Cancer-Associated Thrombosis: An Overview of Mechanisms, Risk Factors, and Treatment


{kind=link}
{kind=link}
Abstract
1. Introduction
2. Types of Cancer-Associated Thrombosis
2.1. Venous Thromboembolism
2.2. Arterial Thrombosis
2.3. Chronic Disseminated Intravascular Coagulation
3. Risk Factors for Cancer-Associated Thrombosis
3.1. Individual Patient Risk Factors
3.1.1. Age
3.1.2. Sex
3.1.3. Race
3.1.4. Comorbidities
3.1.5. Immobility
3.1.6. Previous History of VTE
3.2. Cancer-Associated Risk Factors
3.2.1. Site of Cancer
3.2.2. Stage of Cancer
3.2.3. Histology of Cancer
3.2.4. Time after Diagnosis
3.3. Cancer-Treatment-Associated Risk Factors
3.3.1. Surgery and Hospitalisation
3.3.2. Chemotherapy
3.3.3. Angiogenesis Inhibitors
3.3.4. Central Venous Catheters
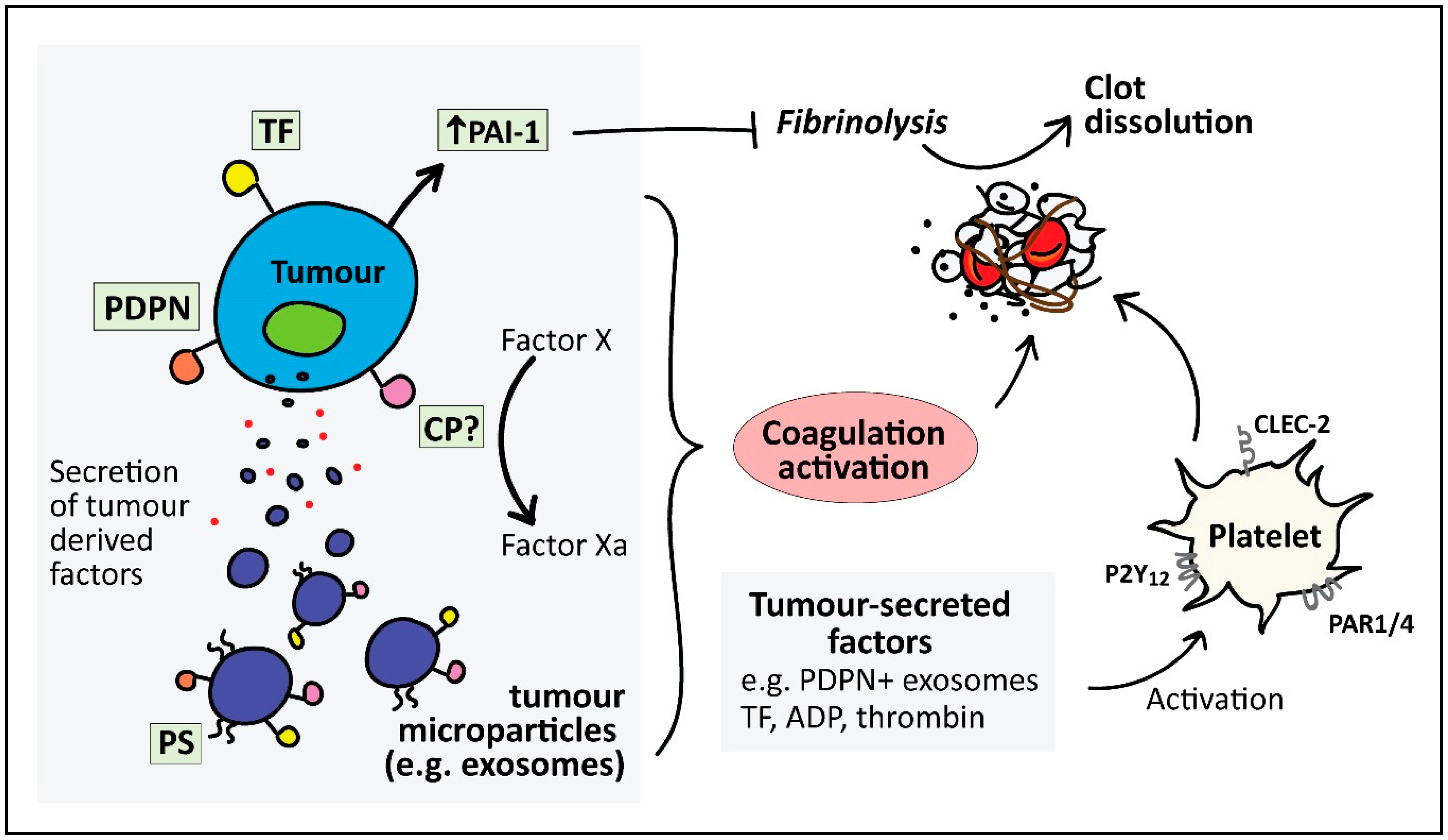
4. Mechanisms of Cancer-Associated Thrombosis
4.1. Direct Mechanisms for Cancer-Associated Thrombosis
4.1.1. Tissue Factor
4.1.2. Microparticles (MP)
4.1.3. Podoplanin
4.1.4. Plasminogen Activator Inhibitor-1 (PAI-1)
4.1.5. Cancer Procoagulant (CP)
4.1.6. Tumour-Derived Platelet Agonists
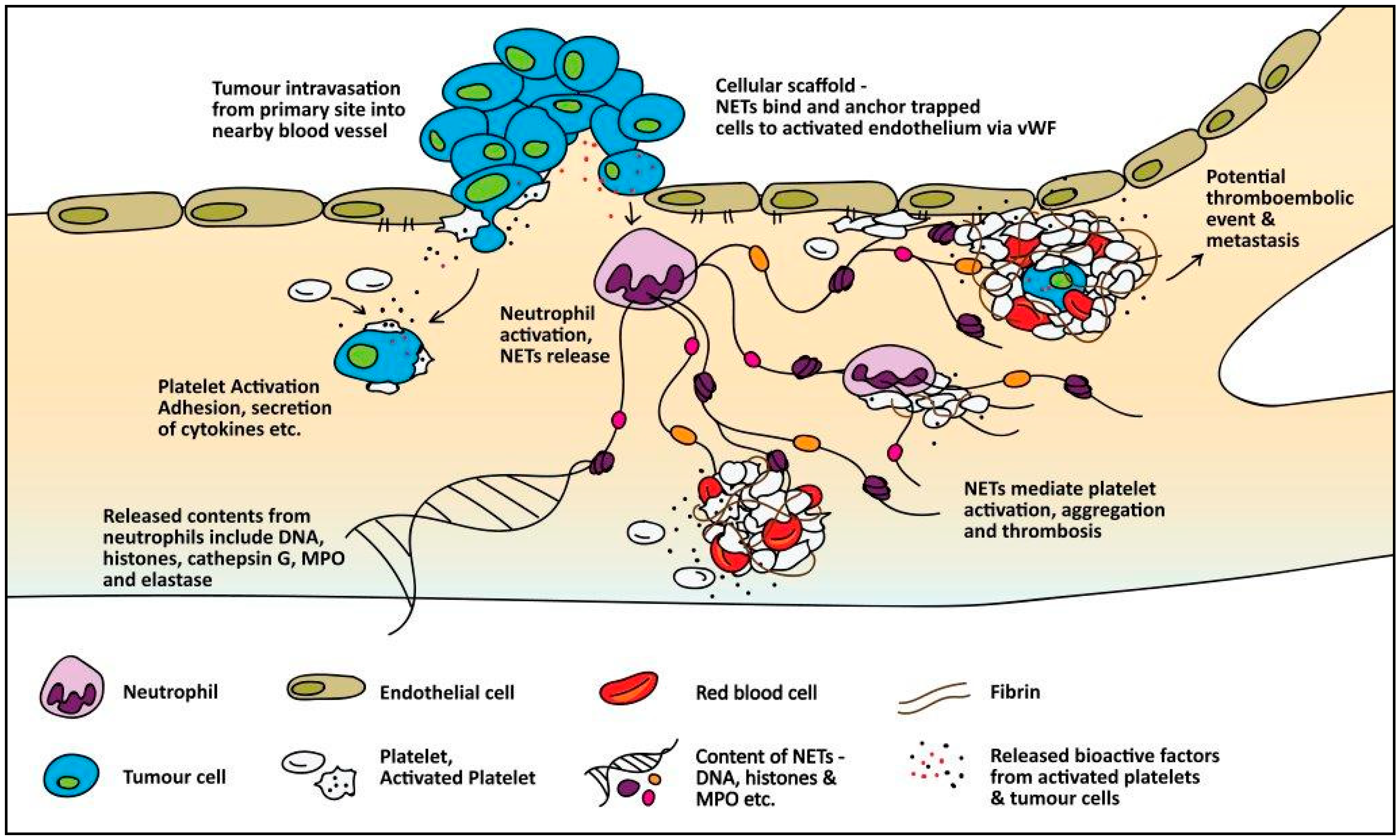
4.2. Indirect Mechanisms of Cancer-Associated Thrombosis
4.2.1. Microparticles
4.2.2. Inflammatory Cytokines
4.2.3. Adhesion Molecules
4.2.4. Neutrophil Extracellular Traps
4.2.5. Mucins
4.2.6. Hypoxia
4.2.7. Damage-Associated Molecular Patterns (DAMPs)
4.2.8. Cancer-Associated Chemotherapy
4.2.9. Coagulation Gene Defects
4.2.10. Decreased Coagulation Inhibitors
5. Patient Management
6. Cancer-Associated Thrombosis Therapy
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Prandoni, P.; Falanga, A.; Piccioli, A. Cancer and venous thromboembolism. Lancet Oncol. 2005, 6, 401–410. [Google Scholar] [CrossRef]
