Role of Gene Therapy in Pancreatic Cancer—A Review

Abstract
1. Introduction
2. Virotherapies
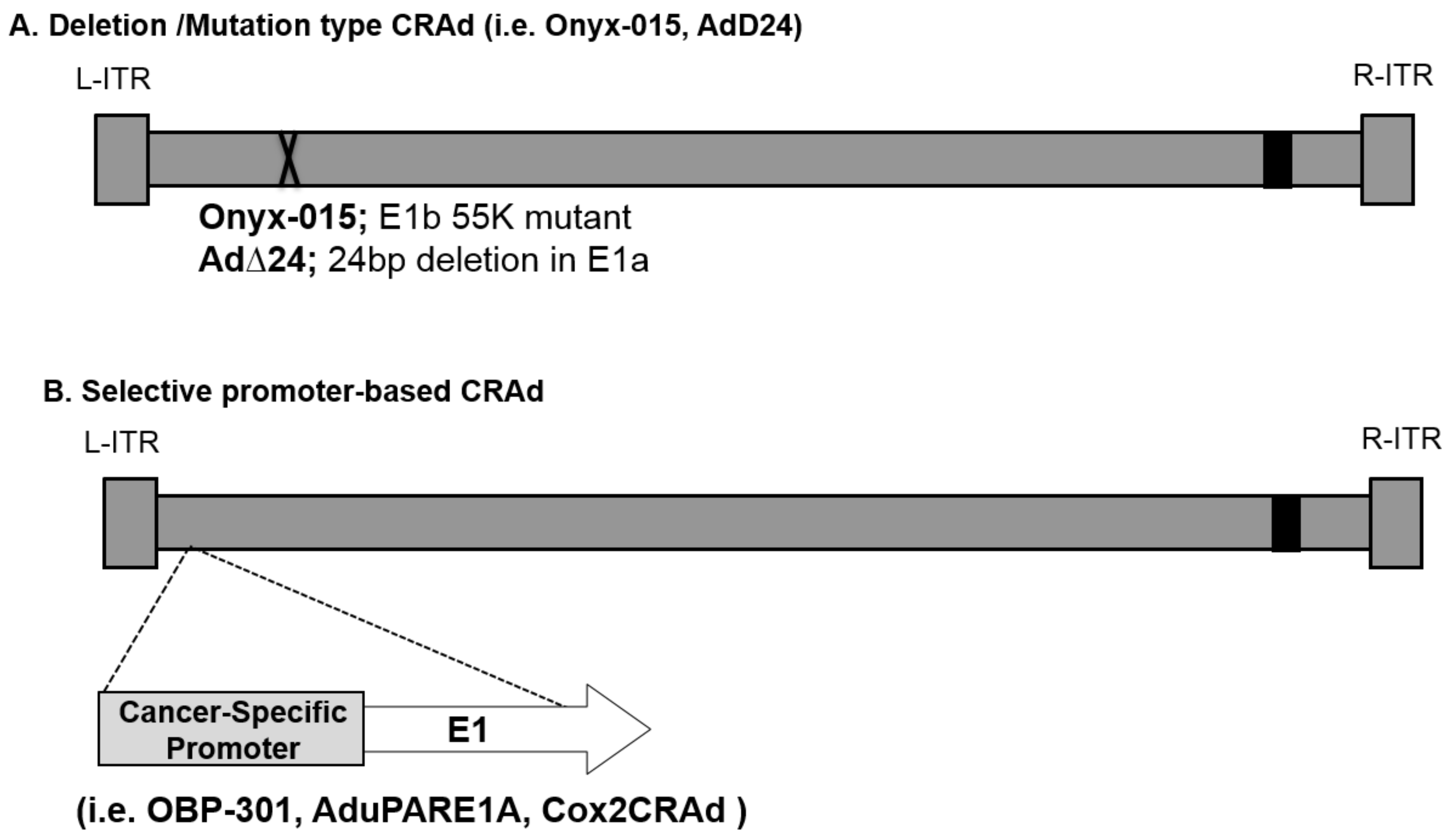
2.1. Replication-Based Control Oncolytic Adenoviruses
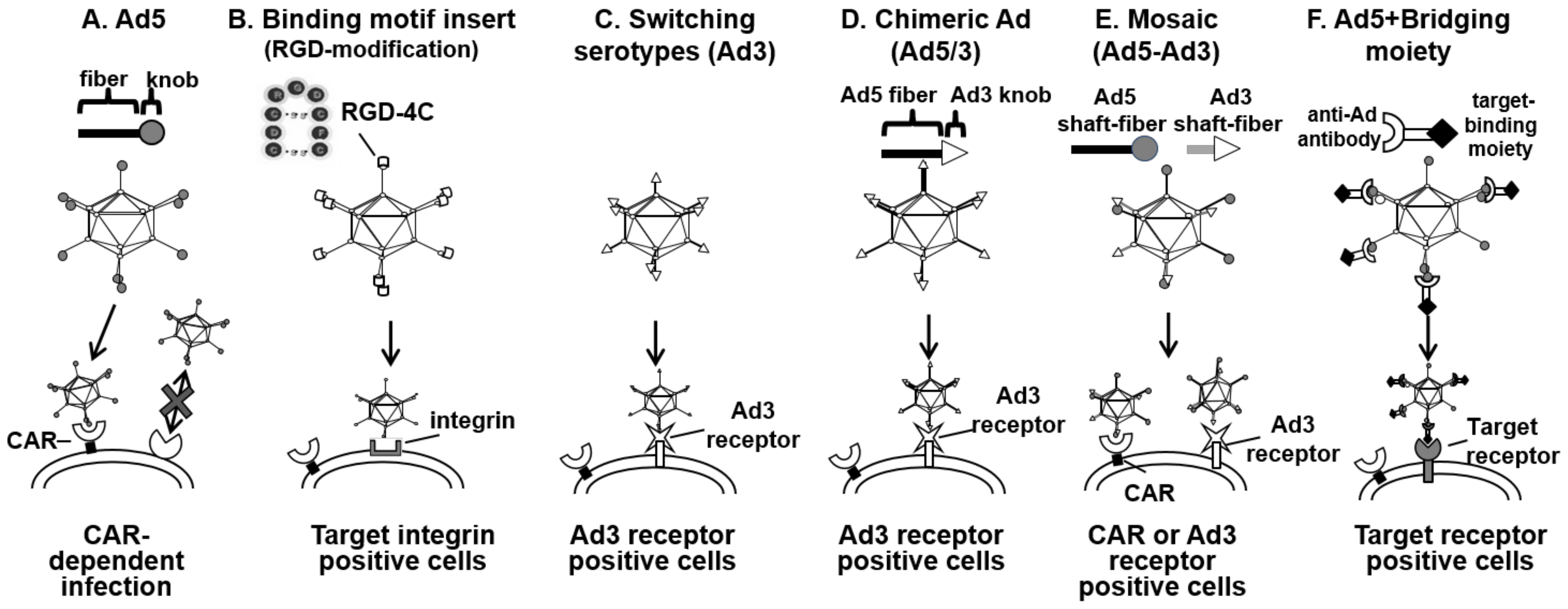
2.2. Enhanced Adenovirus Transduction
2.3. Therapeutic Gene-Expressing Vector
2.4. Combination Therapy with Oncolytic Viruses
3. Non-Viral Gene Therapies
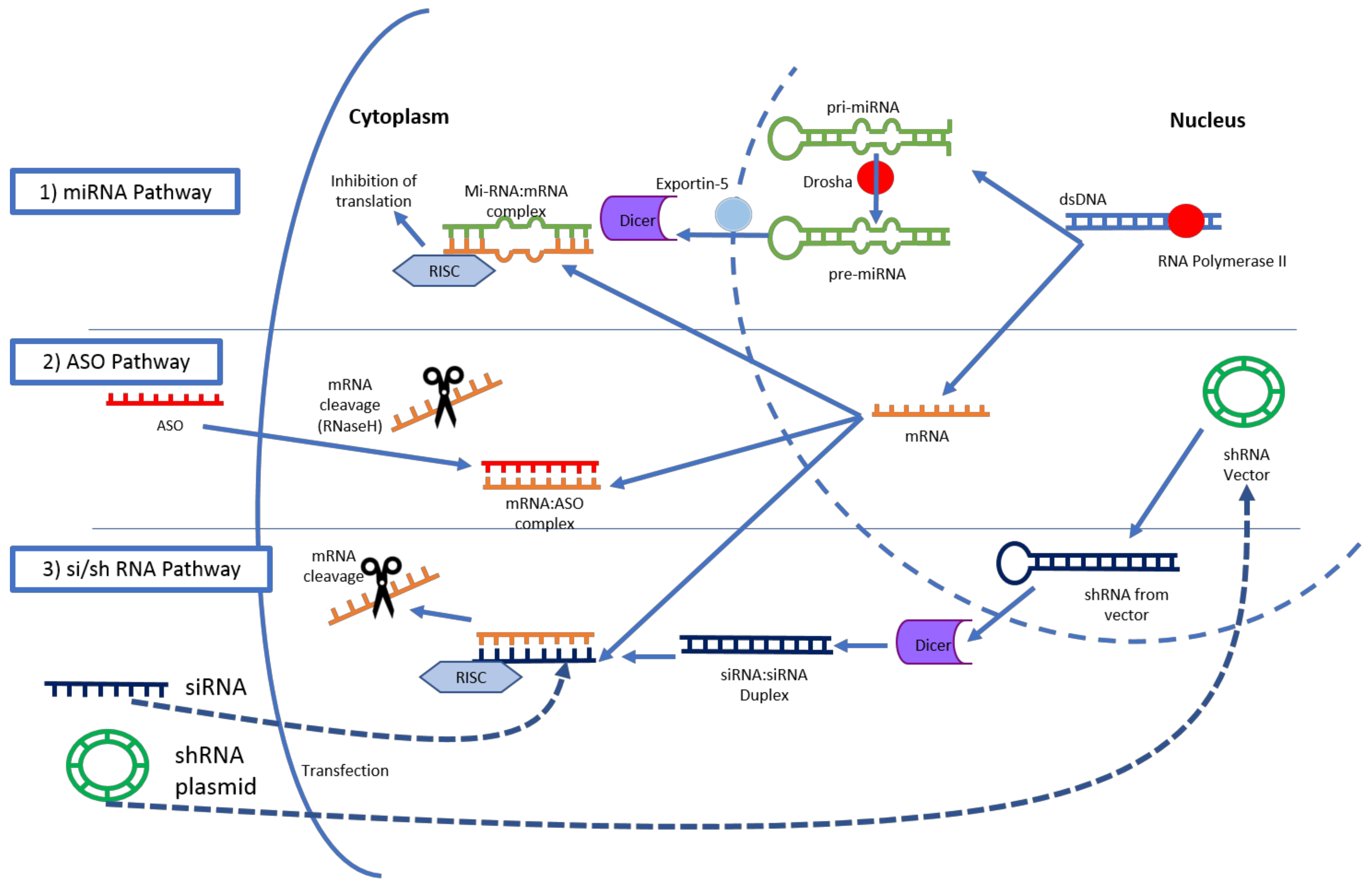
3.1. RNA Interference
3.2. Plasmid DNA
3.3. Gene-Editing Technology
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2017. Cancer J. Clin. 2017, 67, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Kamisawa, T.; Wood, L.D.; Itoi, T.; Takaori, K. Pancreatic cancer. Lancet 2016, 388, 73–85. [Google Scholar] [CrossRef]
- Neoptolemos, J.P.; Palmer, D.H.; Ghaneh, P.; Psarelli, E.E.; Valle, J.W.; Halloran, C.M.; Faluyi, O.; O’Reilly, D.A.; Cunningham, D.; Wadsley, J.; et al. Comparison of adjuvant gemcitabine and capecitabine with gemcitabine monotherapy in patients with resected pancreatic cancer (ESPAC-4): A multicentre, open-label, randomised, phase 3 trial. Lancet 2017, 389, 1011–1024. [Google Scholar] [CrossRef]
- Cunningham, D.; Chau, I.; Stocken, D.D.; Valle, J.W.; Smith, D.; Steward, W.; Harper, P.G.; Dunn, J.; Tudur-Smith, C.; West, J.; et al. Phase III randomized comparison of gemcitabine versus gemcitabine plus capecitabine in patients with advanced pancreatic cancer. J. Clin. Oncol. 2009, 27, 5513–5518. [Google Scholar] [CrossRef] [PubMed]
- Von Hoff, D.D.; Ervin, T.; Arena, F.P.; Chiorean, E.G.; Infante, J.; Moore, M.; Seay, T.; Tjulandin, S.A.; Ma, W.W.; Saleh, M.N.; et al. Increased Survival in Pancreatic Cancer with nab-Paclitaxel plus Gemcitabine. N. Engl. J. Med. 2013, 369, 1691–1703. [Google Scholar] [CrossRef] [PubMed]
- Vaccaro, V.; Sperduti, I.; Milella, M. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N. Engl. J. Med. 2011, 365, 768–769. [Google Scholar] [CrossRef] [PubMed]
- Bilimoria, K.Y.; Bentrem, D.J.; Ko, C.Y.; Tomlinson, J.S.; Stewart, A.K.; Winchester, D.P.; Talamonti, M.S. Multimodality therapy for pancreatic cancer in the U.S.: Utilization, outcomes, and the effect of hospital volume. Cancer 2007, 110, 1227–1234. [Google Scholar] [CrossRef] [PubMed]
- Sohal, D.P.S.; Walsh, R.M.; Ramanathan, R.K.; Khorana, A.A. Pancreatic adenocarcinoma: Treating a systemic disease with systemic therapy. J. Natl. Cancer Inst. 2014, 106, 10–13. [Google Scholar] [CrossRef] [PubMed]
- Bischoff, J.R.; Kirn, D.H.; Williams, A.; Heise, C.; Horn, S.; Muna, M.; Ng, L.; Nye, J.A.; Sampson-Johannes, A.; Fattaey, A.; et al. An Adenovirus Mutant That Replicates Selectively in p53- Deficient Human Tumor Cells. Science 1996, 274, 373–376. [Google Scholar] [CrossRef] [PubMed]
- Kawashima, T.; Kagawa, S.; Kobayashi, N.; Shirakiya, Y.; Umeoka, T.; Teraishi, F.; Taki, M.; Kyo, S.; Tanaka, N.; Fujiwara, T. Telomerase-specific replication-selective virotherapy for human cancer. Clin. Cancer Res. 2004, 10, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Huch, M.; Gros, A.; José, A.; González, J.R.; Alemany, R.; Fillat, C. Urokinase-type plasminogen activator receptor transcriptionally controlled adenoviruses eradicate pancreatic tumors and liver metastasis in mouse models. Neoplasia 2009, 11, 518–528. [Google Scholar] [CrossRef] [PubMed]
- Sobrevals, L.; Mato-Berciano, A.; Urtasun, N.; Mazo, A.; Fillat, C. uPAR-controlled oncolytic adenoviruses eliminate cancer stem cells in human pancreatic tumors. Stem Cell Res. 2014, 12, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, M.; Davydova, J.; Wang, M.; Siegal, G.P.; Krasnykh, V.; Vickers, S.M.; Curiel, D.T. Infectivity enhanced, cyclooxygenase-2 promoter-based conditionally replicative adenovirus for pancreatic cancer. Gastroenterology 2003, 125, 1203–1218. [Google Scholar] [CrossRef]
- Miura, Y.; Yamasaki, S.; Davydova, J.; Brown, E.; Aoki, K.; Vickers, S.; Yamamoto, M. Infectivity-selective oncolytic adenovirus developed by high-throughput screening of adenovirus-formatted library. Mol. Ther. 2013, 21, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Frierson, H.F.; Moskaluk, C.A.; Powell, S.M.; Zhang, H.; Cerilli, L.A.; Stoler, M.H.; Cathro, H.; Hampton, G.M.; Kojima, T.; Oh-eda, M.; et al. Large-scale molecular and tissue microarray analysis of mesothelin expression in common human carcinomas. Hum. Pathol. 2003, 34, 605–609. [Google Scholar] [CrossRef]
