Clinico-Pathological Importance of TGF-β/Phospho-Smad Signaling during Human Hepatic Fibrocarcinogenesis

{kind=link}
{kind=link}
Abstract
1. Introduction
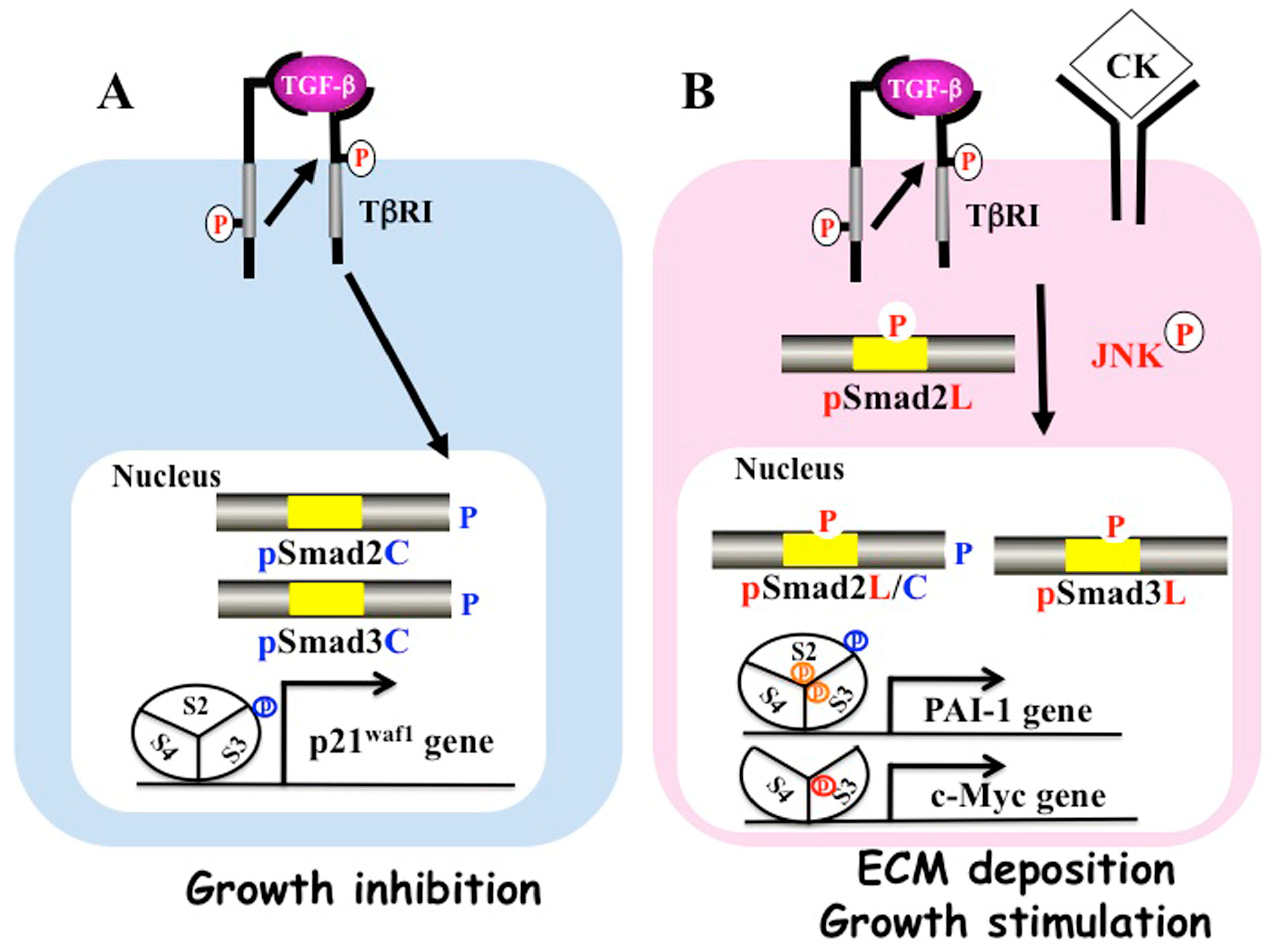
2. Multiple Smad Phospho-Isoforms Signaling Exist
3. PSmad3C Displays Cytostatic Activity
4. PSmad3L (Ser-213) Enhances Mitogenic Pathways
5. PSmad2L/C and pSmad3L/C Promote Pro-Tumorigenic/Fibrogenic Pathways
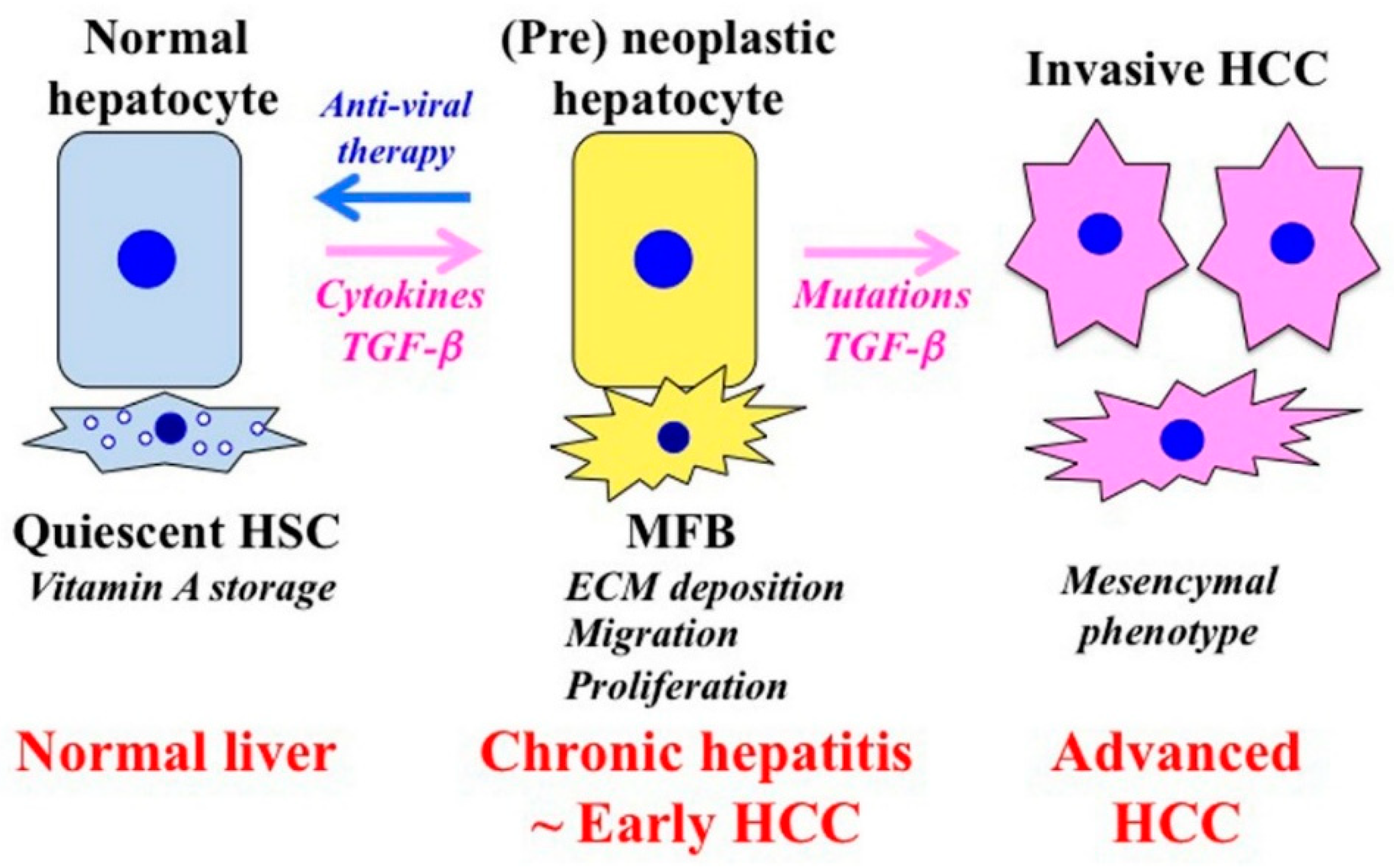
6. A Shift of Hepatocytic Phospho-Smad Signaling from the Tumor-Suppressive pSmad3C Pathway to Carcinogenic pSmad3L and Fibrogenic pSmad2L/C Pathways Observed during Hepatic Fibrocarcinogenesis
7. Effective Antiviral Treatment Can Reverse Phospho-Smad Signaling in Early Stages of Chronic Liver Disease
8. Implications for HCC Prevention and Therapy
9. EMT in Liver Fibrosis and HCC Progression
10. Conclusions
Conflicts of Interest
References
- Dooley, J.S.; Lok, A.F.; Burroughs, A.K.; Heathcote, E.J. Sherlock's Disease of the Liver and Biliary System; Wiley-Blackwell: West Sussex, UK, 2011. [Google Scholar]
- Yoshida, H.; Shiratori, Y.; Moriyama, M.; Arakawa, Y.; Ide, T.; Sata, M.; Inoue, O.; Yano, M.; Tanaka, M.; Fujiyama, S.; et al. Interferon therapy reduces the risk for hepatocellular carcinoma: National surveillance program of cirrhotic and noncirrhotic patients with chronic hepatitis C in Japan. IHIT Study Group. Inhibition of Hepatocarcinogenesis by Interferon Therapy. Ann. Intern. Med. 1999, 131, 174–181. [Google Scholar] [CrossRef] [PubMed]
- Fattovich, G.; Bortolotti, F.; Donato, F. Natural history of chronic hepatitis B: Special emphasis on disease progression and prognostic factors. J. hepatol. 2008, 48, 335–352. [Google Scholar] [CrossRef] [PubMed]
- Inagaki, Y.; Okazaki, I. Emerging insights into Transforming growth factor beta Smad signal in hepatic fibrogenesis. Gut 2007, 56, 284–292. [Google Scholar] [CrossRef] [PubMed]
- Dooley, S.; Ten Dijke, P. TGF-beta in progression of liver disease. Cell Tissue Res. 2012, 347, 245–256. [Google Scholar] [CrossRef] [PubMed]
- Moses, H.L.; Serra, R. Regulation of differentiation by TGF-beta. Curr. Opin. Genet. Dev. 1996, 6, 581–586. [Google Scholar] [CrossRef]
- Rovberts, A.B.; Seyedin, M. The Transforming Growth Factor-bs; Springer: Berlin, Germany, 1990. [Google Scholar]
- Bellam, N.; Pasche, B. Tgf-beta signaling alterations and colon cancer. Cancer Treat. Res. 2010, 155, 85–103. [Google Scholar] [PubMed]
- Matsuzaki, K. Smad phosphoisoform signaling specificity: The right place at the right time. Carcinogenesis 2011, 32, 1578–1588. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaki, K. Smad phosphoisoform signals in acute and chronic liver injury: Similarities and differences between epithelial and mesenchymal cells. Cell Tissue Res. 2012, 347, 225–243. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaki, K. Smad phospho-isoforms direct context-dependent TGF-beta signaling. Cytokine Growth Factor Rev. 2013, 24, 385–399. [Google Scholar] [CrossRef] [PubMed]
- Heldin, C.H.; Miyazono, K.; ten Dijke, P. TGF-beta signalling from cell membrane to nucleus through SMAD proteins. Nature 1997, 390, 465–471. [Google Scholar] [CrossRef] [PubMed]
- Derynck, R.; Miyazono, K. The TGF-b Signaling; Cold Spring Harbor Laboratory Press: New York, NY, USA, 2008. [Google Scholar]
- Guo, X.; Wang, X.F. Signaling cross-talk between TGF-beta/BMP and other pathways. Cell Res. 2009, 19, 71–88. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, H.; Abdollah, S.; Qiu, Y.; Cai, J.; Xu, Y.Y.; Grinnell, B.W.; Richardson, M.A.; Topper, J.N.; Gimbrone, M.A., Jr.; Wrana, J.L.; et al. The MAD-related protein Smad7 associates with the TGFbeta receptor and functions as an antagonist of TGFbeta signaling. Cell 1997, 89, 1165–1173. [Google Scholar] [CrossRef]
- Nakao, A.; Afrakhte, M.; Moren, A.; Nakayama, T.; Christian, J.L.; Heuchel, R.; Itoh, S.; Kawabata, M.; Heldin, N.E.; Heldin, C.H.; et al. Identification of Smad7, a TGFbeta-inducible antagonist of TGF-beta signaling. Nature 1997, 389, 631–635. [Google Scholar] [PubMed]
- Millet, C.; Yamashita, M.; Heller, M.; Yu, L.R.; Veenstra, T.D.; Zhang, Y.E. A negative feedback control of transforming growth factor-beta signaling by glycogen synthase kinase 3-mediated Smad3 linker phosphorylation at Ser-204. J. Biol. Chem. 2009, 284, 19808–19816. [Google Scholar] [CrossRef] [PubMed]
- Massagué, J. TGFβ signalling in context. Nat. Rev. Mol. Cell Biol. 2012, 13, 616–630. [Google Scholar] [CrossRef] [PubMed]
- Liu, F. Smad3 phosphorylation by cyclin-dependent kinases. Cytokine Growth Factor Rev. 2006, 17, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Wrighton, K.H.; Lin, X.; Feng, X.H. Phospho-control of TGF-beta superfamily signaling. Cell Res. 2009, 19, 8–20. [Google Scholar] [CrossRef] [PubMed]
