Selective Inhibition of Histone Deacetylation in Melanoma Increases Targeted Gene Delivery by a Bacteriophage Viral Vector


Abstract
1. Introduction
2. Results
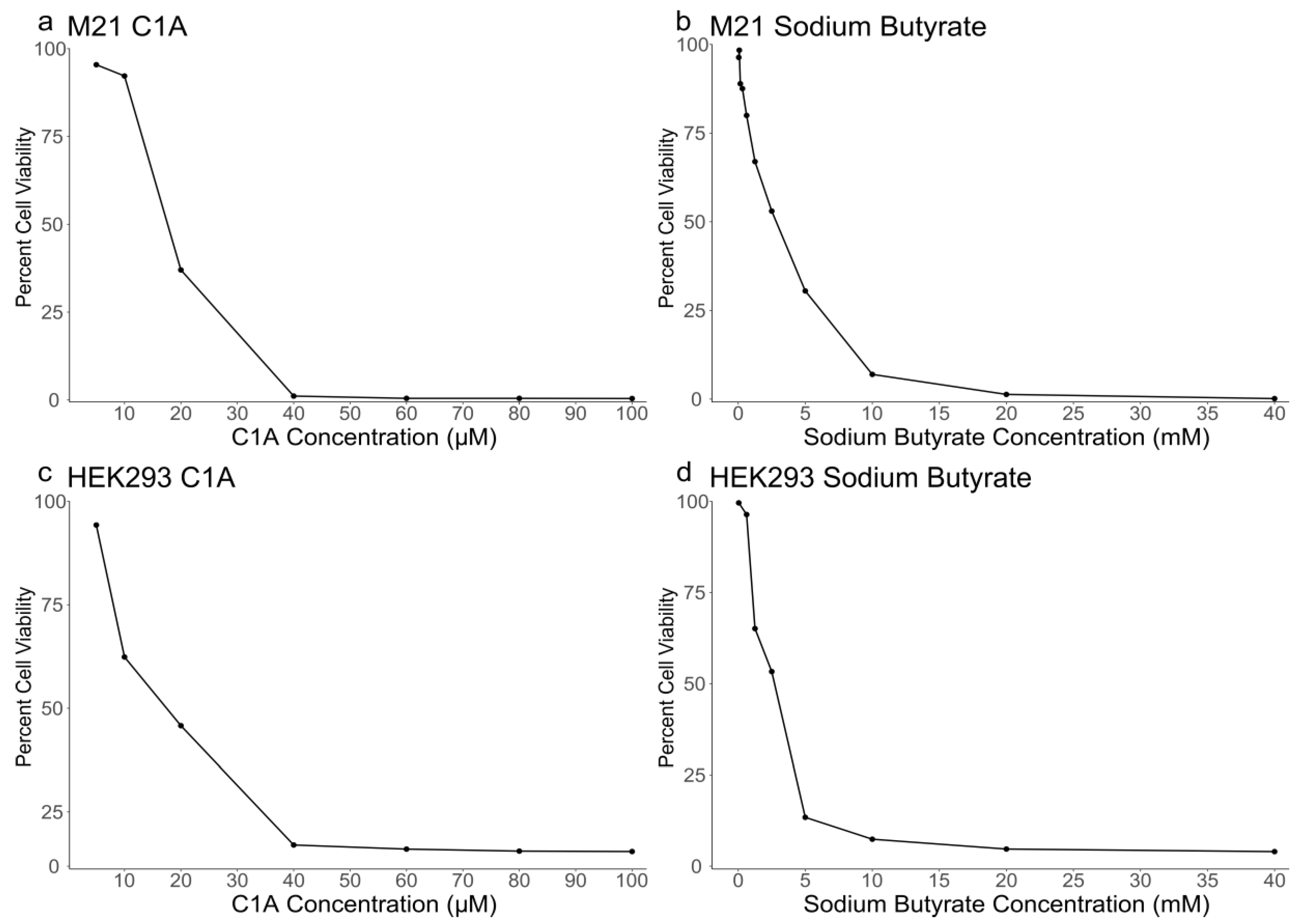
2.1. Sensitivity of M21 and HEK293 Cells to C1A and NaBu
2.2. Targeted Gene Delivery to HEK293 Cells by RGD4C-AAVP in Combination with NaBu and C1A
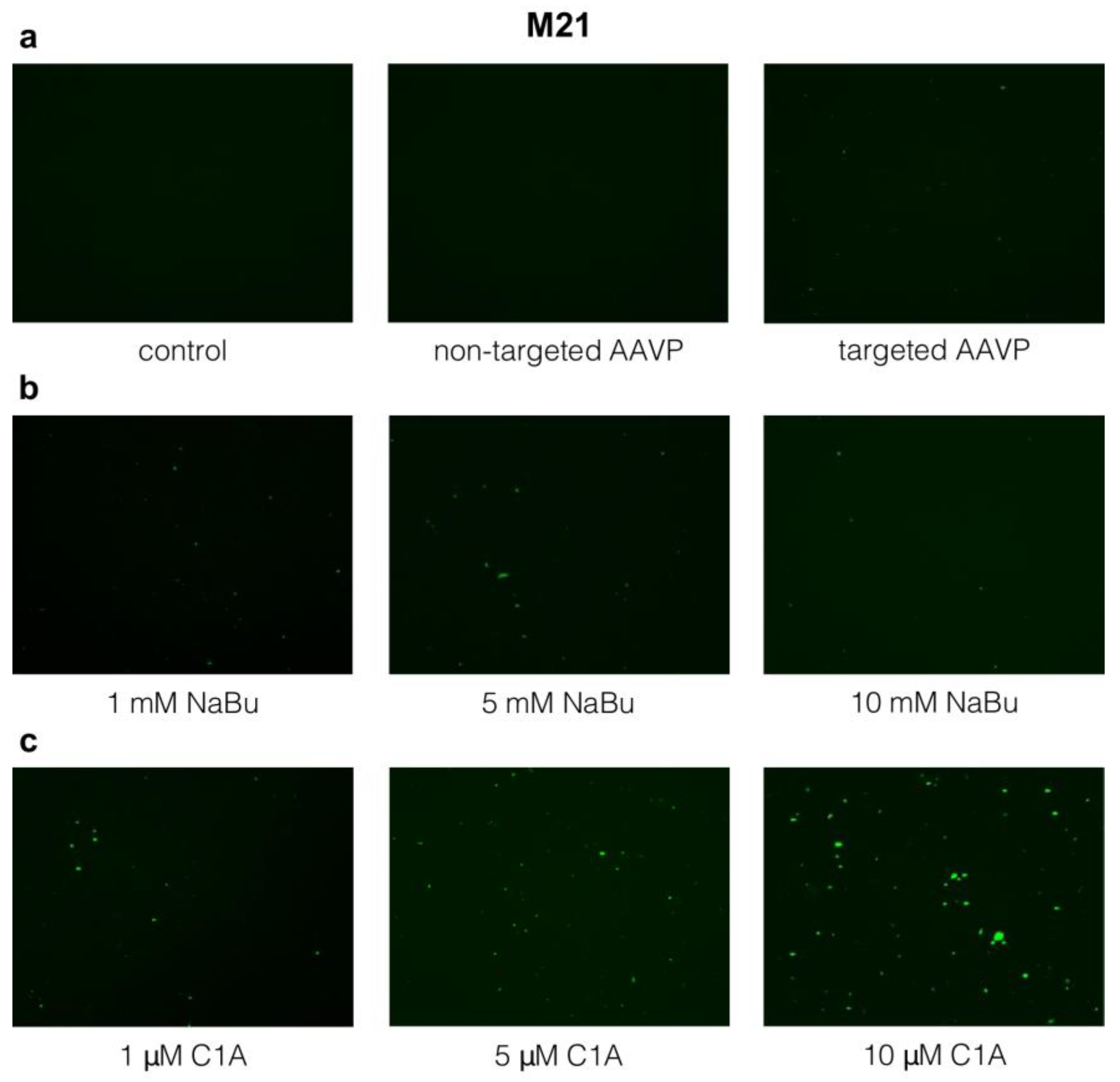
2.3. Targeted Gene Delivery to M21 Cells by RGD4C-AAVP Is Improved by NaBu and C1A
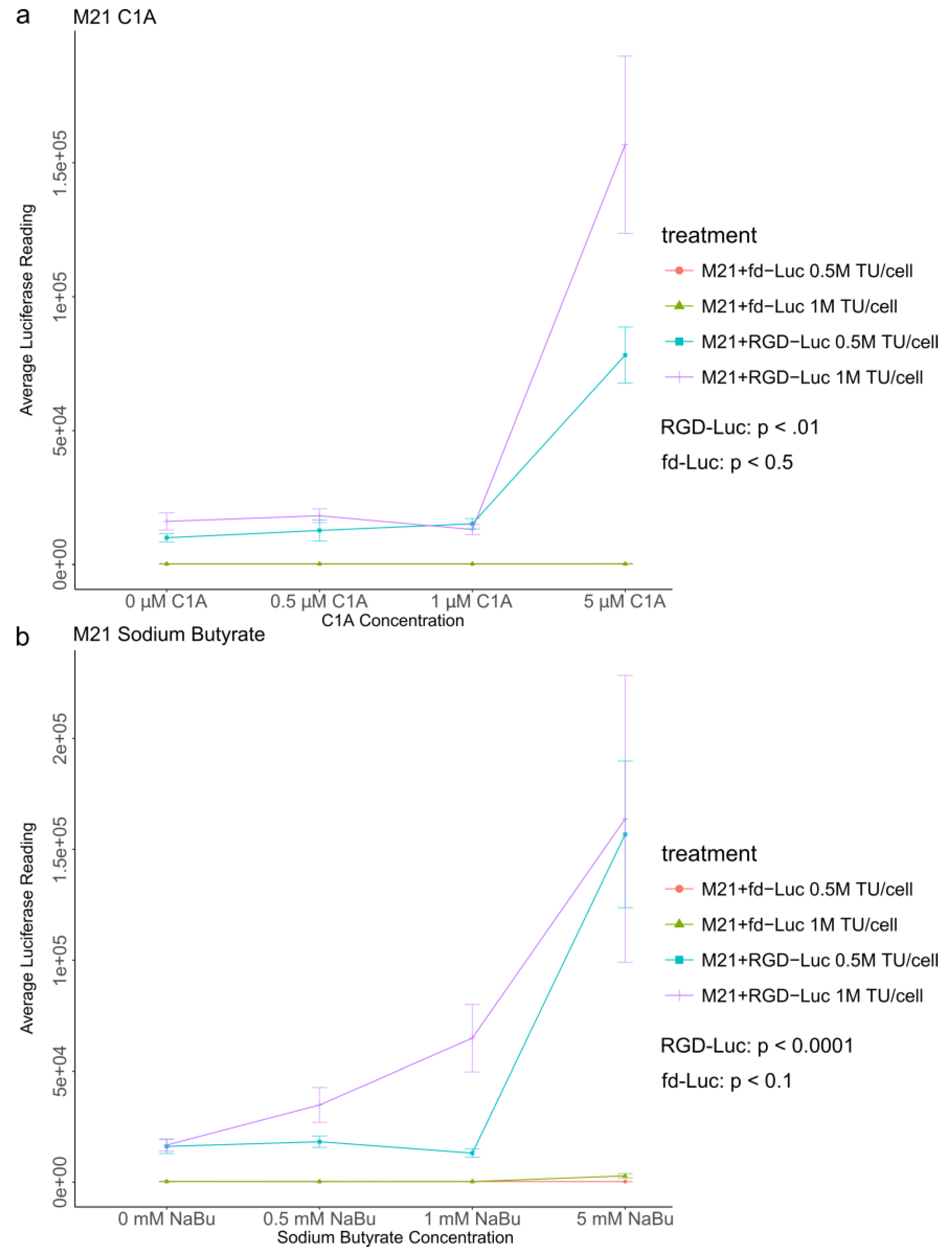
2.4. Quantitative Analysis of C1A and NaBu Effects on AAVP-Mediated Gene Delivery to Human Melanoma
2.5. Quantitative Analysis of C1A and NaBu Effects on AAVP-Mediated Gene Delivery to B16 Melanoma
3. Discussion
4. Materials and Methods
4.1. Cell Lines
4.2. Phage Production
4.3. Adjuvant Treatment and Transduction
4.4. Reporter Genes
4.4.1. GFP
4.4.2. Luciferase
4.5. Cytotoxicity Assay
4.6. Computer Programs and Statistical Analysis
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| AAV | Adeno-associated virus |
| AAVP | Adeno-associated virus/phage |
| B16 | Murine melanoma cell line |
| GFP | Green Fluorescent Protein |
| HDAC | Histone deacetylase |
| HEK293 | Human embryonic kidney 293 cells |
| IC50 | Median lethal concentration, or 50% inhibitory concentration |
| M13 | Filamentous bacteriophage |
| M21 | Human melanoma cell line |
| MuLV | Murine leukemia virus |
| RGD4C | A double cyclic arginylglycylaspartic acid peptide (CDCRGDCFC) that targets αv integrins (principally αvβ3 and αvβ5) |
| RT | Room temperature |
| TU | Transducing unit |
References
- Stewart, B.W.; International Agency for Research on Cancer; World Health Organization. World Cancer Report 2014; International Agency for Research in Cancer: Lyon, France, 2014; ISBN 978-92-832-0429-9. [Google Scholar]
