Epigenetic Modifications as Biomarkers of Tumor Development, Therapy Response, and Recurrence across the Cancer Care Continuum


Abstract
1. Introduction
1.1. DNA Methylation
1.2. Post-Translational Histone Modifications
1.3. Non-Coding RNAs
2. Risk Factors and Screening Strategies
2.1. Breast Cancer
2.2. Nasopharyngeal Carcinoma
2.3. Lung Cancer
3. Diagnostic Biomarkers
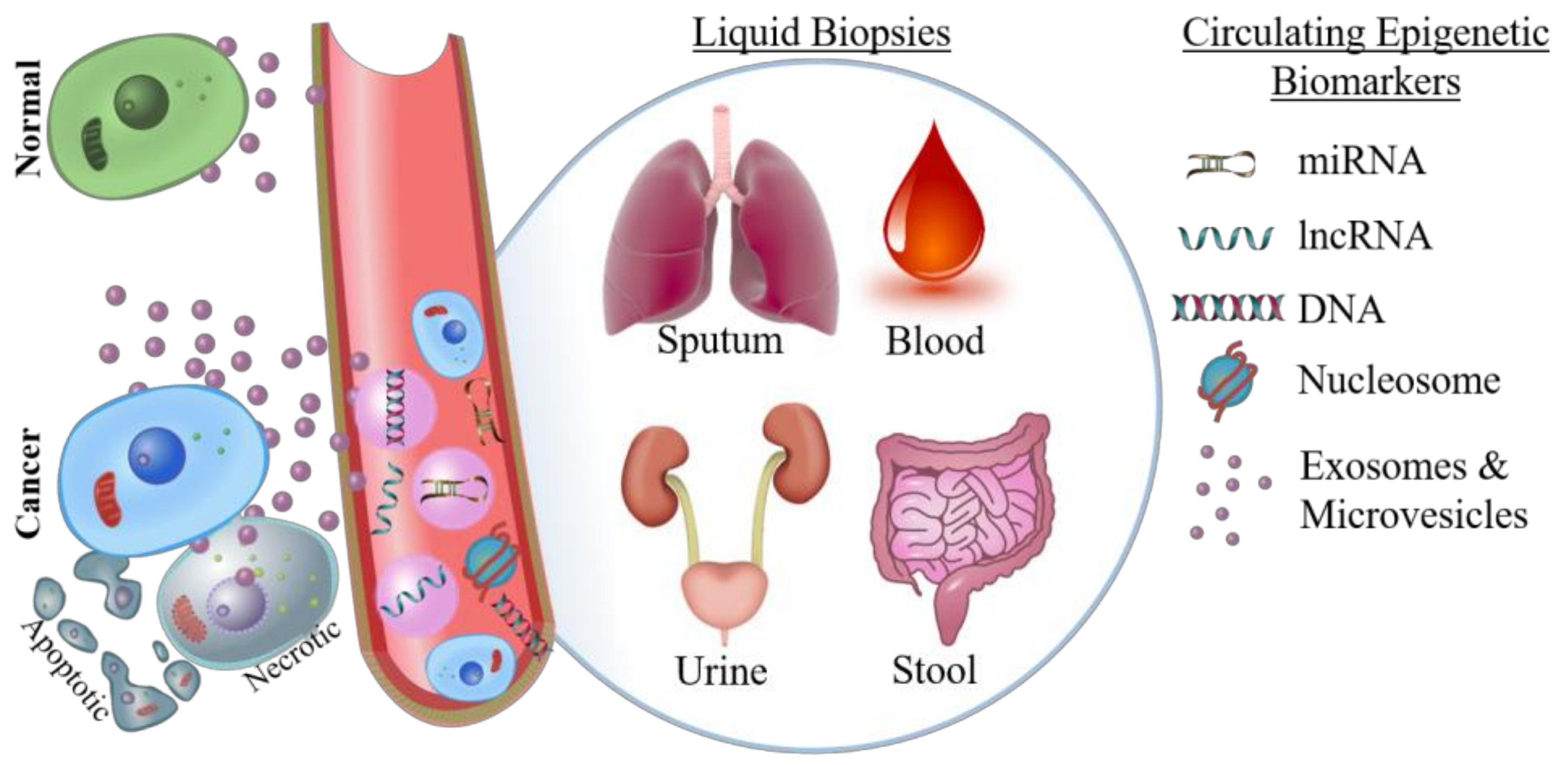
3.1. Need for Liquid Biopsies
3.2. Detecting Circulating Nucleotides and Nucleosomes
3.3. Colorectal Cancer
3.4. Circulating Gene-Specific Promoter Hypermethylation
3.5. Circulating Non-Coding RNAs
3.6. Circulating Histone Modifications
4. Epigenetic Markers Informing Therapeutic Decision-Making
4.1. Epigenetic Prognostic Markers
4.2. Anticipating the Benefit of Adjuvant Therapy
4.3. Epigenetic Biomarkers that Predict Response to Specific Therapies
5. Epigenetic Biomarkers to Monitor Patient Responses
6. Future Directions
Acknowledgments
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A.; Baylin, S.B. The fundamental role of epigenetic events in cancer. Nat. Rev. Genet. 2002, 3, 415. [Google Scholar] [CrossRef] [PubMed]
- Holliday, R. Epigenetics: A historical overview. Epigenetics 2006, 1, 76–80. [Google Scholar] [CrossRef] [PubMed]
- Johnson, T.B.; Coghill, R.D. Researches on pyrimidines. C111. The discovery of 5-methyl-cytosine in tuberculinic acid, the nucleic acid of the Tubercle bacillus. J. Am. Chem. Soc. 1925, 47, 2838–2844. [Google Scholar] [CrossRef]
- Bird, A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002, 16, 6–21. [Google Scholar] [CrossRef] [PubMed]
- Han, H.; Cortez, C.C.; Yang, X.; Nichols, P.W.; Jones, P.A.; Liang, G. DNA methylation directly silences genes with non-CpG island promoters and establishes a nucleosome occupied promoter. Hum. Mol. Genet. 2011, 20, 4299–4310. [Google Scholar] [CrossRef] [PubMed]
- Larsen, F.; Gundersen, G.; Lopez, R.; Prydz, H. CpG islands as gene markers in the human genome. Genomics 1992, 13, 1095–1107. [Google Scholar] [CrossRef]
- Illingworth, R.S.; Bird, A.P. CpG islands—‘A rough guide’. FEBS Lett. 2009, 583, 1713–1720. [Google Scholar] [CrossRef] [PubMed]
- Ehrlich, M. DNA hypomethylation in cancer cells. Epigenomics 2009, 1, 239–259. [Google Scholar] [CrossRef] [PubMed]
- Sheaffer, K.L.; Elliott, E.N.; Kaestner, K.H. DNA hypomethylation contributes to genomic instability and intestinal cancer initiation. Cancer Prev. Res. 2016, 9, 534–546. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; James, S.R.; Kazim, L.; Karpf, A.R. Specific method for the determination of genomic DNA methylation by liquid chromatography-electrospray ionization tandem mass spectrometry. Anal. Chem. 2005, 77, 504–510. [Google Scholar] [CrossRef] [PubMed]
- Yang, A.S.; Estécio, M.R.H.; Doshi, K.; Kondo, Y.; Tajara, E.H.; Issa, J.P.J. A simple method for estimating global DNA methylation using bisulfite PCR of repetitive DNA elements. Nucleic Acids Res. 2004, 32, e38. [Google Scholar] [CrossRef] [PubMed]
- Karimi, M.; Johansson, S.; Ekström, T.J. Using LUMA: A luminometric-based assay for global DNA-methylation. Epigenetics 2006, 1, 45–48. [Google Scholar] [PubMed]
- Tost, J.; Gut, I.G. DNA methylation analysis by pyrosequencing. Nat. Protoc. 2007, 2, 2265–2275. [Google Scholar] [CrossRef] [PubMed]
- Soto, J.; Rodriguez-Antolin, C.; Vallespín, E.; Carpeño, J.D.; de Caceres, I.I. The impact of next-generation sequencing on the DNA methylation–based translational cancer research. Transl. Res. 2016, 169, 1–18.e1. [Google Scholar] [CrossRef] [PubMed]
- Pidsley, R.; Zotenko, E.; Peters, T.J.; Lawrence, M.G.; Risbridger, G.P.; Molloy, P.; van Djik, S.; Muhlhausler, B.; Stirzaker, C.; Clark, S.J. Critical evaluation of the Illumina MethylationEPIC BeadChip microarray for whole-genome DNA methylation profiling. Genome Biol. 2016, 17, 208. [Google Scholar] [CrossRef] [PubMed]
- Herman, J.G.; Graff, J.R.; Myöhänen, S.; Nelkin, B.D.; Baylin, S.B. Methylation-specific PCR: A novel PCR assay for methylation status of CpG islands. Proc. Natl. Acad. Sci. USA 1996, 93, 9821–9826. [Google Scholar] [CrossRef] [PubMed]
- Bednar, J.; Horowitz, R.A.; Grigoryev, S.A.; Carruthers, L.M.; Hansen, J.C.; Koster, A.J.; Woodcock, C.L. Nucleosomes, linker DNA, and linker histone form a unique structural motif that directs the higher-order folding and compaction of chromatin. Proc. Natl. Acad. Sci. USA 1998, 95, 14173–14178. [Google Scholar] [CrossRef] [PubMed]
- Davie, J.R.; Chadee, D.N. Regulation and regulatory parameters of histone modifications. J. Cell. Biochem. Suppl. 1998, 30–31, 203–213. [Google Scholar] [CrossRef]
- Taverna, S.D.; Li, H.; Ruthenburg, A.J.; Allis, C.D.; Patel, D.J. How chromatin-binding modules interpret histone modifications: Lessons from professional pocket pickers. Nat. Struct. Mol. Biol. 2007, 1025–1040. [Google Scholar] [CrossRef] [PubMed]
- Kouzarides, T. Chromatin modifications and their function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Cooper, S.; Brockdorff, N. The interplay of histone modifications—Writers that read. EMBO Rep. 2015, 16, 1467–1481. [Google Scholar] [CrossRef] [PubMed]
- Horikoshi, N.; Kumar, P.; Sharma, G.G.; Chen, M.; Hunt, C.R.; Westover, K.; Chowdhury, S.; Pandita, T.K. Genome-wide distribution of histone H4 Lysine 16 acetylation sites and their relationship to gene expression. Genome Integr. 2013, 4, 3. [Google Scholar] [CrossRef] [PubMed]
- Taylor, G.C.A.; Eskeland, R.; Hekimoglu-Balkan, B.; Pradeepa, M.M.; Bickmore, W.A. H4K16 acetylation marks active genes and enhancers of embryonic stem cells, but does not alter chromatin compaction. Genome Res. 2013, 23, 2053–2065. [Google Scholar] [CrossRef] [PubMed]
- Kurdistani, S.K. Histone modifications in cancer biology and prognosis. Prog. Drug Res. 2011, 67, 91–106. [Google Scholar] [PubMed]
- Fraga, M. F.; Ballestar, E.; Villar-Garea, A.; Boix-Chornet, M.; Espada, J.; Schotta, G.; Bonaldi, T.; Haydon, C.; Ropero, S.; Petrie, K. Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nat. Genet. 2005, 37, 391–400. [Google Scholar] [CrossRef] [PubMed]
- McAnema, P.; Brown, J.A.L.; Kerin, M.J. Circulating nucleosomes and nucleosome modifications as biomarkers in cancer. Cancers (Basel) 2017, 9, 5. [Google Scholar] [CrossRef] [PubMed]