- Yamamoto, Y.; Goto, N.; Miura, K.; Narumi, K.; Ohnami, S.; Uchida, H.; Miura, Y.; Yamamoto, M.; Aoki, K. Development of a Novel Efficient Method To Construct an Adenovirus Library Displaying Random Peptides on the Fiber Knob. Mol. Pharm. 2014, 11, 1069–1074. [Google Scholar] [CrossRef] [PubMed]
- Andtbacka, R.H.I.; Kaufman, H.L.; Collichio, F.; Amatruda, T.; Senzer, N.; Chesney, J.; Delman, K.A.; Spitler, L.E.; Puzanov, I.; Agarwala, S.S.; et al. Talimogene Laherparepvec Improves Durable Response Rate in Patients With Advanced Melanoma. J. Clin. Oncol. 2015, 33, 2780–2788. [Google Scholar] [CrossRef] [PubMed]
- Mahalingam, D.; Patel, S.; Nuovo, G.; Gill, G.; Selvaggi, G.; Coffey, M.; Nawrocki, S.T. The combination of intravenous Reolysin and gemcitabine induces reovirus replication and endoplasmic reticular stress in a patient with KRAS-activated pancreatic cancer. BMC Cancer 2015, 15, 513. [Google Scholar] [CrossRef] [PubMed]
- Nakao, A.; Kasuya, H.; Sahin, T.T.; Nomura, N.; Kanzaki, A.; Misawa, M.; Shirota, T.; Yamada, S.; Fujii, T.; Sugimoto, H.; et al. A phase I dose-escalation clinical trial of intraoperative direct intratumoral injection of HF10 oncolytic virus in non-resectable patients with advanced pancreatic cancer. Cancer Gene Ther. 2011, 18, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Eissa, I.R.; Naoe, Y.; Bustos-Villalobos, I.; Ichinose, T.; Tanaka, M.; Zhiwen, W.; Mukoyama, N.; Morimoto, T.; Miyajima, N.; Hitoki, H.; et al. Genomic Signature of the Natural Oncolytic Herpes Simplex Virus HF10 and Its Therapeutic Role in Preclinical and Clinical Trials. Front. Oncol. 2017, 7, 149. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Garcia, A.; Gimenez-Alejandre, M.; Rojas, J.J.; Moreno, R.; Bazan-Peregrino, M.; Cascallo, M.; Alemany, R. Safety and Efficacy of VCN-01, an Oncolytic Adenovirus Combining Fiber HSG-Binding Domain Replacement with RGD and Hyaluronidase Expression. Clin. Cancer Res. 2015, 21, 1406–1418. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, E.; Milenova, I.; Wenthe, J.; Ståhle, M.; Leja-Jarblad, J.; Ullenhag, G.; Dimberg, A.; Moreno, R.; Alemany, R.; Loskog, A. Shaping the Tumor Stroma and Sparking Immune Activation by CD40 and 4-1BB Signaling Induced by an Armed Oncolytic Virus. Clin. Cancer Res. 2017, 23, 5846–5857. [Google Scholar] [CrossRef] [PubMed]
- Alberts, S.R.; Schroeder, M.; Erlichman, C.; Steen, P.D.; Foster, N.R.; Moore, D.F.; Rowland, K.M.; Nair, S.; Tschetter, L.K.; Fitch, T.R. Gemcitabine and ISIS-2503 for patients with locally advanced or metastatic pancreatic adenocarcinoma: A north central cancer treatment group phase II trial. J. Clin. Oncol. 2004, 22, 4944–4950. [Google Scholar] [CrossRef] [PubMed]
- Adjei, A.A.; Dy, G.K.; Erlichman, C.; Reid, J.M.; Sloan, J.A.; Pitot, H.C.; Alberts, S.R.; Goldberg, R.M.; Hanson, L.J.; Atherton, P.J.; et al. A phase I trial of ISIS 2503, an antisense inhibitor of H-ras, in combination with gemcitabine in patients with advanced cancer. Clin. Cancer Res. 2003, 9, 115–123. [Google Scholar] [PubMed]
- Mahadevan, D.; Chalasani, P.; Rensvold, D.; Kurtin, S.; Pretzinger, C.; Jolivet, J.; Ramanathan, R.K.; Von Hoff, D.D.; Weiss, G.J. Phase I Trial of AEG35156 an Antisense Oligonucleotide to XIAP Plus Gemcitabine in Patients With Metastatic Pancreatic Ductal Adenocarcinoma. Am. J. Clin. Oncol. 2013, 36, 239–243. [Google Scholar] [CrossRef] [PubMed]
- Aleku, M.; Schulz, P.; Keil, O.; Santel, A.; Schaeper, U.; Dieckhoff, B.; Janke, O.; Endruschat, J.; Durieux, B.; Röder, N.; et al. Atu027, a liposomal small interfering RNA formulation targeting protein kinase N3, inhibits cancer progression. Cancer Res. 2008, 68, 9788–9798. [Google Scholar] [CrossRef] [PubMed]
- Leenders, F.; Möpert, K.; Schmiedeknecht, A.; Santel, A.; Czauderna, F.; Aleku, M.; Penschuck, S.; Dames, S.; Sternberger, M.; Röhl, T.; et al. PKN3 is required for malignant prostate cell growth downstream of activated PI 3-kinase. EMBO J. 2004, 23, 3303–3313. [Google Scholar] [CrossRef] [PubMed]