- Lebovits, A.H.; Strain, J.J.; Schleifer, S.J.; Tanaka, J.S.; Bhardwaj, S.; Messe, M.R. Patient noncompliance with self-administered chemotherapy. Cancer 1990, 65, 17–22. [Google Scholar] [CrossRef]
- Sottoriva, A.; Spiteri, I.; Piccirillo, S.G.M.; Touloumis, A.; Collins, V.P.; Marioni, J.C.; Curtis, C.; Watts, C.; Tavaré, S. Intratumor heterogeneity in human glioblastoma reflects cancer evolutionary dynamics. Proc. Natl. Acad. Sci. USA 2013, 110, 4009–4014. [Google Scholar] [CrossRef] [PubMed]
- Friedman, D.L.; Whitton, J.; Leisenring, W.; Mertens, A.C.; Hammond, S.; Stovall, M.; Donaldson, S.S.; Meadows, A.T.; Robison, L.L.; Neglia, J.P. Subsequent neoplasms in 5-year survivors of childhood cancer: The Childhood Cancer Survivor Study. J. Natl. Cancer Inst. 2010, 102, 1083–1095. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, H.E. Radiation therapy-induced neoplams. In Etiology of Cancer in Man; Cancer Growth and Progression; Springer Netherlands: Dordrecht, The Netherlands, 1989; Volume 6, pp. 122–131. ISBN 978-94-009-2532-8. [Google Scholar]
- Wolff, J.A.; Lederberg, J. An early history of gene transfer and therapy. Hum. Gene Ther. 1994, 5, 469–480. [Google Scholar] [CrossRef] [PubMed]
- Tatum, E.L. Molecular biology, nucleic acids, and the future of medicine. Perspect. Biol. Med. 1966, 10, 19–32. [Google Scholar] [CrossRef] [PubMed]
- Sheridan, C. Gene therapy finds its niche. Nat. Biotechnol. 2011, 29, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Hajitou, A.; Trepel, M.; Lilley, C.E.; Soghomonyan, S.; Alauddin, M.M.; Marini, F.C.; Restel, B.H.; Ozawa, M.G.; Moya, C.A.; Rangel, R.; et al. A hybrid vector for ligand-directed tumor targeting and molecular imaging. Cell 2006, 125, 385–398. [Google Scholar] [CrossRef] [PubMed]
- Arap, W.; Pasqualini, R.; Ruoslahti, E. Cancer treatment by targeted drug delivery to tumor vasculature in a mouse model. Science 1998, 279, 377–380. [Google Scholar] [CrossRef] [PubMed]
