RIPK2: New Elements in Modulating Inflammatory Breast Cancer Pathogenesis

 , and
, and
Abstract
1. Introduction
2. Results
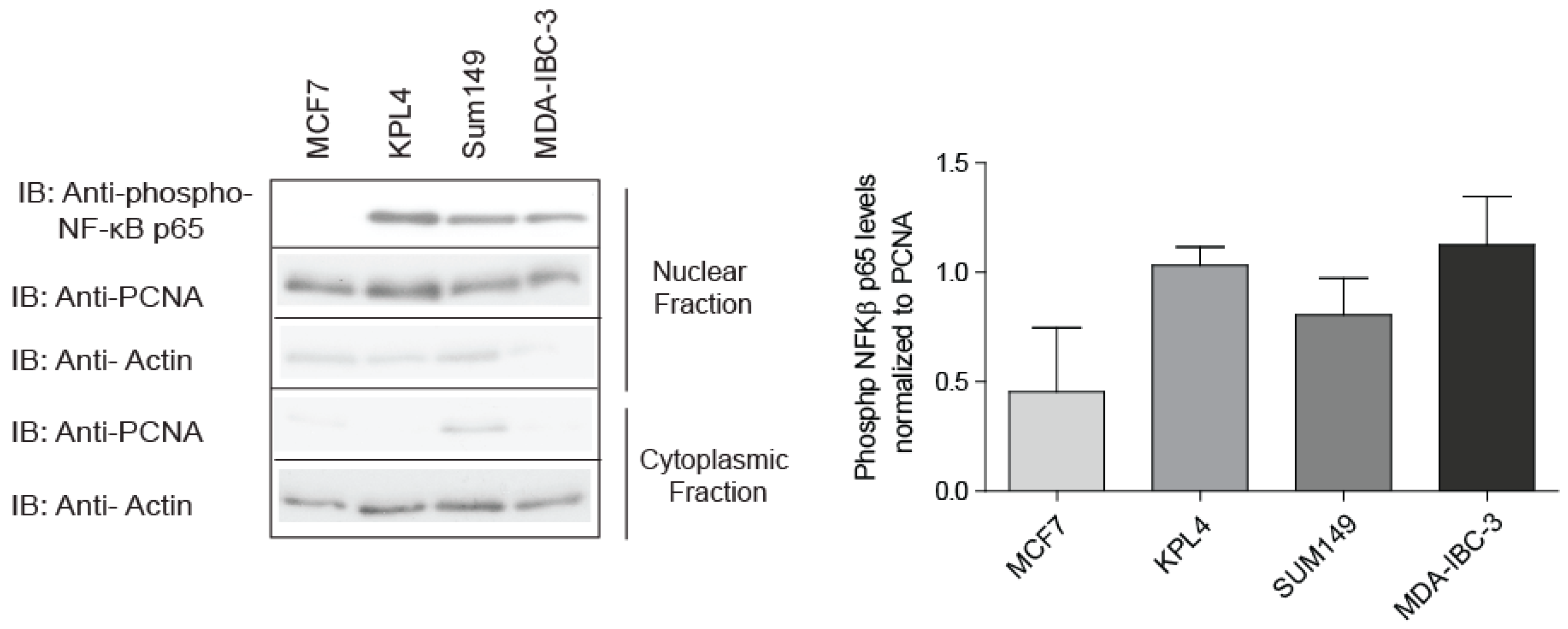
2.1. IBC Cell Lines Exhibit High NF-κB Activity
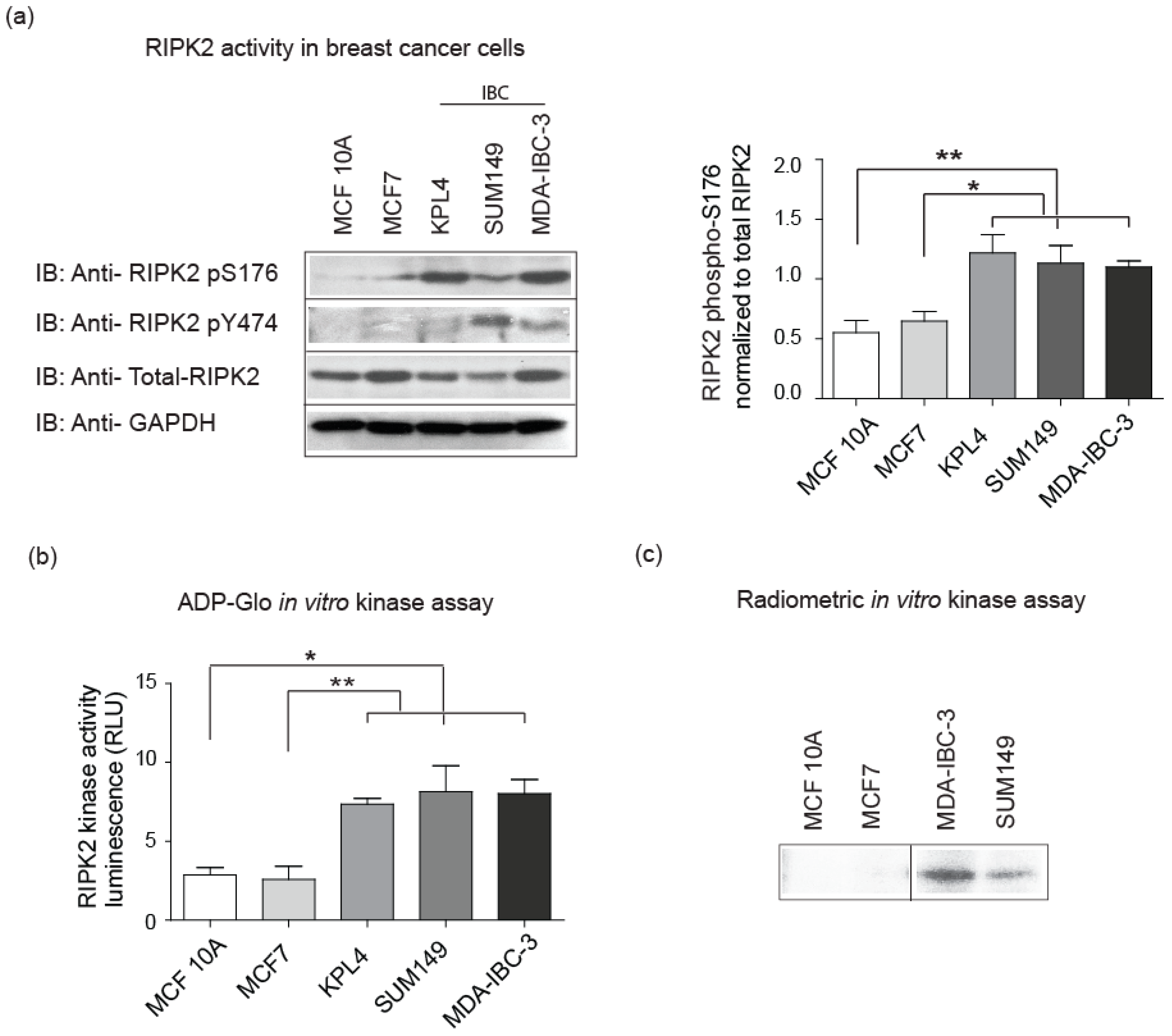
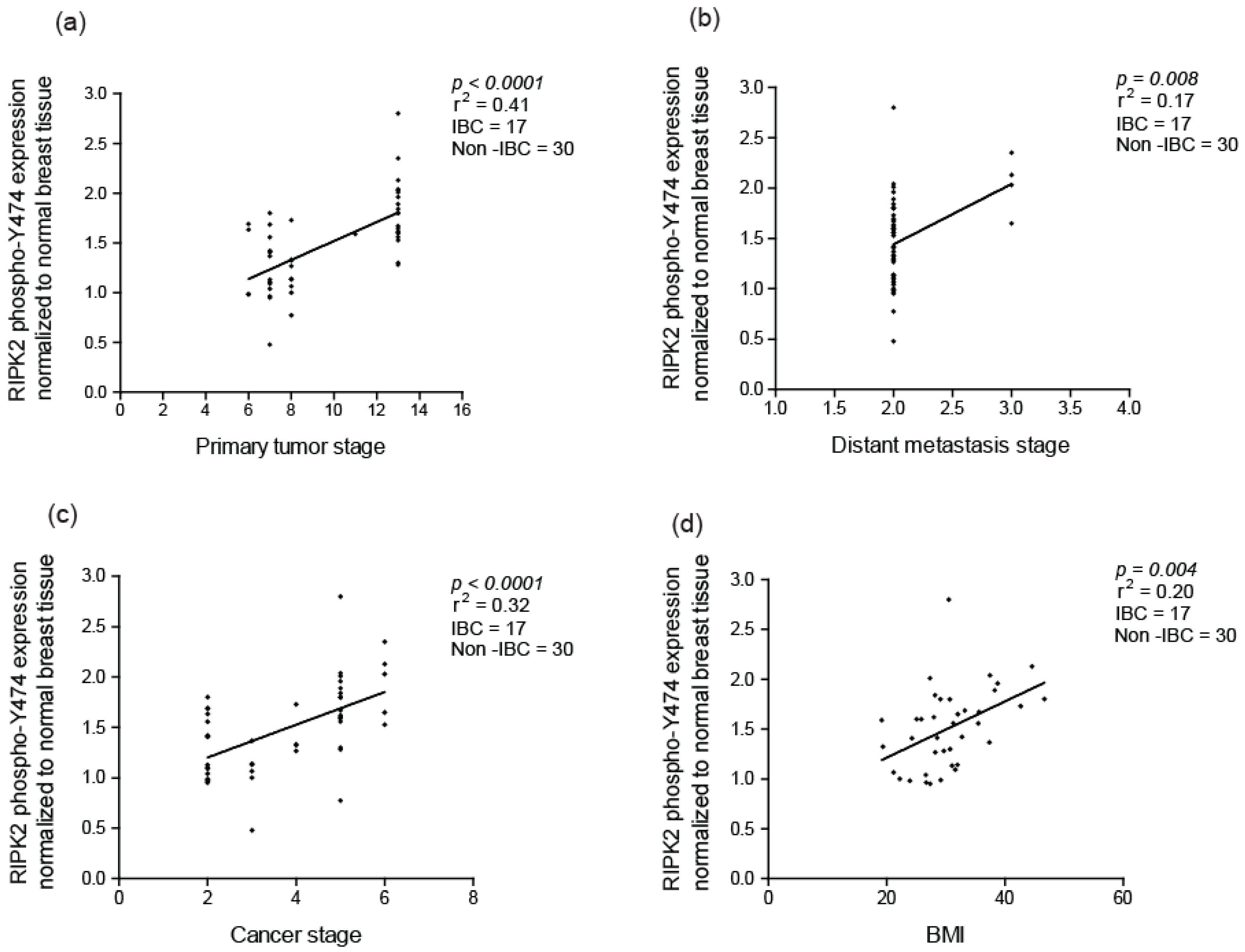
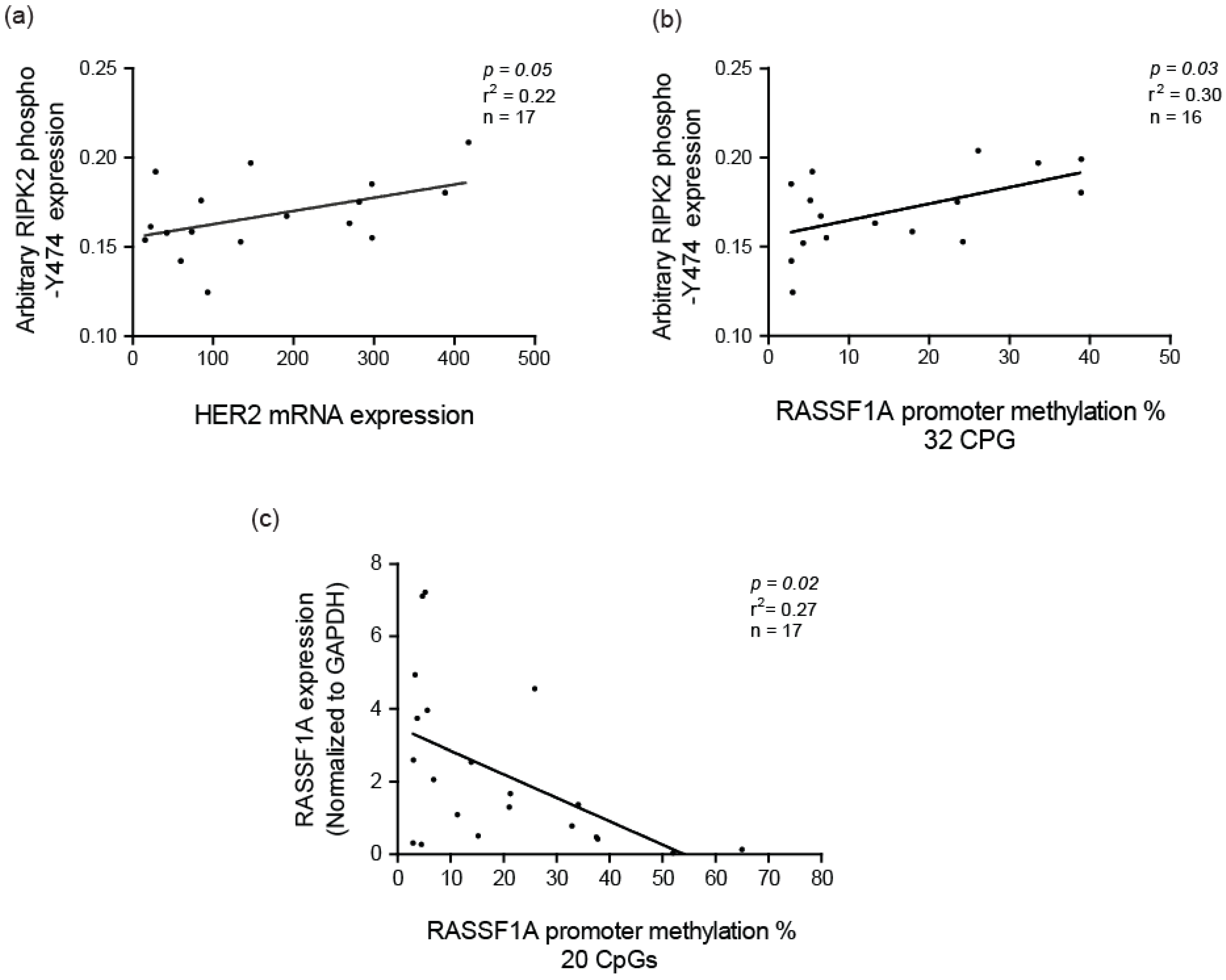
2.2. Elevated RIPK2 Activity Level in IBC Cell Lines and Patient Tissues
2.3. Neoadjuvant Chemotherapy Does Not Inhibit RIPK2 Activity
2.4. RIPK2 Activity as an Independent Prognostic Marker