- Doyen, C.-M.; Montel, F.; Gautier, T.; Menoni, H.; Claudet, C.; Delacour-Larose, M.; Angelov, D.; Hamiche, A.; Bednar, J.; Faivre-Moskalenko, C.; et al. Dissection of the unusual structural and functional properties of the variant H2A.Bbd nucleosome. EMBO J. 2006, 25, 4234–4244. [Google Scholar] [CrossRef] [PubMed]
- Buschbeck, M.; Hake, S.B. Variants of core histones and their roles in cell fate decisions, development and cancer. Nat. Rev. Mol. Cell Biol. 2017, 18, 299–314. [Google Scholar] [CrossRef] [PubMed]
- Hua, S.; Kallen, C.B.; Dhar, R.; Baquero, M.T.; Mason, C.E.; Russell, B.A.; Shah, P.K.; Liu, J.; Khramtsov, A.; Tretiakova, M.S. Genomic analysis of estrogen cascade reveals histone variant H2A.Z associated with breast cancer progression. Mol. Syst. Biol. 2008, 4, 188. [Google Scholar] [CrossRef] [PubMed]
- Vardabasso, C.; Gaspar-Maia, A.; Hasson, D.; Pünzeler, S.; Valle-Garcia, D.; Straub, T.; Keilhauer, E.C.; Strub, T.; Dong, J.; Panda, T.; et al. Histone variant H2A.Z.2 mediates proliferation and drug sensitivity of malignant melanoma. Mol. Cell 2015, 59, 75–88. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.-H.; Miyai, K.; Sporn, J.C.; Luo, L.; Wang, J.Y.J.; Cosman, B.; Ramamoorthy, S. Loss of histone variant macroH2A2 expression associates with progression of anal neoplasm. J. Clin. Pathol. 2016, 69, 627–631. [Google Scholar] [CrossRef] [PubMed]
- Schwartzentruber, J.; Korshunov, A.; Liu, X.-Y.; Jones, D.T.W.; Pfaff, E.; Jacob, K.; Sturm, D.; Fontebasso, A.M.; Quang, D.-A.K.; Tönjes, M.; et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 2012, 482, 226–231. [Google Scholar] [CrossRef] [PubMed]
- Gessi, M.; Gielen, G.H.; Hammes, J.; Dörner, E.; Mühlen, A.Z.; Waha, A.; Pietsch, T. H3.3 G34R mutations in pediatric primitive neuroectodermal tumors of central nervous system (CNS-PNET) and pediatric glioblastomas: Possible diagnostic and therapeutic implications? J. Neurooncol. 2013, 112, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zang, C.; Rosenfeld, J.A.; Schones, D.E.; Barski, A.; Cuddapah, S.; Cui, K.; Roh, T.-Y.; Peng, W.; Zhang, M.Q.; et al. Combinatorial patterns of histone acetylations and methylations in the human genome. Nat. Genet. 2008, 40, 897–903. [Google Scholar] [CrossRef] [PubMed]
- Dai, B.; Giardina, C.; Rasmussen, T.P. Quantitation of nucleosome acetylation and other histone posttranslational modifications using microscale NU-ELISA. Methods Mol. Biol. 2013, 981, 167–176. [Google Scholar] [PubMed]
- Amaral, P.P.; Dinger, M.E.; Mercer, T.R.; Mattick, J.S. The eukaryotic genome as an RNA machine. Science 2008, 319, 1787–1789. [Google Scholar] [CrossRef] [PubMed]
- Gibb, E.A.; Brown, C.J.; Lam, W.L. The functional role of long non-coding RNA in human carcinomas. Mol. Cancer 2011, 10, 38. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Getz, G.; Miska, E.A.; Alvarez-Saavedra, E.; Lamb, J.; Peck, D.; Sweet-Cordero, A.; Ebert, B.L.; Mak, R.H.; Ferrando, A.A.; et al. MicroRNA expression profiles classify human cancers. Nature 2005, 435, 834–838. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.C.; Chang, H.Y. Molecular mechanisms of long noncoding RNAs. Mol. Cell 2011, 43, 904–914. [Google Scholar] [CrossRef] [PubMed]
- Carthew, R.W.; Sontheimer, E.J. Origins and mechanisms of miRNAs and siRNAs. Cell 2009, 136, 642–655. [Google Scholar] [CrossRef] [PubMed]
- Fabris, L.; Calin, G.A. Circulating free xeno-microRNAs—The new kids on the block. Mol. Oncol. 2016, 10, 503–508. [Google Scholar] [CrossRef] [PubMed]
- Haemmerle, M.; Gutschner, T. Long non-coding RNAs in cancer and development: Where do we go from here? Int. J. Mol. Sci. 2015, 16, 1395–1405. [Google Scholar] [CrossRef] [PubMed]
- Szeto, C.Y.-Y.; Lin, C.H.; Choi, S.C.; Yip, T.T.C.; Ngan, R.K.-C.; Tsao, G.S.-W.; Lung, M.L. Integrated mRNA and microRNA transcriptome sequencing characterizes sequence variants and mRNA-microRNA regulatory network in nasopharyngeal carcinoma model systems. FEBS Open Bio 2014, 4, 128–140. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xue, S.; Liu, X.; Liu, H.; Hu, T.; Qiu, X.; Zhang, J.; Lei, M. Analyses of long non-coding RNA and mRNA profiling using RNA sequencing during the pre-implantation phases in pig endometrium. Sci. Rep. 2016, 6, 20238. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.G.; Calin, G.A.; Volinia, S.; Croce, C.M. MicroRNA expression profiling using microarrays. Nat. Protoc. 2008, 3, 563–578. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Shang, J. Long noncoding RNA expression profiling using Arraystar LncRNA microarrays. Methods Mol. Biol. 2016, 1402, 43–61. [Google Scholar] [PubMed]
- Mitchell, P.S.; Parkin, R.K.; Kroh, E.M.; Fritz, B.R.; Wyman, S.K.; Pogosova-Agadjanyan, E.L.; Peterson, A.; Noteboom, J.; O’Briant, K.C.; Allen, A.; et al. Circulating microRNAs as stable blood-based markers for cancer detection. Proc. Natl. Acad. Sci. USA 2008, 105, 10513–10518. [Google Scholar] [CrossRef] [PubMed]
- Reeves, G.K.; Pirie, K.; Beral, V.; Green, J.; Spencer, E.; Bull, D. Cancer incidence and mortality in relation to body mass index in the million women study: Cohort study. BMJ Br. Med. J. 2007, 335, 1134–1139. [Google Scholar] [CrossRef] [PubMed]
- Calle, E.E.; Rodriguez, C.; Walker-Thurmond, K.; Thun, M.J. Overweight, obesity, and mortality from cancer in a prospectively studied cohort of U.S. adults. N. Engl. J. Med. 2003, 348, 1625–1638. [Google Scholar] [CrossRef] [PubMed]
- Giovannucci, E.; Ascherio, A.; Rimm, E.B.; Colditz, G.A.; Stampfer, M.J.; Willett, W.C. Physical activity, obesity, and risk for colon cancer and adenoma in men. Ann. Intern. Med. 1995, 122, 327. [Google Scholar] [CrossRef] [PubMed]
- Smith-Warner, S.A.; Spiegelman, D.; Yaun, S.-S.; van den Brandt, P.A.; Folsom, A.R.; Goldbohm, R.A.; Graham, S.; Holmberg, L.; Howe, G.R.; Marshall, J.R.; et al. Alcohol and breast cancer in women. JAMA 1998, 279, 535. [Google Scholar] [CrossRef] [PubMed]
- Johansson, A.; Flanagan, J.M. Epigenome-wide association studies for breast cancer risk and risk factors. Trends Cancer Res. 2017, 12, 19–28. [Google Scholar] [PubMed]