- Hood, J.D.; Bednarski, M.; Frausto, R.; Guccione, S.; Reisfeld, R.A.; Xiang, R.; Cheresh, D.A. Tumor regression by targeted gene delivery to the neovasculature. Science 2002, 296, 2404–2407. [Google Scholar] [CrossRef] [PubMed]
- Hajitou, A.; Pasqualini, R.; Arap, W. Vascular targeting: Recent advances and therapeutic perspectives. Trends Cardiovasc. Med. 2006, 16, 80–88. [Google Scholar] [CrossRef] [PubMed]
- Kaur, T.; Nafissi, N.; Wasfi, O.; Sheldon, K.; Wettig, S.; Slavcev, R. Immunocompatibility of Bacteriophages as Nanomedicines. J. Nanotechnol. 2012, 2012, 1–13. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Geier, M.R.; Trigg, M.E.; Merril, C.R. Fate of bacteriophage lambda in non-immune germ-free mice. Nature 1973, 246, 221–223. [Google Scholar] [CrossRef] [PubMed]
- Merril, C.R.; Biswas, B.; Carlton, R.; Jensen, N.C.; Creed, G.J.; Zullo, S.; Adhya, S. Long-circulating bacteriophage as antibacterial agents. Proc. Natl. Acad. Sci. USA 1996, 93, 3188–3192. [Google Scholar] [CrossRef] [PubMed]
- Kia, A.; Yata, T.; Hajji, N.; Hajitou, A. Inhibition of Histone Deacetylation and DNA Methylation Improves Gene Expression Mediated by the Adeno-Associated Virus/Phage in Cancer Cells. Viruses 2013, 5, 2561–2572. [Google Scholar] [CrossRef] [PubMed]
- Carew, J.S.; Giles, F.J.; Nawrocki, S.T. Histone deacetylase inhibitors: Mechanisms of cell death and promise in combination cancer therapy. Cancer Lett. 2008, 269, 7–17. [Google Scholar] [CrossRef] [PubMed]
- Kaliszczak, M.; Trousil, S.; Åberg, O.; Perumal, M.; Nguyen, Q.-D.; Aboagye, E.O. A novel small molecule hydroxamate preferentially inhibits HDAC6 activity and tumour growth. Br. J. Cancer 2013, 108, 342–350. [Google Scholar] [CrossRef] [PubMed]