- Golan, T.; Khvalevsky, E.Z.; Hubert, A.; Gabai, R.M.; Hen, N.; Segal, A.; Domb, A.; Harari, G.; David, E.B.; Raskin, S.; et al. RNAi therapy targeting KRAS in combination with chemotherapy for locally advanced pancreatic cancer patients. Oncotarget 2015, 6, 24560–24570. [Google Scholar] [CrossRef] [PubMed]
- Buscail, L.; Bournet, B.; Vernejoul, F.; Cambois, G.; Lulka, H.; Hanoun, N.; Dufresne, M.; Meulle, A.; Vignolle-Vidoni, A.; Ligat, L.; et al. First-in-man phase 1 clinical trial of gene therapy for advanced pancreatic cancer: Safety, biodistribution, and preliminary clinical findings. Mol. Ther. 2015, 23, 779–789. [Google Scholar] [CrossRef] [PubMed]
- Carrere, N.; Vernejoul, F.; Souque, A.; Asnacios, A.; Vaysse, N.; Pradayrol, L.; Susini, C.; Buscail, L.; Cordelier, P. Characterization of the bystander effect of somatostatin receptor sst2 after in vivo gene transfer into human pancreatic cancer cells. Hum. Gene Ther. 2005, 16, 1175–1193. [Google Scholar] [CrossRef] [PubMed]
- Hanna, N.; Ohana, P.; Konikoff, F.M.; Leichtmann, G.; Hubert, A.; Appelbaum, L.; Kopelman, Y.; Czerniak, A.; Hochberg, A. Phase 1/2a, dose-escalation, safety, pharmacokinetic and preliminary efficacy study of intratumoral administration of BC-819 in patients with unresectable pancreatic cancer. Cancer Gene Ther. 2012, 19, 374–381. [Google Scholar] [CrossRef] [PubMed]
- Senzer, N.; Nemunaitis, J.; Nemunaitis, D.; Bedell, C.; Edelman, G.; Barve, M.; Nunan, R.; Pirollo, K.F.; Rait, A.; Chang, E.H. Phase i study of a systemically delivered p53 nanoparticle in advanced solid tumors. Mol. Ther. 2013, 21, 1096–1103. [Google Scholar] [CrossRef] [PubMed]
- Killock, D. Skin cancer: T-VEC oncolytic viral therapy shows promise in melanoma. Nat. Rev. Clin. Oncol. 2015, 12, 438. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, M.; Curiel, D.T. Current issues and future directions of oncolytic adenoviruses. Mol. Ther. 2010, 18, 243–250. [Google Scholar] [CrossRef] [PubMed]
- Alemany, R.; Balagué, C.; Curiel, D.T. Replicative adenoviruses for cancer therapy. Nat. Biotechnol. 2000, 18, 723–727. [Google Scholar] [CrossRef] [PubMed]
- Liebert, M.A.; Hallenbeck, P.L.; Chang, Y.; Hay, C.; Golightly, D.; Art, D.S.; Lin, J.; Phipps, S.; Chiang, Y.L. A novel tumor-specific replication-restricted adenoviral vector for gene therapy of hepatocellular carcinoma. Hum. Gene Ther. 1999, 10, 1721–1733. [Google Scholar]
- Shenk, T. Adenoviridae: The viruses and their replication. In Virology; Fields, B., Knipe, D., Howley, P., Eds.; Lipponcott-Raven: Philadelphia, PA, USA, 1996; Volume 2, pp. 2111–2148. [Google Scholar]
- Rodriguez-Rocha, H.; Gomez-Gutierrez, J.G.; Garcia-Garcia, A.; Rao, X.-M.; Chen, L.; McMasters, K.M.; Zhou, H.S. Adenoviruses induce autophagy to promote virus replication and oncolysis. Virology 2011, 416, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Heise, C.; Hermiston, T.; Johnson, L.; Brooks, G.; Sampson-Johannes, A.; Williams, A.; Hawkins, L.; Kirn, D. An adenovirus E1A mutant that demonstrates potent and selective systemic anti-tumoral efficacy. Nat. Med. 2000, 6, 1134–1139. [Google Scholar] [CrossRef] [PubMed]
- Fueyo, J.; Gomez-Manzano, C.; Alemany, R.; Lee, P.S.; McDonnell, T.J.; Mitlianga, P.; Shi, Y.X.; Levin, V.A.; Yung, W.K.; Kyritsis, A.P. A mutant oncolytic adenovirus targeting the Rb pathway produces anti-glioma effect in vivo. Oncogene 2000, 19, 2–12. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Rao, X.-M.; Gomez-Gutierrez, J.G.; Hao, H.; McMasters, K.M.; Zhou, H.S. Adenovirus E1B55K region is required to enhance cyclin E expression for efficient viral DNA replication. J. Virol. 2008, 82, 3415–3427. [Google Scholar] [CrossRef] [PubMed]
- LaRocca, C.J.; Han, J.; Salzwedel, A.O.; Davydova, J.; Herzberg, M.C.; Gopalakrishnan, R.; Yamamoto, M. Oncolytic adenoviruses targeted to Human Papilloma Virus-positive head and neck squamous cell carcinomas. Oral Oncol. 2016, 56, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Davydova, J.; Le, L.P.; Gavrikova, T.; Wang, M.; Krasnykh, V.; Yamamoto, M. Infectivity-enhanced cyclooxygenase-2-based conditionally replicative adenoviruses for esophageal adenocarcinoma treatment. Cancer Res. 2004, 64, 4319–4327. [Google Scholar] [CrossRef] [PubMed]