- Noble, S.; Pasi, J. Epidemiology and pathophysiology of cancer-associated thrombosis. Br. J. Cancer 2010, 102, S2–S9. [Google Scholar] [CrossRef] [PubMed]
- Levi, M. Cancer-related coagulopathies. Thromb. Res. 2014, 133, S70–S75. [Google Scholar] [CrossRef]
- Eichinger, S. Cancer associated thrombosis: Risk factors and outcomes. Thromb. Res. 2016, 140, S12–S17. [Google Scholar] [CrossRef]
- Falanga, A.; Marchetti, M.; Russo, L. The mechanisms of cancer-associated thrombosis. Thromb. Res. 2015, 135, S8–S11. [Google Scholar] [CrossRef]
- Fuchs, T.A.; Brill, A.; Wagner, D.D. Neutrophil Extracellular Trap (NET) Impact on Deep Vein Thrombosis. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1777–1783. [Google Scholar] [CrossRef] [PubMed]
- Aird, W.C. Vascular bed-specific thrombosis. J. Thromb. Haemost. 2007, 5, 283–291. [Google Scholar] [CrossRef] [PubMed]
- Næss, I.A.; Christiansen, S.C.; Romundstad, P.; Cannegieter, S.C.; Rosendaal, F.R.; Hammerstrøm, J. Incidence and mortality of venous thrombosis: A population-based study. J. Thromb. Haemost. 2007, 5, 692–699. [Google Scholar] [CrossRef] [PubMed]
- Connolly, G.; Francis, C.W. Cancer-associated thrombosis. Hematol. ASH Educ. Prog. 2013, 2013, 684–691. [Google Scholar] [CrossRef] [PubMed]
- Agnelli, G.; Verso, M. Management of venous thromboembolism in patients with cancer. J. Thromb. Haemost. 2011, 9, 316–324. [Google Scholar] [CrossRef] [PubMed]
- Blom, J.W.; Doggen, C.M.; Osanto, S.; Rosendaal, F.R. Malignancies, prothrombotic mutations, and the risk of venous thrombosis. JAMA 2005, 293, 715–722. [Google Scholar] [CrossRef] [PubMed]
- Kwaan, H.C. Coagulation in Cancer; Kwaan, H.C., Green, D., Eds.; Springer: Boston, MA, USA, 2009. [Google Scholar]
- Karimi, M.; Cohan, N. Cancer-Associated thrombosis. Open Cardiovasc. Med. J. 2010, 4, 78–82. [Google Scholar] [CrossRef] [PubMed]