3. Discussion
4. Materials and Methods
4.1. Immunoblotting and Antibodies
4.2. Breast Cancer Samples
4.3. Cell Lines
4.4. RIPK2 In Vitro Kinase Assay
4.5. RIPK2 ADP-Glo Kinase Assay (Promega)
4.6. Immunohistochemical and Immunoblot Staining and Evaluation
4.7. Nuclear and Cytoplasmic Extracts
4.8. Statistical Analysis
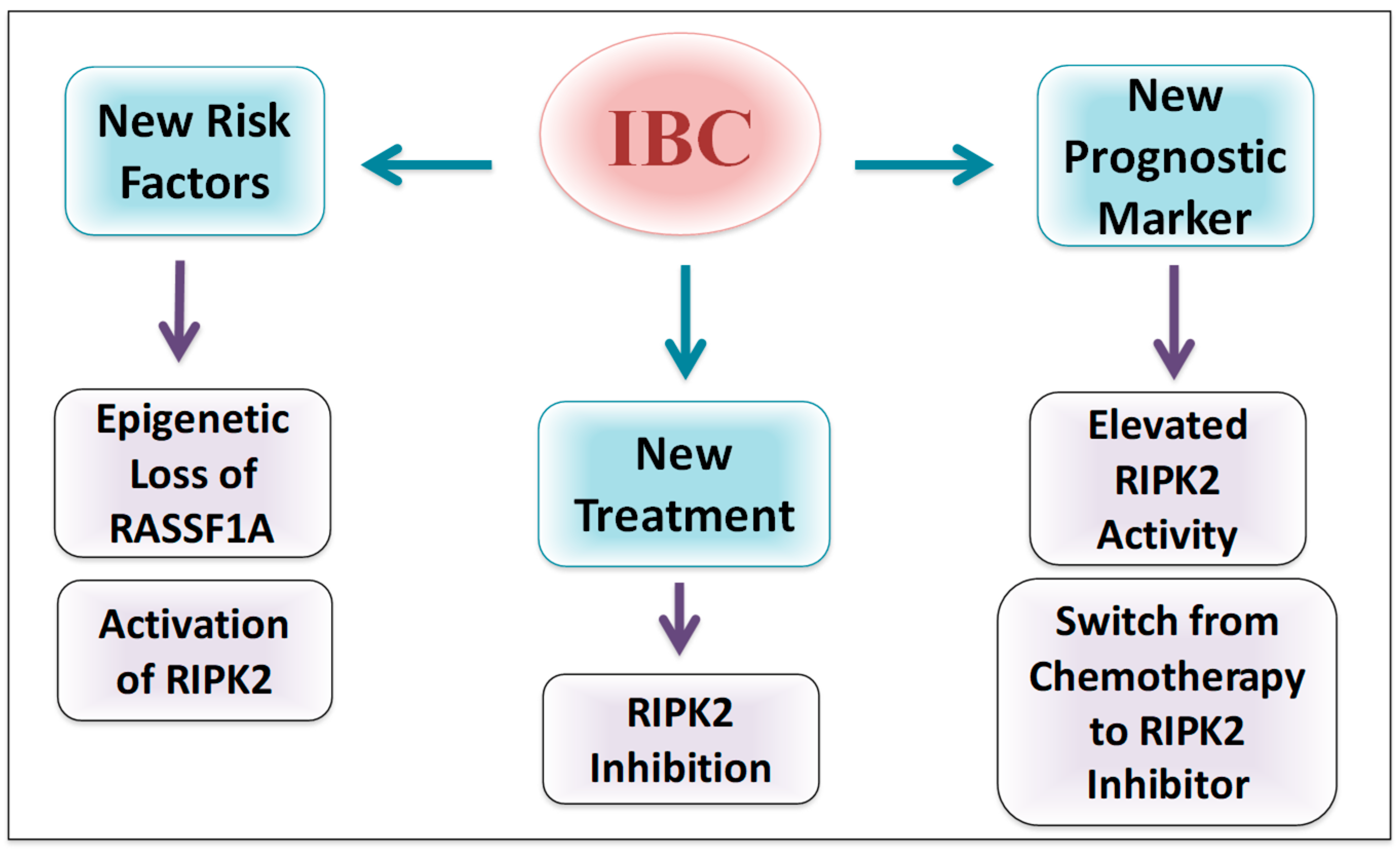
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Singletary, S.E.; Allred, C.; Ashley, P.; Bassett, L.W.; Berry, D.; Bland, K.I.; Borgen, P.I.; Clark, C.G.; Edge, S.B.; Hayes, D.F.; et al. Revision of the American Joint Committee on Cancer staging system for breast cancer. J. Clin. Oncol. 2002, 20, 3628–3636. [Google Scholar] [CrossRef] [PubMed]
- Anderson, W.F.; Schairer, C.; Chen, B.E.; Hance, K.W.; Levine, P.H. Epidemiology of inflammatory breast cancer (IBC). Breast Dis. 2005, 22, 9–23. [Google Scholar] [CrossRef] [PubMed]
- Hance, K.W.; Anderson, W.F.; Devesa, S.S.; Levine, P.H. Trends in inflammatory breast carcinoma incidence and survival: The surveillance, epidemiology, and end results program at the National Cancer Institute. Breast Cancer Res. Treat. 2003, 82, S148. [Google Scholar] [CrossRef] [PubMed]
- Jaiyesimi, I.A.; Buzdar, A.U.; Hortobagyi, G. Inflammatory breast cancer: A review. J. Clin. Oncol. 1992, 10, 1014–1024. [Google Scholar] [CrossRef] [PubMed]
- Kelly, J.C.; Ueno, N.T. Inflammatory Breast Cancer: What You Should Know. Available online: https://www.medscape.com/viewarticle/778334 (accessed on 4 June 2018).