- Kosaka, T.; Davydova, J.; Ono, H.A.; Akiyama, H.; Hirai, S.I.; Ohno, S.; Takeshita, F.; Aoki, K.; Ochiya, T.; Yamamoto, M.; et al. Imaging and antitumoral effect of a cyclo-oxygenase 2-specific replicative adenovirus for small metastatic gastric cancer lesions. Anticancer Res. 2015, 35, 5201–5210. [Google Scholar] [PubMed]
- Wesseling, J.G.; Bosma, P.J.; Krasnykh, V.; Kashentseva, E.A.; Blackwell, J.L.; Reynolds, P.N.; Li, H.; Parameshwar, M.; Vickers, S.M.; Jaffee, E.M.; et al. Improved gene transfer efficiency to primary and established human pancreatic carcinoma target cells via epidermal growth factor receptor and integrin-targeted adenoviral vectors. Gene Ther. 2001, 8, 969–976. [Google Scholar] [CrossRef] [PubMed]
- Krasnykh, V.; Dmitriev, I.; Mikheeva, G.; Miller, C.R.; Belousova, N.; Curiel, D.T. Characterization of an adenovirus vector containing a heterologous peptide epitope in the HI loop of the fiber knob. J. Virol. 1998, 72, 1844–1852. [Google Scholar] [PubMed]
- Dmitriev, I.; Kashentseva, E.; Rogers, B.E.; Krasnykh, V.; Curiel, D.T. Ectodomain of coxsackievirus and adenovirus receptor genetically fused to epidermal growth factor mediates adenovirus targeting to epidermal growth factor receptor-positive cells. J. Virol. 2000, 74, 6875–6884. [Google Scholar] [CrossRef] [PubMed]
- Yao, V.J.; Ozawa, M.G.; Varner, A.S.; Kasman, I.M.; Chanthery, Y.H.; Pasqualini, R.; Arap, W.; McDonald, D.M. Antiangiogenic Therapy Decreases Integrin Expression in Normalized Tumor Blood Vessels. Cancer Res. 2006, 2639–2649. [Google Scholar] [CrossRef] [PubMed]
- Ramírez, P.J.; Vickers, S.M.; Ono, H.A.; Davydova, J.; Takayama, K.; Thompson, T.C.; Curiel, D.T.; Bland, K.I.; Yamamoto, M. Optimization of conditionally replicative adenovirus for pancreatic cancer and its evaluation in an orthotopic murine xenograft model. Am. J. Surg. 2008, 195, 481–490. [Google Scholar] [CrossRef] [PubMed]
- Gaggar, A.; Shayakhmetov, D.M.; Liszewski, M.K.; Atkinson, J.P.; Lieber, A. Localization of regions in CD46 that interact with adenovirus. J. Virol. 2005, 79, 7503–7513. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sirena, D.; Lilienfeld, B.; Eisenhut, M.; Kälin, S.; Boucke, K.; Beerli, R.R.; Vogt, L.; Ruedl, C.; Bachmann, M.F.; Greber, U.F.; Hemmi, S. The human membrane cofactor CD46 is a receptor for species B adenovirus serotype 3. J. Virol. 2004, 78, 4454–4462. [Google Scholar] [CrossRef] [PubMed]
- Curiel, D.T. Strategies to adapt adenoviral vectors for targeted delivery. Gene Ther. 1999, 886, 158–171. [Google Scholar] [CrossRef]
- Tanaka, T. Carcinoembryonic Antigen-Targeted Selective Gene Therapy for Gastric Cancer through FZ33 Fiber-Modified Adenovirus Vectors. Clin. Cancer Res. 2006, 12, 3803–3813. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Douglas, J.T.; Rogers, B.E.; Rosenfeld, M.E.; Michael, S.I.; Feng, M.; Curiel, D.T. Targeted gene delivery by tropism-modified adenoviral vectors. Nat. Biotechnol. 1996, 14, 303–308. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wang, H.; Yumul, R.; Gao, W.; Gambotto, A.; Morita, T.; Baker, A.; Shayakhmetov, D.; Lieber, A. Transduction of liver metastases after intravenous injection of Ad5/35 or Ad35 vectors with and without factor X-binding protein pretreatment. Hum. Gene Ther. 2009, 20, 621–629. [Google Scholar] [CrossRef] [PubMed]
- Liikanen, I.; Monsurrò, V.; Ahtiainen, L.; Raki, M.; Hakkarainen, T.; Diaconu, I.; Escutenaire, S.; Hemminki, O.; Dias, J.D.; Cerullo, V.; et al. Induction of Interferon Pathways Mediates In Vivo Resistance to Oncolytic Adenovirus. Mol. Ther. 2011, 19, 1858–1866. [Google Scholar] [CrossRef] [PubMed]
- Koski, A.; Kangasniemi, L.; Escutenaire, S.; Pesonen, S.; Cerullo, V.; Diaconu, I.; Nokisalmi, P.; Raki, M.; Rajecki, M.; Guse, K.; et al. Treatment of cancer patients with a serotype 5/3 chimeric oncolytic adenovirus expressing GMCSF. Mol. Ther. 2010, 18, 1874–1884. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, I.; Harden, P.; Bauzon, M.; Chartier, C.; Nye, J.; Thorne, S.; Reid, T.; Ni, S.; Lieber, A.; Fisher, K.; et al. Directed evolution generates a novel oncolytic virus for the treatment of colon cancer. PLoS ONE 2008, 3, e2409. [Google Scholar] [CrossRef] [PubMed]