- Kretzschmar, M.; Doody, J.; Timokhina, I.; Massague, J. A mechanism of repression of TGFbeta/Smad signaling by oncogenic Ras. Genes Dev. 1999, 13, 804–816. [Google Scholar] [CrossRef] [PubMed]
- Matsuura, I.; Denissova, N.G.; Wang, G.; He, D.; Long, J.; Liu, F. Cyclin-dependent kinases regulate the antiproliferative function of Smads. Nature 2004, 430, 226–231. [Google Scholar] [CrossRef] [PubMed]
- Sekimoto, G.; Matsuzaki, K.; Yoshida, K.; Mori, S.; Murata, M.; Seki, T.; Matsui, H.; Fujisawa, J.; Okazaki, K. Reversible Smad-dependent signaling between tumor suppression and oncogenesis. Cancer Res. 2007, 67, 5090–5096. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaki, K.; Kitano, C.; Murata, M.; Sekimoto, G.; Yoshida, K.; Uemura, Y.; Seki, T.; Taketani, S.; Fujisawa, J.; Okazaki, K. Smad2 and Smad3 phosphorylated at both linker and COOH-terminal regions transmit malignant TGF-beta signal in later stages of human colorectal cancer. Cancer Res. 2009, 69, 5321–5330. [Google Scholar] [CrossRef] [PubMed]
- Alarcon, C.; Zaromytidou, A.I.; Xi, Q.; Gao, S.; Yu, J.; Fujisawa, S.; Barlas, A.; Miller, A.N.; Manova-Todorova, K.; Macias, M.J.; et al. Nuclear CDKs drive Smad transcriptional activation and turnover in BMP and TGF-beta pathways. Cell 2009, 139, 757–769. [Google Scholar] [CrossRef] [PubMed]
- Mori, S.; Matsuzaki, K.; Yoshida, K.; Furukawa, F.; Tahashi, Y.; Yamagata, H.; Sekimoto, G.; Seki, T.; Matsui, H.; Nishizawa, M.; et al. TGF-beta and HGF transmit the signals through JNK-dependent Smad2/3 phosphorylation at the linker regions. Oncogene 2004, 23, 7416–7429. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaki, K.; Murata, M.; Yoshida, K.; Sekimoto, G.; Uemura, Y.; Sakaida, N.; Kaibori, M.; Kamiyama, Y.; Nishizawa, M.; Fujisawa, J.; et al. Chronic inflammation associated with hepatitis C virus infection perturbs hepatic transforming growth factor beta signaling, promoting cirrhosis and hepatocellular carcinoma. Hepatology 2007, 46, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.; Alarcon, C.; Sapkota, G.; Rahman, S.; Chen, P.Y.; Goerner, N.; Macias, M.J.; Erdjument-Bromage, H.; Tempst, P.; Massague, J. Ubiquitin ligase Nedd4L targets activated Smad2/3 to limit TGF-beta signaling. Mol. Cell. 2009, 36, 457–468. [Google Scholar] [CrossRef] [PubMed]
- Yumoto, K.; Thomas, P.S.; Lane, J.; Matsuzaki, K.; Inagaki, M.; Ninomiya-Tsuji, J.; Scott, G.J.; Ray, M.K.; Ishii, M.; Maxson, R.; et al. TGF-beta-activated kinase 1 (Tak1) mediates agonist-induced Smad activation and linker region phosphorylation in embryonic craniofacial neural crest-derived cells. J. Biol. Chem. 2013, 288, 13467–13480. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.H.; Lin, X.; Derynck, R. Smad2, Smad3 and Smad4 cooperate with Sp1 to induce p15(Ink4B) transcription in response to TGF-beta. EMBO J. 2000, 19, 5178–5193. [Google Scholar] [CrossRef] [PubMed]
- Pardali, K.; Kurisaki, A.; Moren, A.; ten Dijke, P.; Kardassis, D.; Moustakas, A. Role of Smad proteins and transcription factor Sp1 in p21(Waf1/Cip1) regulation by transforming growth factor-beta. J. Biol. Chem. 2000, 275, 29244–29256. [Google Scholar] [CrossRef] [PubMed]
- Frederick, J.P.; Liberati, N.T.; Waddell, D.S.; Shi, Y.; Wang, X.F. Transforming growth factor beta-mediated transcriptional repression of c-myc is dependent on direct binding of Smad3 to a novel repressive Smad binding element. Mol. Cell. Biol. 2004, 24, 2546–2559. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.A.; Zhang, G.M.; Feigenbaum, L.; Zhang, Y.E. Smad3 reduces susceptibility to hepatocarcinoma by sensitizing hepatocytes to apoptosis through downregulation of Bcl-2. Cancer Cell 2006, 9, 445–457. [Google Scholar] [CrossRef] [PubMed]
- Davis, R.J. Signal transduction by the JNK group of MAP kinases. Cell 2000, 103, 239–252. [Google Scholar] [CrossRef]
- Wagner, E.F.; Nebreda, A.R. Signal integration by JNK and p38 MAPK pathways in cancer development. Nat. Rev. Cancer 2009, 9, 537–549. [Google Scholar] [CrossRef] [PubMed]
- Wrana, J.L. Regulation of Smad activity. Cell 2000, 100, 189–192. [Google Scholar] [CrossRef]
- Derynck, R. SMAD proteins and mammalian anatomy. Nature 1998, 393, 737–739. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.H.; Derynck, R. Specificity and versatility in TGF-beta signaling through Smads. Annu. Rev. Cell Dev. Biol. 2005, 21, 659–693. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.; He, W.; Tulley, S.; Gupta, G.P.; Serganova, I.; Chen, C.R.; Manova-Todorova, K.; Blasberg, R.; Gerald, W.L.; Massague, J. Breast cancer bone metastasis mediated by the Smad tumor suppressor pathway. Proc. Natl. Acad. Sci. USA 2005, 102, 13909–13914. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, F.; Matsuzaki, K.; Mori, S.; Tahashi, Y.; Yoshida, K.; Sugano, Y.; Yamagata, H.; Matsushita, M.; Seki, T.; Inagaki, Y.; et al. p38 MAPK mediates fibrogenic signal through Smad3 phosphorylation in rat myofibroblasts. Hepatology 2003, 38, 879–889. [Google Scholar] [CrossRef] [PubMed]