- Yamauchi, H.; Woodward, W.A.; Valero, V.; Alvarez, R.H.; Lucci, A.; Buchholz, T.A.; Iwamoto, T.; Krishnamurthy, S.; Yang, W.; Reuben, J.M.; et al. Inflammatory Breast Cancer: What We Know and What We Need to Learn. Oncologist 2012, 17, 891–899. [Google Scholar] [CrossRef] [PubMed]
- Sobin, L.H.; Compton, C.C. TNM seventh edition: What’s new, what’s changed: Communication from the International Union Against Cancer and the American Joint Committee on Cancer. Cancer 2010, 116, 5336–5339. [Google Scholar] [CrossRef] [PubMed]
- Edge, S.B.; Compton, C.C. The American Joint Committee on Cancer: The 7th edition of the AJCC cancer staging manual and the future of TNM. Ann. Surg. Oncol. 2010, 17, 1471–1474. [Google Scholar] [CrossRef] [PubMed]
- Ueno, N.T.; Buzdar, A.U.; Singletary, S.E.; Ames, F.C.; McNeese, M.D.; Holmes, F.A.; Theriault, R.L.; Strom, E.A.; Wasaff, B.J.; Asmar, L.; et al. Combined-modality treatment of inflammatory breast carcinoma: Twenty years of experience at M.D. Anderson Cancer Center. Cancer Chemother. Pharmacol. 1997, 40, 321–329. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.T.; Le-Petross, H.T.; Macapinlac, H.; Carkaci, S.; Gonzalez-Angulo, A.M.; Dawood, S.; Resetkova, E.; Hortobagyi, G.N.; Cristofanilli, M. Inflammatory breast cancer: PET/CT, MRI, mammography, and sonography findings. Breast Cancer Res. Treat. 2008, 109, 417–426. [Google Scholar] [CrossRef] [PubMed]
- Radunsky, G.S.; van Golen, K.L. The current understanding of the molecular determinants of inflammatory breast cancer metastasis. Clin. Exp. Metastasis 2005, 22, 615–620. [Google Scholar] [CrossRef] [PubMed]
- Lehman, H.L.; Van Laere, S.J.; van Golen, C.M.; Vermeulen, P.B.; Dirix, L.Y.; van Golen, K.L. Regulation of inflammatory breast cancer cell invasion through Akt1/PKB alpha phosphorylation of RhoC GTPase. Mol. Cancer Res. 2012, 10, 1306–1318. [Google Scholar] [CrossRef] [PubMed]
- Overmoyer, B.; Fu, P.; Hoppel, C.; Radivoyevitch, T.; Shenk, R.; Persons, M.; Silverman, P.; Robertson, K.; Ziats, N.P.; Wasman, J.K.; et al. Inflammatory breast cancer as a model disease to study tumor angiogenesis: Results of a phase IB trial of combination SU5416 and doxorubicin. Clin. Cancer Res. 2007, 13, 5862–5868. [Google Scholar] [CrossRef] [PubMed]
- Van Laere, S.J.; Van der Auwera, I.; Van den Eynden, G.G.; Vandenberghe, M.; Benoy, I.H.; Elst, H.J.; Van Dam, P.; Van Marck, E.A.; Dirix, L.Y.; Vermeulen, P.B. Identification of cell-of-origin breast tumour subtypes in inflammatory breast cancer by gene expression profiling. Breast Cancer Res. Treat. 2006, 95, 243–255. [Google Scholar] [CrossRef] [PubMed]
- Korniluk, A.; Koper, O.; Kemona, H.; Dymicka-Piekarska, V. From inflammation to cancer. Ir. J. Med. Sci. 2017, 186, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Colotta, F.; Allavena, P.; Sica, A.; Garlanda, C.; Mantovani, A. Cancer-related inflammation, the seventh hallmark of cancer: Links to genetic instability. Carcinogenesis 2009, 30, 1073–1081. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Liu, Z.; Wang, L.; Zhang, X. NF-kappaB signaling pathway, inflammation and colorectal cancer. Cell. Mol. Immunol. 2009, 6, 327–334. [Google Scholar] [CrossRef] [PubMed]
- Scholz, C.C.; Taylor, C.T. Hydroxylase-dependent regulation of the NF-kappa B pathway. Biol. Chem. 2013, 394, 479–493. [Google Scholar] [CrossRef] [PubMed]
- Dolcet, X.; Llobet, D.; Pallares, J.; Matias-Guiu, X. NF-kB in development and progression of human cancer. Virchows Arch. 2005, 446, 475–482. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.H.; Jeon, Y.T.; Kim, S.H.; Song, Y.S. NF-κ B as a potential molecular target for cancer therapy. Biofactors 2007, 29, 19–35. [Google Scholar] [CrossRef] [PubMed]
- Van Laere, S.J.; Van der Auwera, I.; Van den Eynden, G.G.; Elst, H.J.; Weyler, J.; Harris, A.L.; van Dam, P.; Van Marck, E.A.; Vermeulen, P.B.; Dirix, L.Y. Nuclear factor-κB signature of inflammatory breast cancer by cDNA microarray validated by quantitative real-time reverse transcription-PCR, immunohistochemistry, and nuclear factor-κB DNA-binding. Clin. Cancer Res. 2006, 12, 3249–3256. [Google Scholar] [CrossRef] [PubMed]
- Lerebours, F.; Vacher, S.; Andrieu, C.; Espie, M.; Marty, M.; Lidereau, R.; Bieche, I. NF-κ B genes have a major role in Inflammatory Breast Cancer. BMC Cancer 2008, 8, 41. [Google Scholar] [CrossRef] [PubMed]
- Fouad, T.M.; Kogawa, T.; Reuben, J.M.; Ueno, N.T. The role of inflammation in inflammatory breast cancer. Adv. Exp. Med. Biol. 2014, 816, 53–73. [Google Scholar] [PubMed]