- Di, Y.; Seymour, L.; Fisher, K. Activity of a group B oncolytic adenovirus (ColoAd1) in whole human blood. Gene Ther. 2014, 21, 440–443. [Google Scholar] [CrossRef] [PubMed]
- Boni, V.; De La Portilla, F.; Cubillo, A.; Gil-Martin, M.; Calvo, E.; Salazar, R.; Santos, C.; Sanchez-Gastaldo, A.; Prados, S.; Sanjuan, X.; et al. 1068P A Phase 1 mechanism of action study of intra-tumoural (IT) or intravenous (IV) administration of enadenotucirev, an oncolytic Ad11/Ad3 chimeric group b adenovirus in colon cancer patients undergoing resection of primary tumour. Ann. Oncol. 2014, 25, iv368. [Google Scholar] [CrossRef][Green Version]
- Joung, I.; Harber, G.; Gerecke, K.M.; Carroll, S.L.; Collawn, J.F.; Engler, J.A. Improved gene delivery into neuroglial cells using a fiber-modified adenovirus vector. Biochem. Biophys. Res. Commun. 2005, 328, 1182–1187. [Google Scholar] [CrossRef] [PubMed]
- Nicklin, S. Ablating Adenovirus Type 5 Fiber–CAR Binding and HI Loop Insertion of the SIGYPLP Peptide Generate an Endothelial Cell-Selective Adenovirus. Mol. Ther. 2001, 4, 534–542. [Google Scholar] [CrossRef] [PubMed]
- Laakkonen, P.; Porkka, K.; Hoffman, J.A.; Ruoslahti, E. A tumor-homing peptide with a targeting specificity related to lymphatic vessels. Nat. Med. 2002, 751–755. [Google Scholar] [CrossRef] [PubMed]
- Nishimoto, T.; Yoshida, K.; Miura, Y.; Kobayashi, A.; Hara, H.; Ohnami, S.; Kurisu, K.; Yoshida, T.; Aoki, K. Oncolytic virus therapy for pancreatic cancer using the adenovirus library displaying random peptides on the fiber knob. Gene Ther. 2009, 16, 669–680. [Google Scholar] [CrossRef] [PubMed]
- Shashkova, E.V.; Kuppuswamy, M.N.; Wold, W.S.M.; Doronin, K. Anticancer activity of oncolytic adenovirus vector armed with IFN-α and ADP is enhanced by pharmacologically controlled expression of TRAIL. Cancer Gene Ther. 2008, 15, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Oh, J.Y.; Park, B.H.; Lee, D.E.; Kim, J.S.; Park, H.E.; Roh, M.S.; Je, J.E.; Yoon, J.H.; Thorne, S.H.; et al. Systemic Armed Oncolytic and Immunologic Therapy for Cancer with JX-594, a Targeted Poxvirus Expressing GM-CSF. Mol. Ther. 2006, 14, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Nukui, Y.; Picozzi, V.J.; Traverso, L.W. Interferon-based adjuvant chemoradiation therapy improves survival after pancreaticoduodenectomy for pancreatic adenocarcinoma. Am. J. Surg. 2000, 179, 367–371. [Google Scholar] [CrossRef]
- Schmidt, J.; Patrut, E.M.; Ma, J.; Jäger, D.; Knaebel, H.-P.; Büchler, M.W.; Märten, A. Immunomodulatory impact of interferon-α in combination with chemoradiation of pancreatic adenocarcinoma (CapRI). Cancer Immunol. Immunother. 2006, 55, 1396–1405. [Google Scholar] [CrossRef] [PubMed]
- Talpaz, M.; Chernajovsky, Y.; Troutman Worden, K.; Wetzler, M.; Kantarjian, H.; Gutterman, J.U.; Kurzrock, R. Interferon-stimulated genes in interferon-sensitive and -resistant chronic myelogenous leukemia patients. Cancer Res 1992, 52, 1087–1090. [Google Scholar] [PubMed]
- Holsti, L.R.; Mattson, K.; Niiranen, A.; Standertskiöld-Nordenstam, C.-G.; Stenman, S.; Sovijärvi, A.; Cantell, K. Enhancement of radiation effects by α interferon in the treatment of small cell carcinoma of the lung. Int. J. Radiat. Oncol. 1987, 13, 1161–1166. [Google Scholar] [CrossRef]
- Feig, C.; Gopinathan, A.; Neesse, A.; Chan, D.S.; Cook, N.; Tuveson, D.A. The pancreas cancer microenvironment. Clin. Cancer Res. 2012, 18, 4266–4276. [Google Scholar] [CrossRef] [PubMed]
- Rodallec, M.; Vilgrain, V.; Couvelard, A.; Rufat, P.; O’Toole, D.; Barrau, V.; Sauvanet, A.; Ruszniewski, P.; Menu, Y. Endocrine Pancreatic Tumours and Helical CT: Contrast Enhancement Is Correlated with Microvascular Density, Histoprognostic Factors and Survival. Pancreatology 2006, 6, 77–85. [Google Scholar] [CrossRef] [PubMed]
- LaRocca, C.J.; Han, J.; Gavrikova, T.; Armstrong, L.; Oliveira, A.R.; Shanley, R.; Vickers, S.M.; Yamamoto, M.; Davydova, J. Oncolytic adenovirus expressing interferon α in a syngeneic Syrian hamster model for the treatment of pancreatic cancer. Surgery 2015, 157, 888–898. [Google Scholar] [CrossRef] [PubMed]
- Cerullo, V.; Pesonen, S.; Diaconu, I.; Escutenaire, S.; Arstila, P.T.; Ugolini, M.; Nokisalmi, P.; Raki, M.; Laasonen, L.; Särkioja, M.; et al. Oncolytic adenovirus coding for granulocyte macrophage colony-stimulating factor induces antitumoral immunity in cancer patients. Cancer Res. 2010, 70, 4297–4309. [Google Scholar] [CrossRef] [PubMed]