- Kuhl, C.K.; Schrading, S.; Leutner, C.C.; Morakkabati-Spitz, N.; Wardelmann, E.; Fimmers, R.; Kuhn, W.; Schild, H.H. Mammography, breast ultrasound, and magnetic resonance imaging for surveillance of women at high familial risk for breast cancer. J. Clin. Oncol. 2005, 23, 8469–8676. [Google Scholar] [CrossRef] [PubMed]
- Richert-Boe, K.E.; Humphrey, L.L. Screening for cancers of the cervix and breast. Arch. Intern. Med. 1992, 152, 2405–2411. [Google Scholar] [CrossRef] [PubMed]
- Locke, I.; Kote-Jarai, Z.; Fackler, M.J.; Bancroft, E.; Osin, P.; Nerurkar, A.; Izatt, L.; Pichert, G.; Gui, G.P.; Eeles, R.A. Gene promoter hypermethylation in ductal lavage fluid from healthy BRCA gene mutation carriers and mutation-negative controls. Breast Cancer Res. 2007, 9, R20. [Google Scholar] [CrossRef] [PubMed]
- Bean, G.R.; Bryson, A.D.; Pilie, P.G.; Goldenberg, V.; Baker, J.C.; Ibarra, C.; Brander, D.M.U.; Paisie, C.; Case, N.R.; Gauthier, M.; et al. Morphologically normal-appearing mammary epithelial cells obtained from high-risk women exhibit methylation silencing of INK4a/ARF. Clin. Cancer Res. 2007, 13, 6834–6841. [Google Scholar] [CrossRef] [PubMed]
- Kazarian, A.; Blyuss, O.; Metodieva, G.; Gentry-Maharaj, A.; Ryan, A.; Kiseleva, E.M.; Prytomanova, O.M.; Jacobs, I.J.; Widschwendter, M.; Menon, U.; et al. Testing breast cancer serum biomarkers for early detection and prognosis in pre-diagnosis samples. Br. J. Cancer 2017, 116, 501–508. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Bolick, S.C.E.; DeRoo, L.A.; Weinberg, C.R.; Sandler, D.P.; Taylor, J.A. Epigenome-wide association study of breast cancer using prospectively collected sister study samples. JNCI J. Natl. Cancer Inst. 2013, 105, 694–700. [Google Scholar] [CrossRef] [PubMed]
- Anjum, S.; Fourkala, E.-O.; Zikan, M.; Wong, A.; Gentry-Maharaj, A.; Jones, A.; Hardy, R.; Cibula, D.; Kuh, D.; Jacobs, I.J.; et al. A BRCA1-mutation associated DNA methylation signature in blood cells predicts sporadic breast cancer incidence and survival. Genome Med. 2004, 6, 47. [Google Scholar] [CrossRef] [PubMed]
- Taslim, C.; Weng, D.Y.; Brasky, T.M.; Dumitrescu, R.G.; Huang, K.; Kallakury, B.V.S.; Krishnan, S.; Llanos, A.A.; Marian, C.; McElroy, J.; et al. Discovery and replication of microRNAs for breast cancer risk using genome-wide profiling. Oncotarget 2016, 7, 86457–86468. [Google Scholar] [CrossRef] [PubMed]
- Tay, J.K.; Lim, M.Y.; Kanagalingam, J. Screening in nasopharyngeal carcinoma: Current strategies and future directions. Curr. Otorhinolaryngol. Rep. 2014, 2, 1–7. [Google Scholar] [CrossRef]
- Yang, X.; Dai, W.; Kwong, D.L.; Szeto, C.Y.Y.; Wong, E.H.; Ng, W.T.; Lee, A.W.M.; Ngan, R.K.C.; Yau, C.C.; Tung, S.Y.; et al. Epigenetic markers for noninvasive early detection of nasopharyngeal carcinoma by methylation-sensitive high resolution melting. Int. J. Cancer 2015, 136, E127–E135. [Google Scholar] [CrossRef] [PubMed]
- Hutajulu, S.H.; Indrasari, S.R.; Indrawati, L.P.; Harijadi, A.; Duin, S.; Haryana, S.M.; Steenbergen, R.D.; Greijer, A.E.; Middeldorp, J.M. Epigenetic markers for early detection of nasopharyngeal carcinoma in a high risk population. Mol. Cancer 2011, 10, 48. [Google Scholar] [CrossRef] [PubMed]
- Thunnissen, F.B.J.M. Sputum examination for early detection of lung cancer. J. Clin. Pathol. 2003, 56, 805–810. [Google Scholar] [CrossRef] [PubMed]
- Belinsky, S.A.; Liechty, K.C.; Gentry, F.D.; Wolf, H.J.; Rogers, J.; Vu, K.; Haney, J.; Kennedy, T.C.; Hirsch, F.R.; Miller, Y.; et al. Promoter hypermethylation of multiple genes in sputum precedes lung cancer incidence in a high-risk cohort. Cancer Res. 2006, 6, 3338–3344. [Google Scholar] [CrossRef] [PubMed]
- Cheuk, I.W.Y.; Shin, V.Y.; Kwong, A. Detection of methylated circulating DNA as noninvasive biomarkers for breast cancer diagnosis. J. Breast Cancer 2017, 20, 12. [Google Scholar] [CrossRef] [PubMed]
- Field, M.; Witham, T.F.; Flickinger, J.C.; Kondziolka, D.; Lunsford, L.D. Comprehensive assessment of hemorrhage risks and outcomes after stereotactic brain biopsy. J. Neurosurg. 2001, 94, 545–551. [Google Scholar] [CrossRef] [PubMed]
- Bokhorst, L.P.; Lepistö, I.; Kakehi, Y.; Bangma, C.H.; Pickles, T.; Valdagni, R.; Alberts, A.R.; Semjonow, A.; Strölin, P.; Montesino, M.F.; et al. Complications after prostate biopsies in men on active surveillance and its effects on receiving further biopsies in the Prostate cancer research international: Active surveillance (PRIAS) study. BJU Int. 2016, 118, 366–371. [Google Scholar] [CrossRef] [PubMed]
- Omuro, A.; DeAngelis, L.M. Glioblastoma and other malignant gliomas. JAMA 2013, 310, 1842. [Google Scholar] [CrossRef] [PubMed]
- Stathis, A.; Moore, M.J. Advanced pancreatic carcinoma: Current treatment and future challenges. Nat. Rev. Clin. Oncol. 2010, 7, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Torre, L.A.; Siegel, R.L.; Jemal, A. Lung Cancer Statistics. In Lung Cancer and Personalized Medicine. Advances in Experimental Medicine and Biology; Ahmad, A., Gadgeel, S., Eds.; Springer: Cham, Switzerland, 2016; Volume 893, pp. 1–19. [Google Scholar]
- Islami, F.; Miller, K.D.; Siegel, R.L.; Fedewa, S.A.; Ward, E.M.; Jemal, A. Disparities in liver cancer occurrence in the United States by race/ethnicity and state. CA. Cancer J. Clin. 2017, 67, 273–289. [Google Scholar] [CrossRef] [PubMed]
- Radpour, R.; Barekati, Z.; Kohler, C.; Lv, Q.; Bürki, N.; Diesch, C.; Bitzer, J.; Zheng, H.; Schmid, S.; Zhong, X.Y. Hypermethylation of tumor suppressor genes involved in critical regulatory pathways for developing a blood-based test in breast cancer. PLoS ONE 2011, 6, e16080. [Google Scholar] [CrossRef] [PubMed]
- Leon, S.A.; Shapiro, B.; Sklaroff, D.M.; Yaros, M.J. Free DNA in the serum of cancer patients and the effect of therapy. Cancer Res. 1977, 37, 646–650. [Google Scholar] [PubMed]
- Jahr, S.; Hentze, H.; Englisch, S.; Hardt, D.; Fackelmayer, F.O.; Hesch, R.D.; Knippers, R. DNA fragments in the blood plasma of cancer patients: Quantitations and evidence for their origin from apoptotic and necrotic cells. Cancer Res. 2001, 61, 1659–1665. [Google Scholar] [PubMed]
- Chan, A.K.C.; Chiu, R.W.K.; Lo, Y.M.D. Clinical sciences reviews committee of the association of clinical biochemists. Cell-free nucleic acids in plasma, serum and urine: A new tool in molecular diagnosis. Ann. Clin. Biochem. 2003, 40, 122–130. [Google Scholar] [CrossRef] [PubMed]