- Davie, J.R. Inhibition of histone deacetylase activity by butyrate. J. Nutr. 2003, 133, 2485S–2493S. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Gu, J.; Feng, Z.; Yang, Y.; Zhu, N.; Lu, W.; Qi, F. Both HDAC5 and HDAC6 are required for the proliferation and metastasis of melanoma cells. J. Transl. Med. 2016, 14, 7. [Google Scholar] [CrossRef] [PubMed]
- Harries, M.; Malvehy, J.; Lebbe, C.; Heron, L.; Amelio, J.; Szabo, Z.; Schadendorf, D. Treatment patterns of advanced malignant melanoma (stage III–IV)—A review of current standards in Europe. Eur. J. Cancer 2016, 60, 179–189. [Google Scholar] [CrossRef] [PubMed]
- Geller, A.C.; Annas, G.D. Epidemiology of melanoma and nonmelanoma skin cancer. Semin. Oncol. Nurs. 2003, 19, 2–11. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.C.; Faries, M.B.; Wanek, L.A.; Morton, D.L. Improved Survival for Stage IV Melanoma from an Unknown Primary Site. J. Clin. Oncol. 2009, 27, 3489–3495. [Google Scholar] [CrossRef] [PubMed]
- Sampson, J.H.; Carter, J.H.; Friedman, A.H.; Seigler, H.F. Demographics, prognosis, and therapy in 702 patients with brain metastases from malignant melanoma. J. Neurosurg. 1998, 88, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Krag, D.N.; Shukla, G.S.; Shen, G.-P.; Pero, S.; Ashikaga, T.; Fuller, S.; Weaver, D.L.; Burdette-Radoux, S.; Thomas, C. Selection of tumor-binding ligands in cancer patients with phage display libraries. Cancer Res. 2006, 66, 7724–7733. [Google Scholar] [CrossRef] [PubMed]
- Salemi, L.M.; Almawi, A.W.; Lefebvre, K.J.; Schild-Poulter, C. Aggresome formation is regulated by RanBPM through an interaction with HDAC6. Biol. Open 2014, 3, 418–430. [Google Scholar] [CrossRef] [PubMed]
- Overwijk, W.W.; Restifo, N.P. B16 as a mouse model for human melanoma. Curr. Protoc. Immunol. 2001, 39. [Google Scholar] [CrossRef]