- Moustakas, A.; Pardali, K.; Gaal, A.; Heldin, C.H. Mechanisms of TGF-beta signaling in regulation of cell growth and differentiation. Immunol. Lett. 2002, 82, 85–91. [Google Scholar] [CrossRef]
- Hui, L.; Zatloukal, K.; Scheuch, H.; Stepniak, E.; Wagner, E.F. Proliferation of human HCC cells and chemically induced mouse liver cancers requires JNK1-dependent p21 downregulation. J. Clin. Investig. 2008, 118, 3943–3953. [Google Scholar] [CrossRef] [PubMed]
- Murata, M.; Matsuzaki, K.; Yoshida, K.; Sekimoto, G.; Tahashi, Y.; Mori, S.; Uemura, Y.; Sakaida, N.; Fujisawa, J.; Seki, T.; et al. Hepatitis B virus X protein shifts human hepatic transforming growth factor (TGF)-beta signaling from tumor suppression to oncogenesis in early chronic hepatitis B. Hepatology 2009, 49, 1203–1217. [Google Scholar] [CrossRef] [PubMed]
- Glick, A.B.; Weinberg, W.C.; Wu, I.H.; Quan, W.; Yuspa, S.H. Transforming growth factor beta 1 suppresses genomic instability independent of a G1 arrest. p53, and Rb. Cancer Res. 1996, 56, 3645–3650. [Google Scholar] [PubMed]
- Tremain, R.; Marko, M.; Kinnimulki, V.; Ueno, H.; Bottinger, E.; Glick, A. Defects in TGF-beta signaling overcome senescence of mouse keratinocytes expressing v-Ha-ras. Oncogene 2000, 19, 1698–1709. [Google Scholar] [CrossRef] [PubMed]
- Anzano, M.A.; Roberts, A.B.; Smith, J.M.; Sporn, M.B.; De Larco, J.E. Sarcoma growth factor from conditioned medium of virally transformed cells is composed of both type alpha and type beta transforming growth factors. Proc. Natl. Acad. Sci. USA 1983, 80, 6264–6268. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.D.; DiChiara, M.R.; Anderson, K.R.; Gimbrone, M.A., Jr.; Topper, J.N. MEKK-1, a component of the stress (stress-activated protein kinase/c-Jun N-terminal kinase) pathway, can selectively activate Smad2-mediated transcriptional activation in endothelial cells. J. Biol. Chem. 1999, 274, 8797–8805. [Google Scholar] [CrossRef] [PubMed]
- Zelivianski, S.; Cooley, A.; Kall, R.; Jeruss, J.S. Cyclin-dependent kinase 4-mediated phosphorylation inhibits Smad3 activity in cyclin D-overexpressing breast cancer cells. Mol. Cancer Res. MCR 2010, 8, 1375–1387. [Google Scholar] [CrossRef] [PubMed]
- Cooley, A.; Zelivianski, S.; Jeruss, J.S. Impact of cyclin E overexpression on Smad3 activity in breast cancer cell lines. Cell Cycle 2010, 9, 4900–4907. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.; Geng, Y.; Sicinski, P. Specific protection against breast cancers by cyclin D1 ablation. Nature 2001, 411, 1017–1021. [Google Scholar] [CrossRef] [PubMed]
- Haas, K.; Staller, P.; Geisen, C.; Bartek, J.; Eilers, M.; Moroy, T. Mutual requirement of CDK4 and Myc in malignant transformation: Evidence for cyclin D1/CDK4 and p16INK4A as upstream regulators of Myc. Oncogene 1997, 15, 179–192. [Google Scholar] [CrossRef] [PubMed]
- Matsuura, I.; Chiang, K.N.; Lai, C.Y.; He, D.; Wang, G.; Ramkumar, R.; Uchida, T.; Ryo, A.; Lu, K.; Liu, F. Pin1 promotes transforming growth factor-beta-induced migration and invasion. J. Biol. Chem. 2010, 285, 1754–1764. [Google Scholar] [CrossRef] [PubMed]
- Hirschhorn, T.; Barizilay, L.; Smorodinsky, N.I.; Ehrlich, M. Differential regulation of Smad3 and of the type II transforming growth factor-beta receptor in mitosis: Implications for signaling. PLoS ONE 2012, 7, e43459. [Google Scholar] [CrossRef] [PubMed]
- Dooley, S.; Hamzavi, J.; Breitkopf, K.; Wiercinska, E.; Said, H.M.; Lorenzen, J.; Ten Dijke, P.; Gressner, A.M. Smad7 prevents activation of hepatic stellate cells and liver fibrosis in rats. Gastroenterology 2003, 125, 178–191. [Google Scholar] [CrossRef]
- Weng, H.L.; Liu, Y.; Chen, J.L.; Huang, T.; Xu, L.J.; Godoy, P.; Hu, J.H.; Zhou, C.; Stickel, F.; Marx, A.; et al. The etiology of liver damage imparts cytokines transforming growth factor beta1 or interleukin-13 as driving forces in fibrogenesis. Hepatology 2009, 50, 230–243. [Google Scholar] [CrossRef] [PubMed]
- Pinzani, M.; Macias-Barragan, J. Update on the pathophysiology of liver fibrosis. Expert Rev. Gastroenterol. Hepatol. 2010, 4, 459–472. [Google Scholar] [CrossRef] [PubMed]
- Bedossa, P.; Peltier, E.; Terris, B.; Franco, D.; Poynard, T. Transforming growth factor-beta 1 (TGF-beta 1) and TGF-beta 1 receptors in normal, cirrhotic, and neoplastic human livers. Hepatology 1995, 21, 760–766. [Google Scholar] [PubMed]
- Bissell, D.M.; Wang, S.S.; Jarnagin, W.R.; Roll, F.J. Cell-specific expression of transforming growth factor-beta in rat liver. Evidence for autocrine regulation of hepatocyte proliferation. J. Clin. Investig. 1995, 96, 447–455. [Google Scholar] [CrossRef] [PubMed]