- Lo, Y.M.D.; Chan, K.C.A.; Sun, H.; Chen, E.Z.; Jiang, P.; Lun, F.M.F.; Zheng, Y.W.; Leung, T.Y.; Lau, T.K.; Cantor, C.R.; et al. Maternal plasma DNA sequencing reveals the genome-wide genetic and mutational profile of the fetus. Sci. Transl. Med. 2010, 2, 61ra91. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.Q.; Bonnefoi, H.; Pelte, M.F.; Lyautey, J.; Lederrey, C.; Movarekhi, S.; Schaeffer, P.; Mulcahy, H.E.; Meyer, P.; Stroun, M.; et al. Telomerase RNA as a detection marker in the serum of breast cancer patients. Clin. Cancer Res. 2001, 6, 3823–3826. [Google Scholar]
- Hasselmann, D.O.; Rappl, G.; Rössler, M.; Ugurel, S.; Tilgen, W.; Reinhold, U. Detection of tumor-associated circulating mRNA in serum, plasma and blood cells from patients with disseminated malignant melanoma. Oncol. Rep. 2001, 8, 115–118. [Google Scholar] [CrossRef] [PubMed]
- Kopreski, M.S.; Benko, F.A.; Gocke, C.D. Circulating RNA as a tumor marker: Detection of 5T4 mRNA in breast and lung cancer patient serum. Ann. N. Y. Acad. Sci. 2001, 945, 172–178. [Google Scholar] [CrossRef] [PubMed]
- Kopreski, M.S.; Benko, F.A.; Kwak, L.W.; Gocke, C.D. Detection of tumor messenger RNA in the serum of patients with malignant melanoma. Clin. Cancer Res. 1999, 5, 1961–1965. [Google Scholar] [PubMed]
- Casey, M.C.; Sweeney, K.J.; Brown, J.A.L.; Kerin, M.J. Exploring circulating micro-RNA in the neoadjuvant treatment of breast cancer. Int. J. Cancer 2016, 139, 12–22. [Google Scholar] [CrossRef] [PubMed]
- Schlosser, K.; Hanson, J.; Villeneuve, P.J.; Dimitroulakos, J.; McIntyre, L.; Pilote, L.; Stewart, D.J. Assessment of circulating LncRNAs under physiologic and pathologic conditions in humans reveals potential limitations as biomarkers. Sci. Rep. 2016, 6, 36596. [Google Scholar] [CrossRef] [PubMed]
- Reddy, D.; Khade, B.; Pandya, R.; Gupta, S. A novel method for isolation of histones from serum and its implications in therapeutics and prognosis of solid tumours. Clin. Epigenetics 2017, 9, 30. [Google Scholar] [CrossRef] [PubMed]
- Snyder, M.W.; Kircher, M.; Hill, A.J.; Daza, R.M.; Shendure, J. Cell-free DNA Comprises an in vivo nucleosome footprint that informs its tissues-of-origin. Cell 2016, 164, 57–68. [Google Scholar] [CrossRef] [PubMed]
- Bae, Y.K.; Shim, Y.R.; Choi, J.H.; Kim, M.J.; Gabrielson, E.; Lee, S.J.; Hwang, T.Y.; Shin, S.O. Gene promoter hypermethylation in tumors and plasma of breast cancer patients. Cancer Res. Treat. 2005, 37, 233–240. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.-L.; Leung, W.K.; Chan, M.W.Y.; Ng, E.K.W.; Tong, J.H.M.; Lo, K.-W.; Chung, S.C.S.; Sung, J.J.Y.; To, K.-F. Detection of gene promoter hypermethylation in the tumor and serum of patients with gastric carcinoma. Clin. Cancer Res. 2002, 8, 1761–1766. [Google Scholar] [PubMed]
- Salehi, R.; Salehi, A.; Emami, M.; Mohammadi, M. Methylation pattern of SFRP1 promoter in stool sample is a potential marker for early detection of colorectal cancer. Adv. Biomed. Res. 2012, 1, 87. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Lee, S.; Park, S.; Jeon, H.; Lee, W.; Kim, J.K.; Cho, M.; Kim, M.; Lim, J.; Kang, C.S.; et al. Gastrointestinal tract cancer screening using fecal carcinoembryonic antigen. Ann. Clin. Lab. Sci. 2003, 33, 32–38. [Google Scholar] [PubMed]
- Ned, R.M.; Melillo, S.; Marrone, M. Fecal DNA testing for colorectal cancer screening: The ColoSureTM test. PLoS Curr. 2011, 3, RRN1220. [Google Scholar] [CrossRef] [PubMed]
- Cho, M.H.; Lee, Y.M.; Kim, J.S.; Kim, H.S.; Lee, K.H.; Juhng, S.W.; Lee, J.H. Aberrant promoter methylation of the vimentin gene in colorectal cancer associated with the adenoma-carcinoma sequence. Korean J. Pathol. 2010, 44, 179–186. [Google Scholar] [CrossRef]
- Shirahata, A.; Sakata, M.; Sakuraba, K.; Goto, T.; Mizukami, H.; Saito, M.; Ishibashi, K.; Kigawa, G.; Nemoto, H.; Sanada, Y.; et al. Vimentin methylation as a marker for advanced colorectal carcinoma. Anticancer Res. 2009, 29, 279–281. [Google Scholar] [PubMed]
- Zou, H.; Harrington, J.J.; Shire, A.M.; Rego, R.L.; Wang, L.; Campbell, M.E.; Oberg, A.L.; Ahlquist, D.A. Highly methylated genes in colorectal neoplasia: Implications for screening. Cancer Epidemiol. Biomarkers Prev. 2007, 16, 2686–2696. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.-D.; Han, Z.J.; Skoletsky, J.; Olson, J.; Sah, J.; Myeroff, L.; Platzer, P.; Lu, S.; Dawson, D.; Willis, J.; et al. Detection in fecal DNA of colon cancer—Specific methylation of the nonexpressed vimentin gene. JNCI J. Natl. Cancer Inst. 2005, 97, 1124–1132. [Google Scholar] [CrossRef] [PubMed]
- YLamb, N.; Dhillon, S. Epi proColon® 2.0 CE: A blood-based screening test for colorectal cancer. Mol. Diagn. Ther. 2017, 21, 225–232. [Google Scholar]
- Journal of the American Medical Association (JAMA). A stool DNA test (Cologuard) for colorectal cancer screening. JAMA 2014, 312, 2566. [Google Scholar]
- Imperiale, T.F.; Ransohoff, D.F.; Itzkowitz, S.H.; Levin, T.R.; Lavin, P.; Lidgard, G.P.; Ahlquist, D.A.; Berger, B.M. Multitarget stool DNA testing for colorectal-cancer screening. N. Engl. J. Med. 2014, 370, 1287–1297. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Gan, A.; Chen, X.; Wang, X.; He, W.; Zhang, X.; Huang, R.; Zhou, S.; Song, X.; Xu, A. Diagnostic performance of DNA hypermethylation markers in peripheral blood for the detection of colorectal cancer: A meta-analysis and systematic review. PLoS ONE 2016, 11, e0155095. [Google Scholar] [CrossRef] [PubMed]
- Schroy, P.C.; Heeren, T.C. Patient perceptions of stool-based DNA testing for colorectal cancer screening. Am. J. Prev. Med. 2005, 28, 208–214. [Google Scholar] [CrossRef] [PubMed]
- Imperiale, T.F.; Ransohoff, D.F.; Itzkowitz, S.H.; Turnbull, B.A.; Ross, M.E.; Colorectal cancer study group. Fecal DNA versus fecal occult blood for colorectal-cancer screening in an average-risk population. N. Engl. J. Med. 2004, 351, 2704–2714. [Google Scholar] [CrossRef] [PubMed]
- Molnár, B.; Tóth, K.; Barták, B.K.; Tulassay, Z. Plasma methylated septin 9: A colorectal cancer screening marker. Expert Rev. Mol. Diagn. 2015, 15, 171–184. [Google Scholar] [CrossRef] [PubMed]