- Becker, J.C.; Houben, R.; Schrama, D.; Voigt, H.; Ugurel, S.; Reisfeld, R.A. Mouse models for melanoma: A personal perspective. Exp. Dermatol. 2010, 19, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Kuzu, O.F.; Nguyen, F.D.; Noory, M.A.; Sharma, A. Current State of Animal (Mouse) Modeling in Melanoma Research. Cancer Growth Metastasis 2015, 8, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Tsafa, E.; Al-Bahrani, M.; Bentayebi, K.; Przystal, J.; Suwan, K.; Hajitou, A. The natural dietary genistein boosts bacteriophage-mediated cancer cell killing by improving phage-targeted tumor cell transduction. Oncotarget 2016, 7, 52135–52149. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Mamelak, A.N.; Hockaday, D. 131I-TM-601 SPECT imaging of Human Glioma. In Gliomas: Glioblastoma (Part 2); Tumors of the Central Nervous System; Springer Science+Business Media: Dordrecht, The Netherlands, 2011; Volume 2, pp. 123–130. ISBN 978-94-007-0344-5. [Google Scholar]
- Muñoz-Pinedo, C.; El Mjiyad, N.; Ricci, J.-E. Cancer metabolism: Current perspectives and future directions. Cell Death Dis. 2012, 3, e248. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, H.; Kaufmann, J.K.; Wang, P.-Y.; Nguyen, T.; Speranza, M.-C.; Kasai, K.; Okemoto, K.; Otsuki, A.; Nakano, I.; Fernandez, S.; et al. Histone deacetylase 6 inhibition enhances oncolytic viral replication in glioma. J. Clin. Investig. 2015, 125, 4269–4280. [Google Scholar] [CrossRef] [PubMed]
- Volcy, K.; Dewhurst, S. Proteasome inhibitors enhance bacteriophage lambda (λ) mediated gene transfer in mammalian cells. Virology 2009, 384, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Perazella, M.A. Onco-Nephrology: Renal Toxicities of Chemotherapeutic Agents. Clin. J. Am. Soc. Nephrol. 2012, 7, 1713–1721. [Google Scholar] [CrossRef] [PubMed]
- Hajitou, A.; Rangel, R.; Trepel, M.; Soghomonyan, S.; Gelovani, J.G.; Alauddin, M.M.; Pasqualini, R.; Arap, W. Design and construction of targeted AAVP vectors for mammalian cell transduction. Nat. Protoc. 2007, 2, 523–531. [Google Scholar] [CrossRef] [PubMed]