- Poutou, J.; Bunuales, M.; Gonzalez-Aparicio, M.; Garcia-Aragoncillo, E.; Quetglas, J.I.; Casado, R.; Bravo-Perez, C.; Alzuguren, P.; Hernandez-Alcoceba, R. Safety and antitumor effect of oncolytic and helper-dependent adenoviruses expressing interleukin-12 variants in a hamster pancreatic cancer model. Gene Ther. 2015, 22, 696–706. [Google Scholar] [CrossRef] [PubMed]
- Loskog, A. Immunostimulatory gene therapy using oncolytic viruses as vehicles. Viruses 2015, 7, 5780–5791. [Google Scholar] [CrossRef] [PubMed]
- Reboul, F.; Serin, D.; Martin, D.P.F. Combination radiotherapy and chemotherapy in cancer of the pancreas. Review of the literature and prospects. Bull Cancer 1990, 77, 275–281. [Google Scholar] [PubMed]
- Noordhuis, M.G.; Eijsink, J.J.H.; Roossink, F.; de Graeff, P.; Pras, E.; Schuuring, E.; Wisman, G.B.A.; de Bock, G.H.; van der Zee, A.G.J. Prognostic Cell Biological Markers in Cervical Cancer Patients Primarily Treated With (Chemo)radiation: A Systematic Review. Int. J. Radiat. Oncol. 2011, 79, 325–334. [Google Scholar] [CrossRef] [PubMed]
- Whistance, R.N.; Blazeby, J.M. Systematic review: Quality of life after treatment for upper gastrointestinal cancer. Curr. Opin. Support. Palliat. Care 2011, 5, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Leitner, S.; Sweeney, K.; Oberg, D.; Davies, D.; Miranda, E.; Lemoine, N.R.; Hallden, G. Oncolytic Adenoviral Mutants with E1B19K Gene Deletions Enhance Gemcitabine-induced Apoptosis in Pancreatic Carcinoma Cells and Anti- Tumor Efficacy In vivo. Clin. Cancer Res. 2009, 15, 1730–1740. [Google Scholar] [CrossRef] [PubMed]
- Raki, M.; Kanerva, A.; Ristimaki, A.; Desmond, R.A.; Chen, D.-T.; Ranki, T.; Sarkioja, M.; Kangasniemi, L. Hemminki, a Combination of gemcitabine and Ad5/3-Delta24, a tropism modified conditionally replicating adenovirus, for the treatment of ovarian cancer. Gene Ther. 2005, 12, 1198–1205. [Google Scholar] [CrossRef] [PubMed]
- Dias, J.D.; Hemminki, O.; Diaconu, I.; Hirvinen, M.; Bonetti, A.; Guse, K.; Escutenaire, S.; Kanerva, A.; Pesonen, S.; Löskog, A.; et al. Targeted cancer immunotherapy with oncolytic adenovirus coding for a fully human monoclonal antibody specific for CTLA-4. Gene Ther. 2012, 19, 988–998. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Kanasty, R.L.; Eltoukhy, A.A.; Vegas, A.J.; Dorkin, J.R.; Anderson, D.G. Non-viral vectors for gene-based therapy. Nat. Rev. Genet. 2014, 15, 541–555. [Google Scholar] [CrossRef] [PubMed]
- Dowdy, S.F. Overcoming cellular barriers for RNA therapeutics. Nat. Biotechnol. 2017, 35, 222–229. [Google Scholar] [CrossRef] [PubMed]
- Schultheis, B.; Strumberg, D.; Santel, A.; Vank, C.; Gebhardt, F.; Keil, O.; Lange, C.; Giese, K.; Kaufmann, J.; Khan, M.; et al. First-in-human phase I study of the liposomal RNA interference therapeutic Atu027 in patients with advanced solid tumors. J. Clin. Oncol. 2014, 32, 4141–4148. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Kim, A.; Miyata, K.; Kataoka, K. Recent progress in development of siRNA delivery vehicles for cancer therapy. Adv. Drug Deliv. Rev. 2016, 104, 61–77. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Shigdar, S.; Shamaileh, H.A.; Gantier, M.P.; Yin, W.; Xiang, D.; Wang, L.; Zhou, S.F.; Hou, Y.; Wang, P.; et al. Challenges and opportunities for siRNA-based cancer treatment. Cancer Lett. 2017, 387, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Jinek, M.; Doudna, J.A. A three-dimensional view of the molecular machinery of RNA interference. Nature 2009, 457, 405–412. [Google Scholar] [CrossRef] [PubMed]
- Barata, P.; Sood, A.K.; Hong, D.S. RNA-targeted therapeutics in cancer clinical trials: Current status and future directions. Cancer Treat. Rev. 2016, 50, 35–47. [Google Scholar] [CrossRef] [PubMed]
- Ying, H.; Dey, P.; Yao, W.; Kimmelman, A.C.; Draetta, G.F.; Maitra, A.; DePinho, R.A. Genetics and biology of pancreatic ductal adenocarcinoma. Genes Dev. 2016, 30, 355–385. [Google Scholar] [CrossRef] [PubMed]