- Salehi, R.; Atapour, N.; Vatandoust, N.; Farahani, N.; Ahangari, F.; Salehi, A. Methylation pattern of ALX4 gene promoter as a potential biomarker for blood-based early detection of colorectal cancer. Adv. Biomed. Res. 2015, 4, 252. [Google Scholar] [CrossRef] [PubMed]
- Toiyama, Y.; Takahashi, M.; Hur, K.; Nagasaka, T.; Tanaka, K.; Inoue, Y.; Kusunoki, M.; Boland, C.R.; Goel, A. Serum miR-21 as a diagnostic and prognostic biomarker in colorectal cancer. JNCI J. Natl. Cancer Inst. 2013, 105, 849–859. [Google Scholar] [CrossRef] [PubMed]
- Gezer, U.; Üstek, D.; Yörüker, E.E.; Cakiris, A.; Abaci, N.; Leszinski, G.; Dalay, N.; Holdenrieder, S. Characterization of H3K9me3- and H4K20me3-associated circulating nucleosomal DNA by high-throughput sequencing in colorectal cancer. Tumor Biol. 2013, 34, 329–336. [Google Scholar] [CrossRef] [PubMed]
- Seligson, D.B.; Horvath, S.; McBrian, M.A.; Mah, V.; Yu, H.; Tze, S.; Wang, Q.; Chia, D.; Goodglick, L.; Kurdistani, S.K. Global levels of histone modifications predict prognosis in different cancers. Am. J. Pathol. 2009, 174, 1619–1628. [Google Scholar] [CrossRef] [PubMed]
- Rahier, J.-F.; Druez, A.; Faugeras, L.; Martinet, J.; Géhénot, M.; Josseaux, E.; Herzog, M.; Micallef, J.; George, F.; Delos, M.; et al. Circulating nucleosomes as new blood-based biomarkers for detection of colorectal cancer. Clin. Epigenetics 2017, 9, 53. [Google Scholar] [CrossRef] [PubMed]
- Shinozaki, M.; Hoon, D.S.B.; Giuliano, A.E.; Hansen, N.M.; Wang, H.-J.; Turner, R.; Taback, B. Distinct hypermethylation profile of primary breast cancer is associated with sentinel lymph node metastasis. Clin. Cancer Res. 2005, 11, 2156–2162. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, N.; Nakayama, T.; Kajita, M.; Miyake, T.; Iwamoto, T.; Kim, S.J.; Sakai, A.; Ishihara, H.; Tamaki, Y.; Noguchi, S. Detection of aberrant promoter methylation of GSTP1, RASSF1A, and RARβ2 in serum DNA of patients with breast cancer by a newly established one-step methylation-specific PCR assay. Breast Cancer Res. Treat. 2012, 132, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Dulaimi, E.; Hillinck, J.; de Caceres, I.I.; Al-Saleem, T.; Cairns, P. Tumor suppressor gene promoter hypermethylation in serum of breast cancer patients. Clin. Cancer Res. 2004, 10, 6189–6193. [Google Scholar] [CrossRef] [PubMed]
- Hoque, M.O.; Feng, Q.; Toure, P.; Dem, A.; Critchlow, C.W.; Hawes, S.E.; Wood, T.; Jeronimo, C.; Rosenbaum, E.; Stern, J.; et al. Detection of aberrant methylation of four genes in plasma DNA for the detection of breast cancer. J. Clin. Oncol. 2006, 24, 4262–4269. [Google Scholar] [CrossRef] [PubMed]
- Kajabova, V.; Smolkova, B.; Zmetakova, I.; Sebova, K.; Krivulcik, T.; Bella, V.; Kajo, K.; Machalekova, K.; Fridrichova, I. RASSF1A promoter methylation levels positively correlate with estrogen receptor expression in breast cancer patients. Transl. Oncol. 2013, 6, 297–304. [Google Scholar] [CrossRef] [PubMed]
- Radpour, R.; Barekati, Z.; Kohler, C.; Schumacher, M.M.; Grussenmeyer, T.; Jenoe, P.; Hartmann, N.; Moes, S.; Letzkus, M.; Bitzer, J.; et al. Integrated epigenetics of human breast cancer: Synoptic investigation of targeted genes, microRNAs and proteins upon demethylation treatment. PLoS ONE 2011, 6, e27355. [Google Scholar] [CrossRef] [PubMed]
- Liggett, T.; Melnikov, A.; Yi, Q.; Replogle, C.; Brand, R.; Kaul, K.; Talamonti, M.; Abrams, R.A.; Levenson, V. Differential methylation of cell-free circulating DNA among patients with pancreatic cancer versus chronic pancreatitis. Cancer 2010, 116, 1674–1680. [Google Scholar] [CrossRef] [PubMed]
- Melnikov, A.A.; Scholtens, D.; Talamonti, M.S.; Bentrem, D.J.; Levenson, V.V. Methylation profile of circulating plasma DNA in patients with pancreatic cancer. J. Surg. Oncol. 2009, 99, 119–122. [Google Scholar] [CrossRef] [PubMed]
- Langevin, S.M.; Eliot, M.; Butler, R.A.; Cheong, A.; Zhang, X.; McClean, M.D.; Koestler, D.C.; Kelsey, K.T. CpG island methylation profile in non-invasive oral rinse samples is predictive of oral and pharyngeal carcinoma. Clin. Epigenetics 2015, 7, 125. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, M.; Gonzalgo, M.L.; Yegnasubramanian, S.; Lin, X.; de Marzo, A.M.; Nelson, W.G. GSTP1 CpG island hypermethylation as a molecular biomarker for prostate cancer. J. Cell. Biochem. 2004, 91, 540–552. [Google Scholar] [CrossRef] [PubMed]
- Woodson, K.; O’Reilly, K.J.; Hanson, J.C.; Nelson, D.; Walk, E.L.; Tangrea, J.A. The usefulness of the detection of GSTP1 methylation in urine as a biomarker in the diagnosis of prostate cancer. J. Urol. 2008, 179, 508–512. [Google Scholar] [CrossRef] [PubMed]
- Doufekas, K.; Zheng, S.C.; Ghazali, S.; Wong, M.; Mohamed, Y.; Jones, A.; Reisel, D.; Mould, T.; Olaitan, A.; Macdonald, N.; et al. DNA methylation signatures in vaginal fluid samples for detection of cervical and endometrial cancer. Int. J. Gynecol. Cancer 2016. [Google Scholar] [CrossRef] [PubMed]
- Dietrich, D.; Kneip, C.; Raji, O.; Liloglou, T.; Seegebarth, A.; Schlegel, T.; Flemming, N.; Rausch, S.; Distler, J.; Fleischhacker, M.; et al. Performance evaluation of the DNA methylation biomarker SHOX2 for the aid in diagnosis of lung cancer based on the analysis of bronchial aspirates. Int. J. Oncol. 2011, 40, 825–832. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Zhu, L.; Liu, B.; Yang, L.; Meng, X.; Zhang, W.; Ma, Y.; Xiao, H. Genome-wide microRNA profiles identify miR-378 as a serum biomarker for early detection of gastric cancer. Cancer Lett. 2012, 316, 196–203. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, S.; Ichikawa, D.; Miyamae, M.; Kawaguchi, T.; Morimura, R.; Hirajima, S.; Okajima, W.; Ohashi, T.; Imamura, T.; Konishi, H.; et al. Malignant potential in pancreatic neoplasm; new insights provided by circulating miR-223 in plasma. Expert Opin. Biol. Ther. 2015, 15, 773–785. [Google Scholar] [CrossRef] [PubMed]
- Schultz, N.A.; Dehlendorff, C.; Jensen, B.V.; Bjerregaard, J.K.; Nielsen, K.R.; Bojesen, S.E.; Calatayud, D.; Nielsen, S.E.; Yilmaz, M.; Holländer, N.H.; et al. MicroRNA biomarkers in whole blood for detection of pancreatic cancer. JAMA 2014, 311, 392. [Google Scholar] [CrossRef] [PubMed]