- Teerapong, Y. Bacteriophage: From Bacteria to Targeted Gene Delivery to Mammalian Cells; Imperial College London: London, UK, 2014. [Google Scholar]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2017. [Google Scholar]
- RStudio Team. RStudio: Integrated Development for R; RStudio, Inc.: Boston, MA, USA, 2015. [Google Scholar]
- Wickham, H. Ggplot2: Elegant Graphics for Data Analysis; Use R! Springer: New York, NY, USA, 2009; ISBN 978-0-387-98140-6. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | C1A IC50 | Sodium Butyrate IC50 |
|---|---|---|
| HEK293 | 17.41 μM | 2.7 mM |
| M21 | 18.05 μM | 2.7 mM |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Campbell, S.; Suwan, K.; Waramit, S.; Aboagye, E.O.; Hajitou, A. Selective Inhibition of Histone Deacetylation in Melanoma Increases Targeted Gene Delivery by a Bacteriophage Viral Vector. Cancers 2018, 10, 125. https://doi.org/10.3390/cancers10040125
Campbell S, Suwan K, Waramit S, Aboagye EO, Hajitou A. Selective Inhibition of Histone Deacetylation in Melanoma Increases Targeted Gene Delivery by a Bacteriophage Viral Vector. Cancers. 2018; 10(4):125. https://doi.org/10.3390/cancers10040125
Chicago/Turabian StyleCampbell, Samuel, Keittisak Suwan, Sajee Waramit, Eric Ofori Aboagye, and Amin Hajitou. 2018. "Selective Inhibition of Histone Deacetylation in Melanoma Increases Targeted Gene Delivery by a Bacteriophage Viral Vector" Cancers 10, no. 4: 125. https://doi.org/10.3390/cancers10040125
APA StyleCampbell, S., Suwan, K., Waramit, S., Aboagye, E. O., & Hajitou, A. (2018). Selective Inhibition of Histone Deacetylation in Melanoma Increases Targeted Gene Delivery by a Bacteriophage Viral Vector. Cancers, 10(4), 125. https://doi.org/10.3390/cancers10040125