- Cabral, H.; Matsumoto, Y.; Mizuno, K.; Chen, Q.; Murakami, M.; Kimura, M.; Terada, Y.; Kano, M.R.; Miyazono, K.; Uesaka, M.; et al. Accumulation of sub-100 nm polymeric micelles in poorly permeable tumours depends on size. Nat. Nanotechnol. 2011, 6, 815–823. [Google Scholar] [CrossRef] [PubMed]
- Khvorova, A.; Watts, J.K. The chemical evolution of oligonucleotide therapies of clinical utility. Nat. Biotechnol. 2017, 35, 238–248. [Google Scholar] [CrossRef] [PubMed]
- Choi, M.; Bien, H.; Mofunanya, A.; Powers, S. Challenges in Ras therapeutics in pancreatic cancer. Semin. Cancer Biol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Cao, Z.; Yan, H. Coexistence of High Levels of Apoptotic Signaling and Inhibitor of Apoptosis Proteins in Human Tumor Cells. Cancer Res. 2003, 63, 6815–6824. [Google Scholar] [PubMed]
- Mohammed, A.; Janakiram, N.B.; Pant, S.; Rao, C.V. Molecular targeted intervention for pancreatic cancer. Cancers (Basel) 2015, 7, 1499–1542. [Google Scholar] [CrossRef] [PubMed]
- Zorde Khvalevsky, E.; Gabai, R.; Rachmut, I.H.; Horwitz, E.; Brunschwig, Z.; Orbach, A.; Shemi, A.; Golan, T.; Domb, A.J.; Yavin, E.; et al. Mutant KRAS is a druggable target for pancreatic cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 20723–20728. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Hannon, G.J. MicroRNAs: Small RNAs with a big role in gene regulation. Nat. Rev. Genet. 2004, 5, 522–531. [Google Scholar] [CrossRef] [PubMed]
- Merhautova, J.; Demlova, R.; Slaby, O. MicroRNA-based therapy in animal models of selected gastrointestinal cancers. Front. Pharmacol. 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Karmakar, S.; Kaushik, G.; Nimmakayala, R.; Rachagani, S.; Ponnusamy, M.P.; Batra, S.K. MicroRNA regulation of K-Ras in pancreatic cancer and opportunities for therapeutic intervention. Semin. Cancer Biol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Scholz, C.; Wagner, E. Therapeutic plasmid DNA versus siRNA delivery: Common and different tasks for synthetic carriers. J. Control. Release 2012, 161, 554–565. [Google Scholar] [CrossRef] [PubMed]
- Amrutkar, M.; Gladhaug, I.P. Pancreatic cancer chemoresistance to gemcitabine. Cancers (Basel) 2017, 9, 157. [Google Scholar] [CrossRef] [PubMed]
- Buscail, L.; Saint-Laurent, N.; Chastre, E.; Vaillant, J.C.; Gespach, C.; Capella, G.; Kalthoff, H.; Lluis, F.; Vaysse, N.; Susini, C. Loss of sst2 somatostatin receptor gene expression in human pancreatic and colorectal cancer. Cancer Res. 1996, 56, 1823–1827. [Google Scholar] [PubMed]
- Ohhashi, S.; Ohuchida, K.; Mizumoto, K.; Fujita, H.; Egami, T.; Yu, J.; Toma, H.; Sadatomi, S.; Nagai, E.; Tanaka, M. Down-regulation of deoxycytidine kinase enhances acquired resistance to gemcitabine in pancreatic cancer. Anticancer Res. 2008, 28, 2205–2212. [Google Scholar] [PubMed]
- Ohana, P.; Bibi, O.; Matouk, I.; Levy, C.; Birman, T.; Ariel, I.; Schneider, T.; Ayesh, S.; Giladi, H.; Laster, M.; et al. Use of H19 regulatory sequences for targeted gene therapy in cancer. Int. J. Cancer 2002, 98, 645–650. [Google Scholar] [CrossRef] [PubMed]
- Ventura, A.; Kirsch, D.G.; McLaughlin, M.E.; Tuveson, D.A.; Grimm, J.; Lintault, L.; Newman, J.; Reczek, E.E.; Weissleder, R.; Jacks, T. Restoration of p53 function leads to tumour regression in vivo. Nature 2007, 445, 661–665. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Tang, W.-H.; Huang, C.-C.; Alexander, W.; Xiang, L.-M.; Pirollo, K.F.; Rait, A.; Chang, E.H. Systemic p53 Gene Therapy of Cancer with Immunolipoplexes Targeted by Anti-Transferrin Receptor scFv. Mol. Med. 2001, 7, 723–734. [Google Scholar] [PubMed]
- Deltcheva, E.; Chylinski, K.; Sharma, C.M.; Gonzales, K. CRISPR RNA maturation by trans -encoded small RNA and host factor RNase III. Nature 2011, 471, 602–607. [Google Scholar] [CrossRef] [PubMed]
- Maresch, R.; Mueller, S.; Veltkamp, C.; Öllinger, R.; Friedrich, M.; Heid, I.; Steiger, K.; Weber, J.; Engleitner, T.; Barenboim, M.; et al. Multiplexed pancreatic genome engineering and cancer induction by transfection-based CRISPR/Cas9 delivery in mice. Nat. Commun. 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Vorvis, C.; Hatziapostolou, M.; Mahurkar-Joshi, S.; Koutsioumpa, M.; Williams, J.; Donahue, T.R.; Poultsides, G.A.; Eibl, G.; Iliopoulos, D. Transcriptomic and CRISPR/Cas9 technologies reveal FOXA2 as a tumor suppressor gene in pancreatic cancer. Am. J. Physiol. Gastrointest. Liver Physiol. 2016, 310, G1124–G1137. [Google Scholar] [CrossRef] [PubMed]