- Ito, N.; Kawata, S.; Tamura, S.; Shirai, Y.; Kiso, S.; Tsushima, H.; Matsuzawa, Y. Positive correlation of plasma transforming growth factor-beta 1 levels with tumor vascularity in hepatocellular carcinoma. Cancer Lett. 1995, 89, 45–48. [Google Scholar] [CrossRef]
- Shirai, Y.; Kawata, S.; Tamura, S.; Ito, N.; Tsushima, H.; Takaishi, K.; Kiso, S.; Matsuzawa, Y. Plasma transforming growth factor-beta 1 in patients with hepatocellular carcinoma. Comparison with chronic liver diseases. Cancer 1994, 73, 2275–2279. [Google Scholar] [CrossRef]
- Kawate, S.; Takenoshita, S.; Ohwada, S.; Mogi, A.; Fukusato, T.; Makita, F.; Kuwano, H.; Morishita, Y. Mutation analysis of transforming growth factor beta type II receptor, Smad2, and Smad4 in hepatocellular carcinoma. Int. J. Oncol. 1999, 14, 127–131. [Google Scholar] [CrossRef] [PubMed]
- Yakicier, M.C.; Irmak, M.B.; Romano, A.; Kew, M.; Ozturk, M. Smad2 and Smad4 gene mutations in hepatocellular carcinoma. Oncogene 1999, 18, 4879–4883. [Google Scholar] [CrossRef] [PubMed]
- Kawate, S.; Ohwada, S.; Hamada, K.; Koyama, T.; Takenoshita, S.; Morishita, Y.; Hagiwara, K. Mutational analysis of the Smad6 and Smad7 genes in hepatocellular carcinoma. Int. J. Mol. Med. 2001, 8, 49–52. [Google Scholar] [CrossRef] [PubMed]
- Levy, L.; Hill, C.S. Alterations in components of the TGF-beta superfamily signaling pathways in human cancer. Cytokine Growth Factor Rev. 2006, 17, 41–58. [Google Scholar] [CrossRef] [PubMed]
- Furuta, K.; Misao, S.; Takahashi, K.; Tagaya, T.; Fukuzawa, Y.; Ishikawa, T.; Yoshioka, K.; Kakumu, S. Gene mutation of transforming growth factor beta1 type II receptor in hepatocellular carcinoma. Int. J. Cancer 1999, 81, 851–853. [Google Scholar] [CrossRef]
- Jiang, Z.; Jhunjhunwala, S.; Liu, J.; Haverty, P.M.; Kennemer, M.I.; Guan, Y.; Lee, W.; Carnevali, P.; Stinson, J.; Johnson, S.; et al. The effects of hepatitis B virus integration into the genomes of hepatocellular carcinoma patients. Genome Res. 2012, 22, 593–601. [Google Scholar] [CrossRef] [PubMed]
- Shafritz, D.A.; Shouval, D.; Sherman, H.I.; Hadziyannis, S.J.; Kew, M.C. Integration of hepatitis B virus DNA into the genome of liver cells in chronic liver disease and hepatocellular carcinoma. Studies in percutaneous liver biopsies and post-mortem tissue specimens. N. Engl. J. Med. 1981, 305, 1067–1073. [Google Scholar] [CrossRef] [PubMed]
- Bonilla Guerrero, R.; Roberts, L.R. The role of hepatitis B virus integrations in the pathogenesis of human hepatocellular carcinoma. J. Hepatol. 2005, 42, 760–777. [Google Scholar] [CrossRef] [PubMed]
- Terradillos, O.; Billet, O.; Renard, C.A.; Levy, R.; Molina, T.; Briand, P.; Buendia, M.A. The hepatitis B virus X gene potentiates c-Myc-induced liver oncogenesis in transgenic mice. Oncogene 1997, 14, 395–404. [Google Scholar] [CrossRef] [PubMed]
- Feitelson, M.A.; Lee, J. Hepatitis B virus integration, fragile sites, and hepatocarcinogenesis. Cancer Lett. 2007, 252, 157–170. [Google Scholar] [CrossRef] [PubMed]
- Feitelson, M.A. c-Myc overexpression in hepatocarcinogenesis. Hum. Pathol. 2004, 35, 1299–1302. [Google Scholar] [CrossRef] [PubMed]
- Koike, K.; Moriya, K.; Iino, S.; Yotsuyanagi, H.; Endo, Y.; Miyamura, T.; Kurokawa, K. High-level expression of hepatitis B virus HBx gene and hepatocarcinogenesis in transgenic mice. Hepatology 1994, 19, 810–819. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, J.; Aoki, H.; Kajino, K.; Moriyama, M.; Arakawa, Y.; Hino, O. Hepatitis C virus core protein activates the MAPK/ERK cascade synergistically with tumor promoter TPA, but not with epidermal growth factor or transforming growth factor alpha. Hepatology 2000, 32, 958–961. [Google Scholar] [CrossRef] [PubMed]
- Erhardt, A.; Hassan, M.; Heintges, T.; Haussinger, D. Hepatitis C virus core protein induces cell proliferation and activates ERK, JNK, and p38 MAP kinases together with the MAP kinase phosphatase MKP-1 in a HepG2 Tet-Off cell line. Virology 2002, 292, 272–284. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Nakao, H.; Tan, S.L.; Polyak, S.J.; Neddermann, P.; Vijaysri, S.; Jacobs, B.L.; Katze, M.G. Subversion of cell signaling pathways by hepatitis C virus nonstructural 5A protein via interaction with Grb2 and P85 phosphatidylinositol 3-kinase. J. Virol. 2002, 76, 9207–9217. [Google Scholar] [CrossRef] [PubMed]
- Qadri, I.; Iwahashi, M.; Capasso, J.M.; Hopken, M.W.; Flores, S.; Schaack, J.; Simon, F.R. Induced oxidative stress and activated expression of manganese superoxide dismutase during hepatitis C virus replication: Role of JNK, p38 MAPK and AP-1. Biochem. J. 2004, 378, 919–928. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.J.; Wang, L.; Ren, H.; Cao, J.; Li, L.; Ke, J.S.; Qi, Z.T. Hepatitis C virus E2 protein promotes human hepatoma cell proliferation through the MAPK/ERK signaling pathway via cellular receptors. Exp. Cell Res. 2005, 305, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Hassan, M.; Ghozlan, H.; Abdel-Kader, O. Activation of c-Jun NH2-terminal kinase (JNK) signaling pathway is essential for the stimulation of hepatitis C virus (HCV) non-structural protein 3 (NS3)-mediated cell growth. Virology 2005, 333, 324–336. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.H.; Hwang, S.B. Modulation of the transforming growth factor-beta signal transduction pathway by hepatitis C virus nonstructural 5A protein. J. Biol. Chem. 2006, 281, 7468–7478. [Google Scholar] [CrossRef] [PubMed]