- Fuentes, H.E.; Tafur, A.J.; Caprini, J.A. Cancer-associated thrombosis. Disease-a-Month 2016, 62, 121–158. [Google Scholar] [CrossRef] [PubMed]
- Sud, R.; Khorana, A.A. Cancer-associated thrombosis: Risk factors, candidate biomarkers and a risk model. Thromb. Res. 2009, 123, S18–S21. [Google Scholar] [CrossRef]
- Rigdon, E.E. Trousseau’s syndrome and acute arterial thrombosis. Cardiovasc. Surg. 2000, 8, 214–218. [Google Scholar] [CrossRef]
- Navi, B.B.; Reiner, A.S.; Kamel, H.; Iadecola, C.; Okin, P.M.; Elkind, M.S.V.; Panageas, K.S.; Deangelis, L.M. Risk of Arterial Thromboembolism in Patients with Cancer. J. Am. Coll. Cardiol. 2017, 70, 926–938. [Google Scholar] [CrossRef] [PubMed]
- Rumbaut, R.E.; Thiagarajan, P. Platelet-Vessel Wall Interactions in Hemostasis and Thrombosis; Morgan & Claypool Life Sciences: San Rafael, CA, USA, 2010; pp. 35–41. [Google Scholar]
- Kawano, K.; Yoshino, H.; Aoki, N.; Udagawa, H.; Watanuki, A.; Hioki, Y.; Hasumura, Y.; Yasumura, T.; Homori, M.; Murata, M.; et al. Shear-induced platelet aggregation increases in patients with proximal and severe coronary artery stenosis. Clin. Cardiol. 2002, 25, 154–160. [Google Scholar] [CrossRef] [PubMed]
- Tuzovic, M.; Herrmann, J.; Iliescu, C.; Marmagkiolis, K.; Ziaeian, B.; Yang, E.H. Arterial Thrombosis in Patients with Cancer. Curr. Treat. Options Cardiovasc. Med. 2018, 20, 40. [Google Scholar] [CrossRef] [PubMed]
- Mi, Y.; Yan, S.; Lu, Y.; Liang, Y.; Li, C. Venous thromboembolism has the same risk factors as atherosclerosis: A PRISMA-compliant systemic review and meta-analysis. Medicine (Baltimore) 2016, 95, e4495. [Google Scholar] [CrossRef] [PubMed]
- Levi, M. Management of cancer-associated disseminated intravascular coagulation. Thrombo. Res. 2016, 140, S66–S70. [Google Scholar] [CrossRef]