- Cyranoski, D. CRISPR gene editing tested in a person. Nature 2016, 539, 479. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Zhang, L.; Liu, H.; Cheng, K. Delivery strategies of the CRISPR-Cas9 gene-editing system for therapeutic applications. J. Control. Release 2017, 266, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Glass, Z.; Lee, M.; Li, Y.; Xu, Q. Engineering the Delivery System for CRISPR-Based Genome Editing. Trends Biotechnol. 2018, 36, 173–185. [Google Scholar] [CrossRef] [PubMed]
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting cancer incidence and deaths to 2030: The unexpected burden of thyroid, liver, and pancreas cancers in the united states. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Name | Vector/Delivery System | Route of Delivery * | References |
|---|---|---|---|
| Virus | |||
| ONYX-015 | Conditionally replicative adenovirus (CRAd) mutant dl1520, lacking E1B region | IV | [9] |
| Mechanism: Selective replication in cancer cells with mutated p53 | |||
| OBP-301 | CRAd—E1A-mutation type | PC | [10] |
| Mechanism: Expresses E1A under the control of the human telomerase reverse transcriptase (hTERT) promoter | |||
| AduPARE1A | CRAd—E1A-mutation type | IV | [11,12] |
| Mechanism: Expresses E1A gene under the control of the urokinase-type plasminogen activator receptor (uPAR) promoter | |||
| Cox2CRAd | CRAd—E1A-mutation type | IT | [13] |
| Mechanism: OAd controlled by cyclooxygenase-2 | |||
| MSLN-targeted OAd | Targeted oncolytic adenovirus (OAd) | IV | [14,15] |
| Mechanism: Selectivity for MSLN-expressing pancreatic cancer cells | |||
| AdSur-SYE | Mechanism: Promoter-controlled pancreatic cancer-targeted OAd. | IT | [16] |
| Mehcanism: Displays the targeting sequence on the fiber knob of survivin promoter | |||
| T-VEC | Herpes simplex virus expressing GM-CSF | IT | [17] |
| Mechanism: Sensitize the tumoricidal effects of chemotherapeutic agents (e.g., 5-FU) and radiotherapy | |||
| Reolysin | Unmodified oncolytic reovirus | IV | [18] |
| Mechanism: Replication in Ras-activated cancer cells, trial in combination with gemcitabine | |||
| HF10 | Unmodified oncolytic herpes simplex virus | IT | [19,20] |
| Mechanism: Selective replication in cancer cells | |||
| VCN-01 | Replication-competent adenovirus | IT | [21] |
| Mechanism: Selective replication in cancer cells with defective RB pathway, hyaluronidase expressing | |||
| LOAd703 | Immunostimulatory adenovirus, trimerized CD40L and 4-1BBL | IT | [22] |
| Mechanism: Activates the CD40 and 4-1BB pathways | |||
| RNA | |||
| ISIS-2503 | Antisense oligonucleotide inhibitor of H-ras | IV | [23,24] |
| AEG35156 | Antisense oligonucleotide targeting X-linked inhibitor of apoptosis (XIAP) | IV | [25] |
| ATu027 | siRNA targeting protein kinase 3 (PKN3) mRNA utilizing a liposomal complex (AtuPLEX) carrier | IV | [26,27] |
| si-G12D-LODER | siRNA drug targeted mutant KRAS, utilizing biodegradable polymeric matrix | IT | [28] |
| DNA | |||
| CYL-02 | Plasmid DNA encoding for somatostatin receptor subtype 2 (SSTR2), deoxycytidine kinase (DCK), and uridylate monophosphate kinase (UMK) | IT | [29,30] |
| BC-819/DTA-H19 | Plasmid DNA encoding the diphtheria toxin-A chain under the regulator of the H19 promoter | IT | [31] |
| SGT-53 | Plasmid DNA encoding normal human wild-type p53 utilizing cationic liposome carrier | IV | [32] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sato-Dahlman, M.; Wirth, K.; Yamamoto, M. Role of Gene Therapy in Pancreatic Cancer—A Review. Cancers 2018, 10, 103. https://doi.org/10.3390/cancers10040103
Sato-Dahlman M, Wirth K, Yamamoto M. Role of Gene Therapy in Pancreatic Cancer—A Review. Cancers. 2018; 10(4):103. https://doi.org/10.3390/cancers10040103
Chicago/Turabian StyleSato-Dahlman, Mizuho, Keith Wirth, and Masato Yamamoto. 2018. "Role of Gene Therapy in Pancreatic Cancer—A Review" Cancers 10, no. 4: 103. https://doi.org/10.3390/cancers10040103
APA StyleSato-Dahlman, M., Wirth, K., & Yamamoto, M. (2018). Role of Gene Therapy in Pancreatic Cancer—A Review. Cancers, 10(4), 103. https://doi.org/10.3390/cancers10040103