- Hassan, M.; Selimovic, D.; Ghozlan, H.; Abdel-kader, O. Hepatitis C virus core protein triggers hepatic angiogenesis by a mechanism including multiple pathways. Hepatology 2009, 49, 1469–1482. [Google Scholar] [CrossRef] [PubMed]
- Park, K.J.; Choi, S.H.; Choi, D.H.; Park, J.M.; Yie, S.W.; Lee, S.Y.; Hwang, S.B. 1Hepatitis C virus NS5A protein modulates c-Jun N-terminal kinase through interaction with tumor necrosis factor receptor-associated factor 2. J. Biol. Chem. 2003, 278, 30711–30718. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.; Tsai, W.L.; Shao, R.X.; Wu, G.; Peng, L.F.; Barlow, L.L.; Chung, W.J.; Zhang, L.; Zhao, H.; Jang, J.Y.; et al. Hepatitis C virus regulates transforming growth factor beta1 production through the generation of reactive oxygen species in a nuclear factor kappaB-dependent manner. Gastroenterology 2010, 138, 2509–2518.e1. [Google Scholar] [CrossRef] [PubMed]
- Moriya, K.; Yotsuyanagi, H.; Shintani, Y.; Fujie, H.; Ishibashi, K.; Matsuura, Y.; Miyamura, T.; Koike, K. Hepatitis C virus core protein induces hepatic steatosis in transgenic mice. J. Gen. Virol. 1997, 78, 1527–1531. [Google Scholar] [CrossRef] [PubMed]
- Moriya, K.; Fujie, H.; Shintani, Y.; Yotsuyanagi, H.; Tsutsumi, T.; Ishibashi, K.; Matsuura, Y.; Kimura, S.; Miyamura, T.; Koike, K. The core protein of hepatitis C virus induces hepatocellular carcinoma in transgenic mice. Nat. Med. 1998, 4, 1065–1067. [Google Scholar] [CrossRef] [PubMed]
- Lerat, H.; Honda, M.; Beard, M.R.; Loesch, K.; Sun, J.; Yang, Y.; Okuda, M.; Gosert, R.; Xiao, S.Y.; Weinman, S.A.; et al. Steatosis and liver cancer in transgenic mice expressing the structural and nonstructural proteins of hepatitis C virus. Gastroenterology 2002, 122, 352–365. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.D.; Nakamura, I.; Roberts, L.R. The tumor microenvironment in hepatocellular carcinoma: Current status and therapeutic targets. Semin. Cancer Biol. 2011, 21, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Severi, T.; van Malenstein, H.; Verslype, C.; van Pelt, J.F. Tumor initiation and progression in hepatocellular carcinoma: Risk factors, classification, and therapeutic targets. Acta Pharmacol. Sin. 2010, 31, 1409–1420. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, K.; Matsuzaki, K.; Mori, S.; Tahashi, Y.; Yamagata, H.; Furukawa, F.; Seki, T.; Nishizawa, M.; Fujisawa, J.; Okazaki, K. Transforming growth factor-beta and platelet-derived growth factor signal via c-Jun N-terminal kinase-dependent Smad2/3 phosphorylation in rat hepatic stellate cells after acute liver injury. Am. J. Pathol. 2005, 166, 1029–1039. [Google Scholar] [CrossRef]
- Kluwe, J.; Pradere, J.P.; Gwak, G.Y.; Mencin, A.; De Minicis, S.; Osterreicher, C.H.; Colmenero, J.; Bataller, R.; Schwabe, R.F. Modulation of hepatic fibrosis by c-Jun-N-terminal kinase inhibition. Gastroenterology 2010, 138, 347–359. [Google Scholar] [CrossRef] [PubMed]
- Dzieran, J.; Fabian, J.; Feng, T.; Coulouarn, C.; Ilkavets, I.; Kyselova, A.; Breuhahn, K.; Dooley, S.; Meindl-Beinker, N.M. Comparative analysis of TGF-beta/Smad signaling dependent cytostasis in human hepatocellular carcinoma cell lines. PLoS ONE 2013, 8, e72252. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, I.M.; McHutchison, J.G.; Dusheiko, G.; Di Bisceglie, A.M.; Reddy, K.R.; Bzowej, N.H.; Marcellin, P.; Muir, A.J.; Ferenci, P.; Flisiak, R.; et al. Telaprevir for previously untreated chronic hepatitis C virus infection. N. Engl. J. Med. 2011, 364, 2405–2416. [Google Scholar] [CrossRef] [PubMed]
- Kumada, H.; Suzuki, Y.; Ikeda, K.; Toyota, J.; Karino, Y.; Chayama, K.; Kawakami, Y.; Ido, A.; Yamamoto, K.; Takaguchi, K.; et al. Daclatasvir plus asunaprevir for chronic HCV genotype 1b infection. Hepatology 2014, 59, 2083–2091. [Google Scholar] [CrossRef] [PubMed]
- Lawitz, E.; Poordad, F.F.; Pang, P.S.; Hyland, R.H.; Ding, X.; Mo, H.; Symonds, W.T.; McHutchison, J.G.; Membreno, F.E. Sofosbuvir and ledipasvir fixed-dose combination with and without ribavirin in treatment-naive and previously treated patients with genotype 1 hepatitis C virus infection (LONESTAR): An open-label, randomised, phase 2 trial. Lancet 2014, 383, 515–523. [Google Scholar] [CrossRef]