- Kwaan, H.C.; Gordon, L.I. Thrombotic microangiopathy in the cancer patient. Acta Haematol. 2001, 106, 52–56. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Hwang, I.G.; Jang, J.; Park, S.H.; Kang, J.H.; Oh, S.Y.; Kwon, H.; Lim, D.H.; Park, K.; Lee, S. Treatment outcomes of chemotheraphy for advanced gastric cancer with disseminated intravascular coagulation. J. Clin. Oncol. 2011, 29, e14532. [Google Scholar] [CrossRef]
- Wada, H.; Matsumoto, T.; Suzuki, K.; Imai, H.; Katayama, N.; Iba, T.; Matsumoto, M. Differences and similarities between disseminated intravascular coagulation and thrombotic microangiopathy. Thrombo. J. 2018, 16, 14. [Google Scholar] [CrossRef] [PubMed]
- Thachil, J.; Falanga, A.; Levi, M.; Liebman, H.; Di Nisio, M. Management of cancer-associated disseminated intravascular coagulation: Guidance from the SSC of the ISTH. J. Thromb. Haemost. 2015, 13, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Sallah, S.; Wan, J.Y.; Nguyen, N.P.; Hanrahan, L.R.; Sigounas, G. Disseminated intravascular coagulation in solid tumors: Clinical and pathologic study. Thromb. Haemost. 2001, 86, 828–833. [Google Scholar] [PubMed]
- Barbui, T.; Falanga, A. Disseminated intravascular coagulation in acute leukemia. Semin. Thromb. Hemost. 2001, 27, 593–604. [Google Scholar] [CrossRef] [PubMed]
- Amer, M.H. Cancer-associated thrombosis: Clinical presentation and survival. Cancer Manag. Res. 2013, 5, 165–178. [Google Scholar] [CrossRef] [PubMed]
- Silverstein, M.D.; Heit, J.A.; Mohr, D.N.; Petterson, T.M.; O’Fallon, W.; Melton Iii, L.J. Trends in the incidence of deep vein thrombosis and pulmonary embolism: A 25-year population-based study. Arch. Intern. Med. 1998, 158, 585–593. [Google Scholar] [CrossRef] [PubMed]
- Tsai, A.W.; Cushman, M.; Rosamond, W.D.; Heckbert, S.R.; Polak, J.F.; Folsom, A.R. Cardiovascular risk factors and venous thromboembolism incidence: The longitudinal investigation of thromboembolism etiology. Arch. Intern. Med. 2002, 162, 1182–1189. [Google Scholar] [CrossRef] [PubMed]