- Helenius, M.; Kyrylenko, S.; Vehvilainen, P.; Salminen, A. Characterization of aging-associated up-regulation of constitutive nuclear factor-κ B binding activity. Antioxid. Redox Signal. 2001, 3, 147–156. [Google Scholar] [CrossRef] [PubMed]
- Strober, W.; Murray, P.J.; Kitani, A.; Watanabe, T. Signalling pathways and molecular interactions of NOD1 and NOD2. Nat. Rev. Immunol. 2006, 6, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Tigno-Aranjuez, J.T.; Asara, J.M.; Abbott, D.W. Inhibition of RIP2’s tyrosine kinase activity limits NOD2-driven cytokine responses. Genes Dev. 2010, 24, 2666–2677. [Google Scholar] [CrossRef] [PubMed]
- Dorsch, M.; Wang, A.; Cheng, H.; Lu, C.; Bielecki, A.; Charron, K.; Clauser, K.; Ren, H.; Polakiewicz, R.D.; Parsons, T.; et al. Identification of a regulatory autophosphorylation site in the serine-threonine kinase RIP2. Cell Signal. 2006, 18, 2223–2229. [Google Scholar] [CrossRef] [PubMed]
- Inohara, N.; del Peso, L.; Koseki, T.; Chen, S.; Nunez, G. RICK, a novel protein kinase containing a caspase recruitment domain, interacts with CLARP and regulates CD95-mediated apoptosis. J. Biol. Chem. 1998, 273, 12296–12300. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, J.V.; Ni, J.; Dixit, V.M. RIP2 is a novel NF-κ B-activating and cell death-inducing kinase. J. Biol. Chem. 1998, 273, 16968–16975. [Google Scholar] [CrossRef] [PubMed]
- Thome, M.; Hofmann, K.; Burns, K.; Martinon, F.; Bodmer, J.L.; Mattmann, C.; Tschopp, J. Identification of CARDIAK, a RIP-like kinase that associates with caspase-1. Curr. Biol. 1998, 8, 885–888. [Google Scholar] [CrossRef]
- Park, J.H.; Kim, Y.G.; McDonald, C.; Kanneganti, T.D.; Hasegawa, M.; Body-Malapel, M.; Inohara, N.; Nunez, G. RICK/RIP2 mediates innate immune responses induced through Nod1 and Nod2 but not TLRs. J. Immunol. 2007, 178, 2380–2386. [Google Scholar] [CrossRef] [PubMed]
- Hoesel, B.; Schmid, J.A. The complexity of NF-κB signaling in inflammation and cancer. Mol. Cancer 2013, 12, 86. [Google Scholar] [CrossRef] [PubMed]
- Goh, F.Y.; Cook, K.L.; Upton, N.; Tao, L.; Lah, L.C.; Leung, B.P.; Wong, W.S. Receptor-interacting protein 2 gene silencing attenuates allergic airway inflammation. J. Immunol. 2013, 191, 2691–2699. [Google Scholar] [CrossRef] [PubMed]
- Shaw, P.J.; Barr, M.J.; Lukens, J.R.; McGargill, M.A.; Chi, H.B.; Mak, T.W.; Kanneganti, T.D. Signaling via the RIP2 Adaptor Protein in Central Nervous System-Infiltrating Dendritic Cells Promotes Inflammation and Autoimmunity. Immunity 2011, 34, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Caruso, R.; Warner, N.; Inohara, N.; Nunez, G. NOD1 and NOD2: Signaling, host defense, and inflammatory disease. Immunity 2014, 41, 898–908. [Google Scholar] [CrossRef] [PubMed]
- Duggan, B.M.; Foley, K.P.; Henriksbo, B.D.; Cavallari, J.F.; Tamrakar, A.K.; Schertzer, J.D. Tyrosine kinase inhibitors of Ripk2 attenuate bacterial cell wall-mediated lipolysis, inflammation and dysglycemia. Sci. Rep. 2017, 7, 1578. [Google Scholar] [CrossRef] [PubMed]
- Tervaniemi, M.H.; Katayama, S.; Skoog, T.; Siitonen, H.A.; Vuola, J.; Nuutila, K.; Sormunen, R.; Johnsson, A.; Linnarsson, S.; Suomela, S.; et al. NOD-like receptor signaling and inflammasome-related pathways are highlighted in psoriatic epidermis. Sci. Rep. 2016, 6, 22745. [Google Scholar] [CrossRef] [PubMed]
- Canning, P.; Ruan, Q.; Schwerd, T.; Hrdinka, M.; Maki, J.L.; Saleh, D.; Suebsuwong, C.; Ray, S.; Brennan, P.E.; Cuny, G.D.; et al. Inflammatory Signaling by NOD-RIPK2 Is Inhibited by Clinically Relevant Type II Kinase Inhibitors. Chem. Biol. 2015, 22, 1174–1184. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; LaFortune, T.A.; Krishnamurthy, S.; Esteva, F.J.; Cristofanilli, M.; Liu, P.; Lucci, A.; Singh, B.; Hung, M.C.; Hortobagyi, G.N.; et al. Epidermal growth factor receptor tyrosine kinase inhibitor reverses mesenchymal to epithelial phenotype and inhibits metastasis in inflammatory breast cancer. Clin. Cancer Res. 2009, 15, 6639–6648. [Google Scholar] [CrossRef] [PubMed]
- Christian, F.; Smith, E.L.; Carmody, R.J. The Regulation of NF-κB Subunits by Phosphorylation. Cells 2016, 5, 12. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, M.M.; Al-Raawi, D.; Sabet, S.F.; El-Shinawi, M. Inflammatory breast cancer: New factors contribute to disease etiology: A review. J. Adv. Res. 2014, 5, 525–536. [Google Scholar] [CrossRef] [PubMed]
- Lerebours, F.; Bieche, I.; Lidereau, R. Update on inflammatory breast cancer. Breast Cancer Res. 2005, 7, 52–58. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, S.; Shairaz, B. (University of Alberta, AB, Canada). Personal communication, 2018.