- Walker, D.R.; Pedrosa, M.C.; Manthena, S.R.; Patel, N.; Marx, S.E. Early View of the Effectiveness of New Direct-Acting Antiviral (DAA) Regimens in Patients with Hepatitis C Virus (HCV). Adv. Ther. 2015, 32, 1117–1127. [Google Scholar] [CrossRef] [PubMed]
- Puoti, M.; Foster, G.R.; Wang, S.; Mutimer, D.; Gane, E.; Moreno, C.; Chang, T.T.; Lee, S.S.; Marinho, R.; DuFour, J.F.; et al. High SVR12 with 8-week and 12-week Glecaprevir/Pibrentasvir: Integrated Analysis of HCV Genotype 1-6 Patients Without Cirrhosis. J. Hepatol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Dienstag, J.L.; Schiff, E.R.; Wright, T.L.; Perrillo, R.P.; Hann, H.W.; Goodman, Z.; Crowther, L.; Condreay, L.D.; Woessner, M.; Rubin, M.; et al. Lamivudine as initial treatment for chronic hepatitis B in the United States. N. Engl. J. Med. 1999, 341, 1256–1263. [Google Scholar] [CrossRef] [PubMed]
- Marcellin, P.; Chang, T.T.; Lim, S.G.; Tong, M.J.; Sievert, W.; Shiffman, M.L.; Jeffers, L.; Goodman, Z.; Wulfsohn, M.S.; Xiong, S.; et al. Adefovir dipivoxil for the treatment of hepatitis B e antigen-positive chronic hepatitis B. N. Engl. J. Med. 2003, 348, 808–816. [Google Scholar] [CrossRef] [PubMed]
- Chang, T.T.; Gish, R.G.; de Man, R.; Gadano, A.; Sollano, J.; Chao, Y.C.; Lok, A.S.; Han, K.H.; Goodman, Z.; Zhu, J.; et al. A comparison of entecavir and lamivudine for HBeAg-positive chronic hepatitis B. N. Engl. J. Med. 2006, 354, 1001–1010. [Google Scholar] [CrossRef] [PubMed]
- Lai, C.L.; Gane, E.; Liaw, Y.F.; Hsu, C.W.; Thongsawat, S.; Wang, Y.; Chen, Y.; Heathcote, E.J.; Rasenack, J.; Bzowej, N.; et al. Telbivudine versus lamivudine in patients with chronic hepatitis B. N. Engl. J. Med. 2007, 357, 2576–2588. [Google Scholar] [CrossRef] [PubMed]
- Lou, L. Advances in Nucleotide Antiviral Development from Scientific Discovery to Clinical Applications: Tenofovir Disoproxil Fumarate for Hepatitis B. J. Clin. Transl. Hepatol. 2013, 1, 33–38. [Google Scholar] [PubMed]
- Shiratori, Y.; Imazeki, F.; Moriyama, M.; Yano, M.; Arakawa, Y.; Yokosuka, O.; Kuroki, T.; Nishiguchi, S.; Sata, M.; Yamada, G.; et al. Histologic improvement of fibrosis in patients with hepatitis C who have sustained response to interferon therapy. Ann. Intern. Med. 2000, 132, 517–524. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, Y.; Kumada, H.; Ikeda, K.; Chayama, K.; Arase, Y.; Saitoh, S.; Tsubota, A.; Kobayashi, M.; Koike, M.; Ogawa, N.; et al. Histological changes in liver biopsies after one year of lamivudine treatment in patients with chronic hepatitis B infection. J. Hepatol. 1999, 30, 743–748. [Google Scholar] [CrossRef]
- Deng, Y.R.; Yoshida, K.; Jin, Q.; Murata, M.; Yamaguchi, T.; Tsuneyama, K.; Moritoki, Y.; Niu, J.; Matsuzaki, K.; Lian, Z.X. Reversible phospho-Smad3 signaling between tumor-suppression and fibro-carcinogenesis in chronic hepatitis B infection. Clin. Exp. Immunol. 2014, 176, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, T.; Matsuzaki, K.; Inokuchi, R.; Kawamura, R.; Yoshida, K.; Murata, M.; Fujisawa, J.; Fukushima, N.; Sata, M.; Kage, M.; et al. Phosphorylated Smad2 and Smad3 signaling: Shifting between tumor suppression and fibro-carcinogenesis in chronic hepatitis C. Hepatol. Res. 2013, 43, 1327–1342. [Google Scholar] [CrossRef] [PubMed]
- Morgan, T.R.; Ghany, M.G.; Kim, H.Y.; Snow, K.K.; Shiffman, M.L.; De Santo, J.L.; Lee, W.M.; Di Bisceglie, A.M.; Bonkovsky, H.L.; Dienstag, J.L.; et al. Outcome of sustained virological responders with histologically advanced chronic hepatitis C. Hepatology 2010, 52, 833–844. [Google Scholar] [CrossRef] [PubMed]
- Kodama, Y.; Kisseleva, T.; Iwaisako, K.; Miura, K.; Taura, K.; De Minicis, S.; Osterreicher, C.H.; Schnabl, B.; Seki, E.; Brenner, D.A. c-Jun N-terminal kinase-1 from hematopoietic cells mediates progression from hepatic steatosis to steatohepatitis and fibrosis in mice. Gastroenterology 2009, 137, 1467–1477.e5. [Google Scholar] [CrossRef] [PubMed]
- Felsher, D.W. Reversibility of oncogene-induced cancer. Curr. Opin. Genet. Dev. 2004, 14, 37–42. [Google Scholar] [CrossRef] [PubMed]
- Nagata, H.; Hatano, E.; Tada, M.; Murata, M.; Kitamura, K.; Asechi, H.; Narita, M.; Yanagida, A.; Tamaki, N.; Yagi, S.; et al. Inhibition of c-Jun NH2-terminal kinase switches Smad3 signaling from oncogenesis to tumor- suppression in rat hepatocellular carcinoma. Hepatology 2009, 49, 1944–1953. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Neilson, E.G. Epithelial-mesenchymal transition and its implications for fibrosis. J. Clin. Investig. 2003, 112, 1776–1784. [Google Scholar] [CrossRef] [PubMed]
- Greenburg, G.; Hay, E.D. Epithelia suspended in collagen gels can lose polarity and express characteristics of migrating mesenchymal cells. J. Cell Biol. 1982, 95, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [PubMed]
- Zavadil, J.; Bottinger, E.P. TGF-beta and epithelial-to-mesenchymal transitions. Oncogene 2005, 24, 5764–5774. [Google Scholar] [CrossRef] [PubMed]
