [Google Scholar] [CrossRef] [PubMed]
- Foster, A.E.; Dotti, G.; Lu, A.; Khalil, M.; Brenner, M.K.; Heslop, H.E.; Rooney, C.M.; Bollard, C.M. Antitumor activity of EBV-specific T lymphocytes transduced with a dominant negative TGF-β receptor. J. Immunother. 2008, 31, 500–505. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Helfand, B.T.; Carneiro, B.A.; Qin, W.; Yang, X.J.; Lee, C.; Zhang, W.; Giles, F.J.; Cristofanilli, M.; Kuzel, T.M. Efficacy Against Human Prostate Cancer by Prostate-specific Membrane Antigen-specific, Transforming Growth Factor-β Insensitive Genetically Targeted CD8(+) T-cells Derived from Patients with Metastatic Castrate-resistant Disease. Eur. Urol. 2018, 73, 648–652. [Google Scholar] [CrossRef] [PubMed]
- Bollard, C.M.; Tripic, T.; Cruz, C.R.; Dotti, G.; Gottschalk, S.; Torrano, V.; Dakhova, O.; Carrum, G.; Ramos, C.A.; Liu, H.; et al. Tumor-specific T-cells engineered to overcome tumor immune evasion induce clinical responses in patients with relapsed hodgkin lymphoma. J. Clin. Oncol. 2018, 36, 1128–1139. [Google Scholar] [CrossRef] [PubMed]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Cowey, C.L.; Lao, C.D.; Schadendorf, D.; Dummer, R.; Smylie, M.; Rutkowski, P.; et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N. Engl. J. Med. 2015, 373, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Nizard, M.; Roussel, H.; Diniz, M.O.; Karaki, S.; Tran, T.; Voron, T.; Dransart, E.; Sandoval, F.; Riquet, M.; Rance, B.; et al. Induction of resident memory T cells enhances the efficacy of cancer vaccine. Nat. Commun. 2017, 8, 15221. [Google Scholar] [CrossRef] [PubMed]


© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dahmani, A.; Delisle, J.-S. TGF-β in T Cell Biology: Implications for Cancer Immunotherapy. Cancers 2018, 10, 194. https://doi.org/10.3390/cancers10060194
Dahmani A, Delisle J-S. TGF-β in T Cell Biology: Implications for Cancer Immunotherapy. Cancers. 2018; 10(6):194. https://doi.org/10.3390/cancers10060194
Chicago/Turabian StyleDahmani, Amina, and Jean-Sébastien Delisle. 2018. "TGF-β in T Cell Biology: Implications for Cancer Immunotherapy" Cancers 10, no. 6: 194. https://doi.org/10.3390/cancers10060194
APA StyleDahmani, A., & Delisle, J.-S. (2018). TGF-β in T Cell Biology: Implications for Cancer Immunotherapy. Cancers, 10(6), 194. https://doi.org/10.3390/cancers10060194