- Gutsche, K.; Randi, E.B.; Blank, V.; Fink, D.; Wenger, R.H.; Leo, C.; Scholz, C.C. Intermittent hypoxia confers pro-metastatic gene expression selectively through NF-κ B in inflammatory breast cancer cells. Free Radic. Biol. Med. 2016, 101, 129–142. [Google Scholar] [CrossRef] [PubMed]
- Caamano, J.; Hunter, C.A. NF-κ B family of transcription factors: Central regulators of innate and adaptive immune functions. Clin. Microbiol. Rev. 2002, 15, 414–429. [Google Scholar] [CrossRef] [PubMed]
- Hussein, M.Z.; Al Fikky, A.; Abdel Bar, I.; Attia, O. Serum IL-6 and IL-12 levels in breast cancer patients. Egypt. J. Immunol. 2004, 11, 165–170. [Google Scholar] [PubMed]
- Singel, S.M.; Batten, K.; Cornelius, C.; Jia, G.X.; Fasciani, G.; Barron, S.L.; Wright, W.E.; Shay, J.W. Receptor-interacting protein kinase 2 promotes triple-negative breast cancer cell migration and invasion via activation of nuclear factor-κB and c-Jun N-terminal kinase pathways. Breast Cancer Res. 2014, 16, R28. [Google Scholar] [CrossRef] [PubMed]
- Tigno-Aranjuez, J.T.; Benderitter, P.; Rombouts, F.; Deroose, F.; Bai, X.D.; Mattioli, B.; Cominelli, F.; Pizarro, T.T.; Hoflack, J.; Abbott, D.W. In Vivo Inhibition of RIPK2 Kinase Alleviates Inflammatory Disease. J. Biol. Chem. 2014, 289, 29651–29664. [Google Scholar] [CrossRef] [PubMed]
- Nachbur, U.; Stafford, C.A.; Bankovacki, A.; Zhan, Y.; Lindqvist, L.M.; Fiil, B.K.; Khakham, Y.; Ko, H.J.; Sandow, J.J.; Falk, H.; et al. A RIPK2 inhibitor delays NOD signalling events yet prevents inflammatory cytokine production. Nat. Commun. 2015, 6, 6442. [Google Scholar] [CrossRef] [PubMed]
- Salla, M.; Aguayo-Ortiz, R.; Danmaliki, G.I.; Zare, A.; Said, A.; Moore, J.; Pandya, V.; Manaloor, R.; Fong, S.; Blankstein, A.R.; et al. Identification and Characterization of Novel Receptor-Interacting Serine/Threonine-Protein Kinase 2 Inhibitors Using Structural Similarity Analysis. J. Pharmacol. Exp. Ther. 2018, 365, 354–367. [Google Scholar] [CrossRef] [PubMed]
- Vyas, D.; Laput, G.; Vyas, A.K. Chemotherapy-enhanced inflammation may lead to the failure of therapy and metastasis. Oncotargets Ther. 2014, 7, 1015–1023. [Google Scholar] [CrossRef] [PubMed]
- Sobin, L.H. TNM: Principles, history, and relation to other prognostic factors. Cancer 2001, 91, 1589–1592. [Google Scholar] [CrossRef]
- Park, Y.H.; Lee, S.J.; Cho, E.Y.; Choi, Y.L.; Lee, J.E.; Nam, S.J.; Yang, J.H.; Shin, J.H.; Ko, E.Y.; Han, B.K.; et al. Clinical relevance of TNM staging system according to breast cancer subtypes. Ann. Oncol. 2011, 22, 1554–1560. [Google Scholar] [CrossRef] [PubMed]
- Dong, G.Z.; Wang, D.G.; Liang, X.L.; Gao, H.Q.; Wang, L.; Yu, X.J.; Liu, J.J. Factors related to survival rates for breast cancer patients. Int. J. Clin. Exp. Med. 2014, 7, 3719–3724. [Google Scholar] [PubMed]
- Hayes, D.F. Tumor, Node, Metastasis (TNM) Staging Classification for Breast Cancer; UpToDate, Inc.: Waltham, MA, USA, 2018. [Google Scholar]
- Bennett, L.; Mallon, E.A.; Horgan, P.G.; Paul, A.; McMillan, D.C.; Edwards, J. The relationship between members of the canonical NF-kB pathway, components of tumour microenvironment and survival in patients with invasive ductal breast cancer. Oncotarget 2017, 8, 33002–33013. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.; Buzdar, A.U.; Hursting, S.D. Inflammatory breast cancer and body mass index. J. Clin. Oncol. 1998, 16, 3731–3735. [Google Scholar] [CrossRef] [PubMed]
- Dietze, E.C.; Chavez, T.A.; Seewaldt, V.L. Obesity and Triple-Negative Breast Cancer: Disparities, Controversies, and Biology. Am. J. Pathol. 2018, 188, 280–290. [Google Scholar] [CrossRef] [PubMed]
- Feola, A.; Ricci, S.; Kouidhi, S.; Rizzo, A.; Penon, A.; Formisano, P.; Giordano, A.; Di Carlo, A.; Di Domenico, M. Multifaceted Breast Cancer: The Molecular Connection With Obesity. J. Cell. Physiol. 2017, 232, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Huang, A.; Cao, S.S.; Tang, L.L. The tumor microenvironment and inflammatory breast cancer. J. Cancer 2017, 8, 1884–1891. [Google Scholar] [CrossRef] [PubMed]
- Chirieleison, S.M.; Kertesy, S.B.; Abbott, D.W. Synthetic Biology Reveals the Uniqueness of the RIP Kinase Domain. J. Immunol. 2016, 196, 4291–4297. [Google Scholar] [CrossRef] [PubMed]
- FC-IBC-02: A New in vitro-in vivo Model of Triple Negative Inflammatory Breast Cancer. Available online: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE40464 (accessed on 29 May 2018).