- Cicchini, C.; Filippini, D.; Coen, S.; Marchetti, A.; Cavallari, C.; Laudadio, I.; Spagnoli, F.M.; Alonzi, T.; Tripodi, M. Snail controls differentiation of hepatocytes by repressing HNF4alpha expression. J. Cell. Physiol. 2006, 209, 230–238. [Google Scholar] [CrossRef] [PubMed]
- Kaimori, A.; Potter, J.; Kaimori, J.Y.; Wang, C.; Mezey, E.; Koteish, A. Transforming growth factor-beta1 induces an epithelial-to-mesenchymal transition state in mouse hepatocytes in vitro. J. Biol. Chem. 2007, 282, 22089–22101. [Google Scholar] [CrossRef] [PubMed]
- Franco, D.L.; Mainez, J.; Vega, S.; Sancho, P.; Murillo, M.M.; de Frutos, C.A.; Del Castillo, G.; Lopez-Blau, C.; Fabregat, I.; Nieto, M.A. Snail1 suppresses TGF-beta-induced apoptosis and is sufficient to trigger EMT in hepatocytes. J. Cell Sci. 2010, 123, 3467–3477. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Eischeid, A.N.; Chen, X.M. Col1A1 production and apoptotic resistance in TGF-beta1-induced epithelial-to-mesenchymal transition-like phenotype of 603B cells. PLoS ONE 2012, 7, e51371. [Google Scholar]
- Brenner, D.A.; Waterboer, T.; Choi, S.K.; Lindquist, J.N.; Stefanovic, B.; Burchardt, E.; Yamauchi, M.; Gillan, A.; Rippe, R.A. New aspects of hepatic fibrosis. J. Hepatol. 2000, 32, 32–38. [Google Scholar] [CrossRef]
- Reimann, T.; Hempel, U.; Krautwald, S.; Axmann, A.; Scheibe, R.; Seidel, D.; Wenzel, K.W. Transforming growth factor-beta1 induces activation of Ras, Raf-1, MEK and MAPK in rat hepatic stellate cells. FEBS Lett. 1997, 403, 57–60. [Google Scholar] [CrossRef]
- Pinzani, M.; Gesualdo, L.; Sabbah, G.M.; Abboud, H.E. Effects of platelet-derived growth factor and other polypeptide mitogens on DNA synthesis and growth of cultured rat liver fat-storing cells. J. Clin. Investig. 1989, 84, 1786–1793. [Google Scholar] [CrossRef] [PubMed]
- Rockey, D.C.; Fouassier, L.; Chung, J.J.; Carayon, A.; Vallee, P.; Rey, C.; Housset, C. Cellular localization of endothelin-1 and increased production in liver injury in the rat: Potential for autocrine and paracrine effects on stellate cells. Hepatology 1998, 27, 472–480. [Google Scholar] [CrossRef] [PubMed]
- Marra, F.; Arrighi, M.C.; Fazi, M.; Caligiuri, A.; Pinzani, M.; Romanelli, R.G.; Efsen, E.; Laffi, G.; Gentilini, P. Extracellular signal-regulated kinase activation differentially regulates platelet-derived growth factor’s actions in hepatic stellate cells, and is induced by in vivo liver injury in the rat. Hepatology 1999, 30, 951–958. [Google Scholar] [CrossRef] [PubMed]
- Tahashi, Y.; Matsuzaki, K.; Date, M.; Yoshida, K.; Furukawa, F.; Sugano, Y.; Matsushita, M.; Himeno, Y.; Inagaki, Y.; Inoue, K. Differential regulation of TGF-beta signal in hepatic stellate cells between acute and chronic rat liver injury. Hepatology 2002, 35, 49–61. [Google Scholar] [CrossRef] [PubMed]
- Nouchi, T.; Tanaka, Y.; Tsukada, T.; Sato, C.; Marumo, F. Appearance of alpha-smooth-muscle-actin-positive cells in hepatic fibrosis. Liver 1991, 11, 100–105. [Google Scholar] [CrossRef] [PubMed]
- Schmitt-Graff, A.; Kruger, S.; Bochard, F.; Gabbiani, G.; Denk, H. Modulation of alpha smooth muscle actin and desmin expression in perisinusoidal cells of normal and diseased human livers. Am. J. Pathol. 1991, 138, 1233–1242. [Google Scholar] [PubMed]
- Guarino, M.; Rubino, B.; Ballabio, G. The role of epithelial-mesenchymal transition in cancer pathology. Pathology 2007, 39, 305–318. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.P.; Deng, J.; Xia, W.; Xu, J.; Li, Y.M.; Gunduz, M.; Hung, M.C. Dual regulation of Snail by GSK-3beta-mediated phosphorylation in control of epithelial-mesenchymal transition. Nat. Cell Biol. 2004, 6, 931–940. [Google Scholar] [CrossRef] [PubMed]
- Larue, L.; Bellacosa, A. Epithelial-mesenchymal transition in development and cancer: Role of phosphatidylinositol 3′ kinase/AKT pathways. Oncogene 2005, 24, 7443–7454. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.; Ding, J.; Chen, C.; Sun, W.; Ning, B.F.; Wen, W.; Huang, L.; Han, T.; Yang, W.; Wang, C.; et al. Hepatic transforming growth factor beta gives rise to tumor-initiating cells and promotes liver cancer development. Hepatology 2012, 56, 2255–2267. [Google Scholar] [CrossRef] [PubMed]
- Fan, Q.M.; Jing, Y.Y.; Yu, G.F.; Kou, X.R.; Ye, F.; Gao, L.; Li, R.; Zhao, Q.D.; Yang, Y.; Lu, Z.H.; et al. Tumor-associated macrophages promote cancer stem cell-like properties via transforming growth factor-beta1-induced epithelial-mesenchymal transition in hepatocellular carcinoma. Cancer Lett. 2014, 352, 160–168. [Google Scholar] [CrossRef] [PubMed]
- Fernando, J.; Malfettone, A.; Cepeda, E.B.; Vilarrasa-Blasi, R.; Bertran, E.; Raimondi, G.; Fabra, A.; Alvarez-Barrientos, A.; Fernandez-Salguero, P.; Fernandez-Rodriguez, C.M.; et al. A mesenchymal-like phenotype and expression of CD44 predict lack of apoptotic response to sorafenib in liver tumor cells. Int. J. Cancer 2015, 136, E161–E172. [Google Scholar] [CrossRef] [PubMed]