- Kojima, M.; Sudo, H.; Kawauchi, J.; Takizawa, S.; Kondou, S.; Nobumasa, H.; Ochiai, A. MicroRNA markers for the diagnosis of pancreatic and biliary-tract cancers. PLoS ONE 2015, 10, e0118220. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, Z.; Zheng, S.; Zhou, Y.; Zhao, L.; Ye, H.; Zhao, X.; Gao, W.; Fu, Z.; Zhou, Q.; et al. Expression profile of long non-coding RNAs in pancreatic cancer and their clinical significance as biomarkers. Oncotarget 2015, 6, 35684–35698. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Yin, C.; Dang, Y.; Ye, F.; Zhang, G. Identification of the long non-coding RNA H19 in plasma as a novel biomarker for diagnosis of gastric cancer. Sci. Rep. 2015, 5, 11516. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Shao, Y.; Zhang, X.; Zheng, T.; Miao, M.; Qin, L.; Wang, B.; Ye, G.; Xiao, B.; Guo, J. Plasma long noncoding RNA protected by exosomes as a potential stable biomarker for gastric cancer. Tumor Biol. 2015, 36, 2007–2012. [Google Scholar] [CrossRef] [PubMed]
- Elsheikh, S.E.; Green, A.R.; Rakha, E.A.; Powe, D.G.; Ahmed, R.A.; Collins, H.M.; Soria, D.; Garibaldi, J.M.; Paish, C.E.; Ammar, A.A.; et al. Global histone modifications in breast cancer correlate with tumor phenotypes, prognostic factors, and patient outcome. Cancer Res. 2009, 69, 3802–3809. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, J.; Chen, Y.-Y.; Scott, G.K.; DeVries, S.; Chin, K.; Benz, C.C.; Waldman, F.M.; Hwang, E.S. Protein acetylation and histone deacetylase expression associated with malignant breast cancer progression. Clin. Cancer Res. 2009, 15, 3163–3171. [Google Scholar] [CrossRef] [PubMed]
- Deligezer, U.; Akisik, E.E.; Erten, N.; Dalay, N. Sequence-specific histone methylation is detectable on circulating nucleosomes in plasma. Clin. Chem. 2008, 54, 1125–1131. [Google Scholar] [CrossRef] [PubMed]
- Leszinski, G.; Gezer, U.; Siegele, B.; Stoetzer, O.; Holdenrieder, S. Relevance of histone marks H3K9me3 and H4K20me3 in cancer. Anticancer Res. 2012, 32, 2199–2205. [Google Scholar] [PubMed]
- Bauden, M.; Pamart, D.; Ansari, D.; Herzog, M.; Eccleston, M.; Micallef, J.; Andersson, B.; Andersson, R. Circulating nucleosomes as epigenetic biomarkers in pancreatic cancer. Clin. Epigenetics 2015, 7, 106. [Google Scholar] [CrossRef] [PubMed]
- Harvey, J.M.; Clark, G.M.; Osborne, C.K.; Allred, D.C. Estrogen receptor status by immunohistochemistry is superior to the ligand-binding assay for predicting response to adjuvant endocrine therapy in breast cancer. J. Clin. Oncol. 1999, 17, 1474–1481. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.; Janzer, A.; Becker, A.; Zimmer, A.; Schüle, R.; Buettner, R.; Kirfel, J. Lysine-specific demethylase 1 (LSD1) is highly expressed in ER-negative breast cancers and a biomarker predicting aggressive biology. Carcinogenesis 2010, 31, 512–520. [Google Scholar] [CrossRef] [PubMed]
- Van Grembergen, O.; Bizet, M.; de Bony, E.J.; Calonne, E.; Putmans, P.; Brohée, S.; Olsen, C.; Guo, M.; Bontempi, G.; Sotiriou, C.; et al. Portraying breast cancers with long noncoding RNAs. Sci. Adv. 2016, 2, e1600220. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Li, Z.; Yang, Y.; Xiang, T.; Song, W.; Liu, S. Microarray expression profiling of dysregulated long non-coding RNAs in triple-negative breast cancer. Cancer Biol. Ther. 2015, 16, 856–865. [Google Scholar] [CrossRef] [PubMed]
- Toyota, M.; Ahuja, N.; Ohe-Toyota, M.; Herman, J.G.; Baylin, S.B.; Issa, J.P. CpG island methylator phenotype in colorectal cancer. Proc. Natl. Acad. Sci. USA 1999, 96, 8681–8686. [Google Scholar] [CrossRef] [PubMed]
- Jia, M.; Jansen, L.; Walter, V.; Tagscherer, K.; Roth, W.; Herpel, E.; Kloor, M.; Bläker, H.; Chang-Claude, J.; Brenner, H.; et al. No association of CpG island methylator phenotype and colorectal cancer survival: Population-based study. Br. J. Cancer 2016, 115, 1359–1366. [Google Scholar] [CrossRef] [PubMed]
- Kong, X.; Du, Y.; Wang, G.; Gao, J.; Gong, Y.; Li, L.; Zhang, Z.; Zhu, J.; Jing, Q.; Qin, Y.; et al. Detection of differentially expressed microRNAs in serum of pancreatic ductal adenocarcinoma patients: miR-196a could be a potential marker for poor prognosis. Dig. Dis. Sci. 2011, 56, 602–609. [Google Scholar] [CrossRef] [PubMed]
- Kolacinska, A.; Morawiec, J.; Fendler, W.; Malachowska, B.; Morawiec, Z.; Szemraj, J.; Pawlowska, Z.; Chowdhury, D.; Choi, Y.E.; Kubiak, R.; et al. Association of microRNAs and pathologic response to preoperative chemotherapy in triple negative breast cancer: Preliminary report. Mol. Biol. Rep. 2014, 41, 2851–2857. [Google Scholar] [CrossRef] [PubMed]
- Kheirelseid, E.A.H.; Miller, N.; Chang, K.H.; Curran, C.; Hennessey, E.; Sheehan, M.; Newell, J.; Lemetre, C.; Balls, G.; Kerin, M.J. miRNA expressions in rectal cancer as predictors of response to neoadjuvant chemoradiation therapy. Int. J. Colorectal Dis. 2013, 28, 247–260. [Google Scholar] [CrossRef] [PubMed]
- Shen, S.; Wang, G.; Shi, Q.; Zhang, R.; Zhao, Y.; Wei, Y.; Chen, F.; Christiani, D.C. Seven-CpG-based prognostic signature coupled with gene expression predicts survival of oral squamous cell carcinoma. Clin. Epigenetics 2017, 9, 88. [Google Scholar] [CrossRef] [PubMed]
- Barlési, F.; Giaccone, G.; Gallegos-Ruiz, M.I.; Loundou, A.; Span, S.W.; Lefesvre, P.; Kruyt, F.A.E.; Rodriguez, J.A. Global histone modifications predict prognosis of resected non-small-cell lung cancer. J. Clin. Oncol. 2007, 25, 4358–4364. [Google Scholar] [CrossRef] [PubMed]
- Yates, D.R.; Rehman, I.; Abbod, M.F.; Meuth, M.; Cross, S.S.; Linkens, D.A.; Hamdy, F.C.; Catto, J.W.F. Promoter hypermethylation identifies progression risk in bladder cancer. Clin. Cancer Res. 2007, 13, 2046–2053. [Google Scholar] [CrossRef] [PubMed]
- Catto, J.W.F.; Hartmann, A.; Stoehr, R.; Bolderson, E.; Rehman, I.; Rosario, D.J.; Hamdy, F.C.; Meuth, M. Multifocal urothelial cancers with the mutator phenotype are of monoclonal origin and require panurothelial treatment for tumor clearance. J. Urol. 2006, 175, 2323–2330. [Google Scholar] [CrossRef]
- Dulaimi, E.; Uzzo, R.G.; Greenberg, R.E.; Al-Saleem, T.; Cairns, P. Detection of bladder cancer in urine by a tumor suppressor gene hypermethylation panel. Clin. Cancer Res. 2004, 10, 1887–1893. [Google Scholar] [CrossRef] [PubMed]