- Woodward, W.A.; Krishnamurthy, S.; Yamauchi, H.; El-Zein, R.; Dai, O.; Eri, K.; Shin-ichiro, N.; Massimo, C.; Peter, V.; Luc, D.; et al. Genomic and expression analysis of microdissected inflammatory breast cancer. Breast Cancer Res. Treat. 2013, 138, 761–772. [Google Scholar] [CrossRef] [PubMed]
- Mertins, P.; Mani, D.R.; Ruggles, K.V.; Gillette, M.A.; Clauser, K.R.; Wang, P.; Wang, X.L.; Qiao, J.W.; Cao, S.; Petralia, F.; et al. Proteogenomics connects somatic mutations to signalling in breast cancer. Nature 2016, 534, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Shi, M.; Duan, C.; Chen, H.; Hu, Y.; Yang, Z.; Duan, H.; Guo, N. Downregulation of Erbin in Her2-overexpressing breast cancer cells promotes cell migration and induces trastuzumab resistance. Mol. Immunol. 2013, 56, 104–112. [Google Scholar] [CrossRef] [PubMed]
- Kufer, T.A.; Kremmer, E.; Banks, D.J.; Philpott, D.J. Role for erbin in bacterial activation of Nod2. Infect. Immun. 2006, 74, 3115–3124. [Google Scholar] [CrossRef] [PubMed]
- Volodko, N.; Salla, M.; Zare, A.; Abulghasem, E.A.; Vincent, K.; Benesch, M.G.K.; McMullen, T.P.W.; Bathe, O.F.; Postovit, L.; Baksh, S. RASSF1A Site-Specific Methylation Hotspots in Cancer and Correlation with RASSF1C and MOAP-1. Cancers 2016, 8, 55. [Google Scholar] [CrossRef] [PubMed]
- Pogo, B.G.; Holland, J.F.; Levine, P.H. Human mammary tumor virus in inflammatory breast cancer. Cancer 2010, 116, 2741–2744. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, J.M. The Origin of Human Milk Bacteria: Is There a Bacterial Entero-Mammary Pathway during Late Pregnancy and Lactation? Adv. Nutr. 2014, 5, 779–784. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, R.L.; El-Zein, R.; Valero, V.; Lucci, A.; Bevers, T.B.; Fouad, T.; Liao, W.Q.; Ueno, N.T.; Woodward, W.A.; Brewster, A.M. Epidemiological risk factors associated with inflammatory breast cancer subtypes. Cancer Cause Control 2016, 27, 359–366. [Google Scholar] [CrossRef] [PubMed]
- Bertucci, F.; Fekih, M.; Autret, A.; Petit, T.; Dalenc, F.; Levy, C.; Romieu, G.; Bonneterre, J.; Ferrero, J.M.; Kerbrat, P.; et al. Bevacizumab plus neoadjuvant chemotherapy in patients with HER2-negative inflammatory breast cancer (BEVERLY-1): A multicentre, single-arm, phase 2 study. Lancet Oncol. 2016, 17, 600–611. [Google Scholar] [CrossRef]
- Jolly, M.K.; Boareto, M.; Debeb, B.G.; Aceto, N.; Farach-Carson, M.C.; Woodward, W.A.; Levine, H. Inflammatory breast cancer: A model for investigating cluster-based dissemination. NPJ Breast Cancer 2017, 3, 21. [Google Scholar] [CrossRef] [PubMed]
- Roberts, I. Bone-Marrow Transplantation in Children—Current Results and Controversies. Bone Marrow Transpl. 1994, 14, 197–199. [Google Scholar]
- Brouckaert, O.; Wildiers, H.; Floris, G.; Neven, P. Update on triple-negative breast cancer: Prognosis and management strategies. Int. J. Womens Health 2012, 4, 511–520. [Google Scholar] [PubMed]
- StataCorp. Stata 13 Base Reference Manual; Stata Press: College Station, TX, USA, 2013. [Google Scholar]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variable | n (%) | Variable | n (%) |
|---|---|---|---|
| Age | TNBC | ||
| ≤45 | 11 (22) | Yes | 10 (20) |
| >45 | 39 (78) | No | 40 (80) |
| Grade | Tumor Size | ||
| I | 11 (22) | ≤3 cm | 29 (58) |
| III | 35 (70) | >3 cm | 13 (26) |
| Unknown | 4 (8) | Unknown | 8 (16) |
| ER | PR | ||
| Positive | 20 (40) | Positive | 28 (56) |
| Negative | 27 (54) | Negative | 19 (38) |
| Unknown | 3 (6) | Unknown | 2 (4) |
| TNM | HER2 | ||
| I | 17 (34) | Positive | 30 (60) |
| II | 13 (26) | Negative | 18 (36) |
| III | 16 (32) | Unknown | 2 (4) |
| IV | 4 (8) | ||
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zare, A.; Petrova, A.; Agoumi, M.; Armstrong, H.; Bigras, G.; Tonkin, K.; Wine, E.; Baksh, S. RIPK2: New Elements in Modulating Inflammatory Breast Cancer Pathogenesis. Cancers 2018, 10, 184. https://doi.org/10.3390/cancers10060184
Zare A, Petrova A, Agoumi M, Armstrong H, Bigras G, Tonkin K, Wine E, Baksh S. RIPK2: New Elements in Modulating Inflammatory Breast Cancer Pathogenesis. Cancers. 2018; 10(6):184. https://doi.org/10.3390/cancers10060184
Chicago/Turabian StyleZare, Alaa, Alexandra Petrova, Mehdi Agoumi, Heather Armstrong, Gilbert Bigras, Katia Tonkin, Eytan Wine, and Shairaz Baksh. 2018. "RIPK2: New Elements in Modulating Inflammatory Breast Cancer Pathogenesis" Cancers 10, no. 6: 184. https://doi.org/10.3390/cancers10060184
APA StyleZare, A., Petrova, A., Agoumi, M., Armstrong, H., Bigras, G., Tonkin, K., Wine, E., & Baksh, S. (2018). RIPK2: New Elements in Modulating Inflammatory Breast Cancer Pathogenesis. Cancers, 10(6), 184. https://doi.org/10.3390/cancers10060184