- Vincent, T.; Neve, E.P.; Johnson, J.R.; Kukalev, A.; Rojo, F.; Albanell, J.; Pietras, K.; Virtanen, I.; Philipson, L.; Leopold, P.L.; et al. A SNAIL1-SMAD3/4 transcriptional repressor complex promotes TGF-beta mediated epithelial-mesenchymal transition. Nat. Cell Biol. 2009, 11, 943–950. [Google Scholar] [CrossRef] [PubMed]
- Xue, J.; Lin, X.; Chiu, W.T.; Chen, Y.H.; Yu, G.; Liu, M.; Feng, X.H.; Sawaya, R.; Medema, R.H.; Hung, M.C.; et al. Sustained activation of SMAD3/SMAD4 by FOXM1 promotes TGF-beta-dependent cancer metastasis. J. Clin. Investig. 2014, 124, 564–579. [Google Scholar] [CrossRef] [PubMed]
- Lindemann, R.K.; Nordheim, A.; Dittmer, J. Interfering with TGFbeta-induced Smad3 nuclear accumulation differentially affects TGFbeta-dependent gene expression. Mol. Cancer 2003, 2, 20. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.N.; Ding, W.Q.; Guo, X.J.; Yuan, X.W.; Wang, D.M.; Song, J.G. Epigenetic regulation of Smad2 and Smad3 by profilin-2 promotes lung cancer growth and metastasis. Nat. Commun. 2015, 6, 8230. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Liu, Q.; Li, Z.; Zhang, R.; Jia, C.; Yang, Z.; Zhao, H.; Ya, S.; Mao, R.; Ailijiang, T.; et al. GP73 promotes epithelial-mesenchymal transition and invasion partly by activating TGF-beta1/Smad2 signaling in hepatocellular carcinoma. Carcinogenesis 2018. [Google Scholar] [CrossRef] [PubMed]
- Dayti, P.; Rezaei, H.B.; Sharifat, N.; Kamoto, D.; Little, P.J. G protein coupled receptors can transduce signals through carboxy terminal and linker region phosphorylation of Smad transcription factors. Life Sci. 2018, 199, 10–15. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Shen, M.; Lu, P.; Li, X.; Zhu, S.; Yue, S. NEDD9 may regulate hepatocellular carcinoma cell metastasis by promoting epithelial-mesenchymal-transition and stemness via repressing Smad7. Oncotarget 2017, 8, 1714–1724. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.H.; Chen, L.; Liang, H.F.; Li, G.Z.; Zhang, B.X.; Chen, X.P. Smad3 Sensitizes Hepatocelluar Carcinoma Cells to Cisplatin by Repressing Phosphorylation of AKT. Int. J. Mol. Sci. 2016, 17, 610. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.N.; Zeng, Q.; Wang, H.Y.; Zhang, B.; Li, S.T.; Nan, X.; Cao, N.; Fu, C.J.; Yan, X.L.; Jia, Y.L.; et al. MicroRNA-125b attenuates epithelial-mesenchymal transitions and targets stem-like liver cancer cells through small mothers against decapentaplegic 2 and 4. Hepatology 2015, 62, 801–815. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Li, S.; Wu, Y.; Gao, F. miRNA-708 functions as a tumour suppressor in hepatocellular carcinoma by targeting SMAD3. Oncology Lett. 2017, 14, 2552–2558. [Google Scholar] [CrossRef] [PubMed]
- Kan, H.; Guo, W.; Huang, Y.; Liu, D. MicroRNA-520g induces epithelial-mesenchymal transition and promotes metastasis of hepatocellular carcinoma by targeting SMAD7. FEBS Lett. 2015, 589, 102–109. [Google Scholar] [CrossRef] [PubMed]
- Talati, N.; Kamoto, D.; Piva, T.J.; Little, P.J.; Osman, N. Thrombin promotes PAI-1 expression and migration in keratinocytes via ERK dependent Smad linker region phosphorylation. Cell Signal. 2018, 47, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Rostam, M.A.; Shajimoon, A.; Kamoto, D.; Mitra, P.; Piva, T.J.; Getachew, R.; Cao, Y.; Zheng, W.; Osman, N.; Little, P.J. Flavopiridol inhibits TGF-β-stimulated biglycan synthesis by blocking linker region phosphorylation and nuclear translocation of Smad2. J. Pharmacol. Exp. Ther. 2018, 365, 156–164. [Google Scholar] [CrossRef] [PubMed]


© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yoshida, K.; Matsuzaki, K.; Murata, M.; Yamaguchi, T.; Suwa, K.; Okazaki, K. Clinico-Pathological Importance of TGF-β/Phospho-Smad Signaling during Human Hepatic Fibrocarcinogenesis. Cancers 2018, 10, 183. https://doi.org/10.3390/cancers10060183
Yoshida K, Matsuzaki K, Murata M, Yamaguchi T, Suwa K, Okazaki K. Clinico-Pathological Importance of TGF-β/Phospho-Smad Signaling during Human Hepatic Fibrocarcinogenesis. Cancers. 2018; 10(6):183. https://doi.org/10.3390/cancers10060183
Chicago/Turabian StyleYoshida, Katsunori, Koichi Matsuzaki, Miki Murata, Takashi Yamaguchi, Kanehiko Suwa, and Kazuichi Okazaki. 2018. "Clinico-Pathological Importance of TGF-β/Phospho-Smad Signaling during Human Hepatic Fibrocarcinogenesis" Cancers 10, no. 6: 183. https://doi.org/10.3390/cancers10060183
APA StyleYoshida, K., Matsuzaki, K., Murata, M., Yamaguchi, T., Suwa, K., & Okazaki, K. (2018). Clinico-Pathological Importance of TGF-β/Phospho-Smad Signaling during Human Hepatic Fibrocarcinogenesis. Cancers, 10(6), 183. https://doi.org/10.3390/cancers10060183