- Gaudet, M.M.; Campan, M.; Figueroa, J.D.; Yang, X.R.; Lissowska, J.; Peplonska, B.; Brinton, L.A.; Rimm, D.L.; Laird, P.W.; Garcia-Closas, M.; et al. DNA hypermethylation of ESR1 and PGR in breast cancer: pathologic and epidemiologic associations. Cancer Epidemiol. Biomarkers Prev. 2009, 18, 3036–3043. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Galán, J.; Torres-Torres, B.; Núñez, M.I.; López-Peñalver, J.; del Moral, R.; de Almodóvar, J.M.R.; Menjón, S.; Concha, Á.; Chamorro, C.; Ríos, S.; et al. ESR1 gene promoter region methylation in free circulating DNA and its correlation with estrogen receptor protein expression in tumor tissue in breast cancer patients. BMC Cancer 2014, 14, 59. [Google Scholar] [CrossRef] [PubMed]
- Widschwendter, M.; Siegmund, K.D.; Müller, H.M.; Fiegl, H.; Marth, C.; Müller-Holzner, E.; Jones, P.A.; Laird, P.W. Association of breast cancer DNA methylation profiles with hormone receptor status and response to tamoxifen. Cancer Res. 2004, 64, 3807–3813. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Liu, J.; Jing, H.; Dong, S.X.; Wu, J. The diagnostic and prognostic value of CHFR hypermethylation in colorectal cancer, a meta-analysis and literature review. Oncotarget 2017, 8, 89142–89148. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Liu, F.; Zhang, H.; Sun, J.; Ma, Y. CHFR hypermethylation, a frequent event in acute myeloid leukemia, is independently associated with an adverse outcome. Genes Chromosom. Cancer 2016, 55, 158–168. [Google Scholar] [CrossRef] [PubMed]
- Derks, S.; Cleven, A.H.G.; Melotte, V.; Smits, K.M.; Brandes, J.C.; Azad, N.; van Criekinge, W.; de Bruïne, A.P.; Herman, J.G.; van Engeland, M. Emerging evidence for CHFR as a cancer biomarker: From tumor biology to precision medicine. Cancer Metastasis Rev. 2013, 33, 161–171. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Li, W.; Li, H.; Ma, Y.; He, G.; Tan, G. Genomic methylation profiling combined with gene expression microarray reveals the aberrant methylation mechanism involved in nasopharyngeal carcinoma taxol resistance. Anticancer Drugs 2012, 23, 856–864. [Google Scholar] [CrossRef] [PubMed]
- Pelosof, L.; Yerram, S.R.; Ahuja, N.; Delmas, A.; Danilova, L.; Herman, J.G.; Azad, N.S. CHFR silencing or microsatellite instability is associated with increased antitumor activity of docetaxel or gemcitabine in colorectal cancer. Int. J. Cancer 2014, 134, 596–605. [Google Scholar] [CrossRef] [PubMed]
- Yun, T.; Liu, Y.; Gao, D.; Linghu, E.; Brock, M.V.; Yin, D.; Zhan, Q.; Herman, J.G.; Guo, M. Methylation of CHFR sensitizes esophageal squamous cell cancer to docetaxel and paclitaxel. Genes Cancer 2015, 6, 38–48. [Google Scholar] [PubMed]
- Wang, X.; Yang, Y.; Xu, C.; Xiao, L.; Shen, H.; Zhang, X.; Li, T.; Li, X. CHFR suppression by hypermethylation sensitizes endometrial cancer cells to paclitaxel. Int. J. Gynecol. Cancer 2011, 21, 996–1003. [Google Scholar] [CrossRef] [PubMed]
- Satoh, A.; Toyota, M.; Itoh, F.; Sasaki, Y.; Suzuki, H.; Ogi, K.; Kikuchi, T.; Mita, H.; Yamashita, T.; Kojima, T. Epigenetic inactivation of CHFR and sensitivity to microtubule inhibitors in gastric cancer. Cancer Res. 2003, 63, 8606–8613. [Google Scholar] [PubMed]
- Yoshida, K.; Hamai, Y.; Suzuki, T.; Sanada, Y.; Oue, N.; Yasui, W. DNA methylation of CHFR is not a predictor of the response to docetaxel and paclitaxel in advanced and recurrent gastric cancer. Anticancer Res. 2006, 26, 49–54. [Google Scholar] [PubMed]
- Kelly, R.J.; Wrangle, J.; Hales, R.K.; Molena, D.; Yang, S.C.; Rodgers, K.; Lang, M.; Reynolds, J.; Beckman, T.; Choflet, A.; et al. An interim analysis of a phase II study using an epigenetic biomarker (CHFR methylation status) to personalize chemotherapy in patients with operable esophageal cancer. J. Clin. Oncol. 2014, 32, 76. [Google Scholar] [CrossRef]
- Esteller, M.; Garcia-Foncillas, J.; Andion, E.; Goodman, S.N.; Hidalgo, O.F.; Vanaclocha, V.; Baylin, S.B.; Herman, J.G. Inactivation of the DNA-repair gene MGMT and the clinical response of gliomas to alkylating agents. N. Engl. J. Med. 2000, 343, 1350–1354. [Google Scholar] [CrossRef] [PubMed]
- Christmann, M.; Naumann, S.; Roos, W.P. MGMT: Key node in the battle against genotoxicity, carcinogenicity and apoptosis induced by alkylating agents. DNA Repair (Amst). 2007, 6, 1079–1099. [Google Scholar]
- Hegi, M.E.; Diserens, A.-C.; Gorlia, T.; Hamou, M.-F.; de Tribolet, N.; Weller, M.; Kros, J.M.; Hainfellner, J.A.; Mason, W.; Mariani, L.; et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N. Engl. J. Med. 2005, 352, 997–1003. [Google Scholar] [CrossRef] [PubMed]
- Hegi, M.E.; Diserens, A.-C.; Godard, S.; Dietrich, P.-Y.; Regli, L.; Ostermann, S.; Otten, P.; van Melle, G.; de Tribolet, N.; Stupp, R. Clinical trial substantiates the predictive value of O-6-methylguanine-DNA methyltransferase promoter methylation in glioblastoma patients treated with temozolomide. Clin. Cancer Res. 2004, 10, 1871–1874. [Google Scholar] [CrossRef] [PubMed]
- Weller, M.; Stupp, R.; Reifenberger, G.; Brandes, A.A.; van den Bent, M.J.; Wick, W.; Hegi, M.E. MGMT promoter methylation in malignant gliomas: Ready for personalized medicine? Nat. Rev. Neurol. 2010, 6, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Von Minckwitz, G.; Kummel, S.; Vogel, P.; Hanusch, C.; Eidtmann, H.; Hilfrich, J.; Gerber, B.; Huober, J.; Costa, S.D.; Jackisch, C.; et al. Intensified neoadjuvant chemotherapy in early-responding breast cancer: phase III randomized GeparTrio study. JNCI J. Natl. Cancer Inst. 2008, 100, 552–562. [Google Scholar] [CrossRef] [PubMed]
- Holdenrieder, S.; Stieber, P.; von Pawel, J.; Raith, H.; Nagel, D.; Feldmann, K.; Seidel, D. Circulating nucleosomes predict the response to chemotherapy in patients with advanced non-small cell lung cancer. Clin. Cancer Res. 2004, 10, 5981–5987. [Google Scholar] [CrossRef] [PubMed]
- Phé, V.; Cussenot, O.; Rouprêt, M. Interest of methylated genes as biomarkers in urothelial cell carcinomas of the urinary tract. BJU Int. 2009, 104, 896–901. [Google Scholar] [CrossRef] [PubMed]
- Hiraki, M.; Kitajima, Y.; Koga, Y.; Tanaka, T.; Nakamura, J.; Hashiguchi, K.; Noshiro, H.; Miyazaki, K. Aberrant gene methylation is a biomarker for the detection of cancer cells in peritoneal wash samples from advanced gastric cancer patients. Ann. Surg. Oncol. 2011, 18, 3013–3019. [Google Scholar] [CrossRef] [PubMed]
- Hiraki, M.; Kitajima, Y.; Sato, S.; Mitsuno, M.; Koga, Y.; Nakamura, J.; Hashiguchi, K.; Noshiro, H.; Miyazaki, K. Aberrant gene methylation in the lymph nodes provides a possible marker for diagnosing micrometastasis in gastric cancer. Ann. Surg. Oncol. 2010, 17, 1177–1186. [Google Scholar] [CrossRef] [PubMed]
- Van Otterdijk, S.D.; Norden, J.; Dickinson, A.M.; Pearce, M.S.; Relton, C.L.; Mathers, J.C.; Strathdee, G. Aberrations in DNA methylation are detectable during remission of acute lymphoblastic leukemia and predict patient outcome. Epigenomics 2015, 7, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Furlan, C.; Polesel, J.; Barzan, L.; Franchin, G.; Sulfaro, S.; Romeo, S.; Colizzi, F.; Rizzo, A.; Baggio, V.; Giacomarra, V.; et al. Prognostic significance of LINE-1 hypomethylation in oropharyngeal squamous cell carcinoma. Clin. Epigenetics 2017, 9, 58. [Google Scholar] [CrossRef] [PubMed]
- Sueta, A.; Yamamoto, Y.; Tomiguchi, M.; Takeshita, T.; Yamamoto-Ibusuki, M.; Iwase, H. Differential expression of exosomal miRNAs between breast cancer patients with and without recurrence. Oncotarget 2017, 8, 69934–69944. [Google Scholar] [CrossRef] [PubMed]
- Imamura, T.; Komatsu, S.; Ichikawa, D.; Kawaguchi, T.; Miyamae, M.; Okajima, W.; Ohashi, T.; Arita, T.; Konishi, H.; Shiozaki, A.; et al. Liquid biopsy in patients with pancreatic cancer: Circulating tumor cells and cell-free nucleic acids. World J. Gastroenterol. 2016, 22, 5627. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Yuan, Y.; Cho, J.H.; McClarty, S.; Baxter, D.; Galas, D.J. Comparing the MicroRNA Spectrum between serum and plasma. PLoS ONE 2012, 7, e41561. [Google Scholar] [CrossRef] [PubMed]
- Langevin, S.M.; Houseman, E.A.; Accomando, W.P.; Koestler, D.C.; Christensen, B.C.; Nelson, H.H.; Karagas, M.R.; Marsit, C.J.; Wiencke, J.K.; Kelsey, K.T. Leukocyte-adjusted epigenome-wide association studies of blood from solid tumor patients. Epigenetics 2014, 9, 884–895. [Google Scholar] [CrossRef] [PubMed]
- Koestler, D.C.; Marsit, C.J.; Christensen, B.C.; Accomando, W.; Langevin, S.M.; Houseman, E.A.; Nelson, H.H.; Karagas, M.R.; Wiencke, J.K.; Kelsey, K.T. Peripheral blood immune cell methylation profiles are associated with nonhematopoietic cancers. Cancer Epidemiol. Biomarkers Prev. 2012, 21, 1293–1302. [Google Scholar] [CrossRef] [PubMed]
- Shabalin, A.A.; Aberg, K.A.; van den Oord, E.J. Candidate gene methylation studies are at high risk of erroneous conclusions. Epigenomics 2015, 7, 13–15. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.S.; Wu, H.; Ji, X.; Stelzer, Y.; Wu, X.; Czauderna, S.; Shu, J.; Dadon, D.; Young, R.A.; Jaenisch, R. Editing DNA methylation in the mammalian genome. Cell 2016, 167, 233–247.e17. [Google Scholar] [CrossRef] [PubMed]
- Vojta, A.; Dobrinić, P.; Tadić, V.; Bočkor, L.; Korać, P.; Julg, B.; Klasić, M.; Zoldoš, V. Repurposing the CRISPR-Cas9 system for targeted DNA methylation. Nucleic Acids Res. 2016, 44, 5615–5628. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Clinical Trial ID | Marker | Test | Sample | Neoplasm | Biomarker Type | Result/Status |
|---|---|---|---|---|---|---|
| NCT01329718 | Hypermethylated Septin-9 (SEPT9) | Epi proColon | Blood | Colorectal cancer | Diagnostic | Completed |
| NCT03218423 | Colorectal cancer | Longitudinal (Diagnostic) | Recruiting | |||
| NCT00696345 | Colorectal cancer | Diagnostic | Completed | |||
| NCT02198092 | Hereditary colorectal cancer | Diagnostic | Recruiting | |||
| NCT03311152 | Hepatocellular carcinoma | Diagnostic | Recruiting | |||
| NCT02540850 | Hypermethylated SEPT9 | Blood | Colorectal cancer | Diagnostic | Completed | |
| NCT00855348 | Colorectal cancer | Diagnostic | Completed | |||
| NCT02419716 | Hypermethylated NDRG family member 4 (NDRG4), bone morphogenetic protein 3 (BMP3) | ColoGuard | Stool | Colorectal cancer | Diagnostic | Active/Not Recruiting |
| NCT02715141 | Recruiting | |||||
| NCT01793207 | 7 Cpgs | Blood | Colorectal cancer | Diagnostic | Completed | |
| NCT03146520 | Hypermethylated syndecan 3 (SDC2) | EarlyTect | Stool | Colorectal cancer | Diagnostic | Enrolling by invitation |
| NCT02159339 | Hypermethylated cyclin dependent kinase inhibitor 2A (p16) | Tumor | Gastric cancer | Prognostic- Metastasis | Completed | |
| NCT00835341 | Biopsy | Oral cancer | Diagnostic | Completed | ||
| NCT01695018 | Mucosal Biopsy | Oral cancer | Diagnostic | Completed | ||
| NCT01774266 | Hypermethylated TP53-dependent G2 arrest mediator homolog (REPRIMO) | Serum | Gastric cancer | Diagnostic | Recruiting | |
| NCT01715233 | Hypermethylated checkpoint with forkhead and ring finger domains (CHFR) | Biopsy | Esophageal, gastroesophageal, gastric cancer | Predictive-Taxane Response | Recruiting | |
| NCT01372202 | Biopsy | Esophageal cancer | Active/Not Recruiting | |||
| NCT03217097 | Hypermethylated O-6-methylguanine-DNA methyltransferase (MGMT) | Biopsy | Neuroendocrine tumors | Predictive-Oxaliplatin/Alkylating Agent Response | Not Yet Recruiting | |
| NCT02700464 | 15 CpGs | EpiCheck | Urine | Bladder eurothelial cell carcinoma | Recurrence | Recruiting |
| NCT02647112 | Recruiting | |||||
| NCT02688491 | 5 CpGs | Surgical Specimen | Clear cell renal carcinoma (Stage III) | Predictive-Sunitinib Response | Not Yet Recruiting |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thomas, M.L.; Marcato, P. Epigenetic Modifications as Biomarkers of Tumor Development, Therapy Response, and Recurrence across the Cancer Care Continuum. Cancers 2018, 10, 101. https://doi.org/10.3390/cancers10040101
Thomas ML, Marcato P. Epigenetic Modifications as Biomarkers of Tumor Development, Therapy Response, and Recurrence across the Cancer Care Continuum. Cancers. 2018; 10(4):101. https://doi.org/10.3390/cancers10040101
Chicago/Turabian StyleThomas, Margaret L., and Paola Marcato. 2018. "Epigenetic Modifications as Biomarkers of Tumor Development, Therapy Response, and Recurrence across the Cancer Care Continuum" Cancers 10, no. 4: 101. https://doi.org/10.3390/cancers10040101
APA StyleThomas, M. L., & Marcato, P. (2018). Epigenetic Modifications as Biomarkers of Tumor Development, Therapy Response, and Recurrence across the Cancer Care Continuum. Cancers, 10(4), 101. https://doi.org/10.3390/cancers10040101