- Khorana, A.A.; Francis, C.W.; Culakova, E.; Kuderer, N.M.; Lyman, G.H. Frequency, risk factors, and trends for venous thromboembolism among hospitalized cancer patients. Cancer 2007, 110, 2339–2346. [Google Scholar] [CrossRef] [PubMed]
- Khorana, A.A.; Francis, C.W.; Culakova, E.; Fisher, R.I.; Kuderer, N.M.; Lyman, G.H. Thromboembolism in hospitalized neutropenic cancer patients. J. Clin. Oncol. 2006, 24, 484. [Google Scholar] [CrossRef] [PubMed]
- Vergati, M.; Della-Morte, D.; Ferroni, P.; Cereda, V.; Tosetto, L.; La Farina, F.; Guadagni, F.; Roselli, M. Increased Risk of Chemotherapy-Associated Venous Thromboembolism in Elderly Patients with Cancer. Rejuvenation Res. 2013, 16, 224–231. [Google Scholar] [CrossRef] [PubMed]
- Previtali, E.; Bucciarelli, P.; Passamonti, S.M.; Martinelli, I. Risk factors for venous and arterial thrombosis. Blood Transf. 2011, 9, 120–138. [Google Scholar]
- Chew, H.K.; Wun, T.; Harvey, D.; Zhou, H.; White, R.H. Incidence of venous thromboembolism and its effect on survival among patients with common cancers. Arch. Intern. Med. 2006, 166, 458–464. [Google Scholar] [CrossRef] [PubMed]
- Stein, P.D.; Beemath, A.; Meyers, F.A.; Skaf, E.; Sanchez, J.; Olson, R.E. Incidence of Venous Thromboembolism in Patients Hospitalized with Cancer. Am. J. Med. 2006, 119, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Khorana, A.A.; Francis, C.W.; Culakova, E.; Lyman, G.H. Risk factors for chemotherapy-associated venous thromboembolism in a prospective observational study. Cancer 2005, 104, 2822–2829. [Google Scholar] [CrossRef] [PubMed]
- Al Diab, A.I. Cancer-related venous thromboembolism: Insight into underestimated risk factors. Hematol. Oncol. Stem Cell Ther. 2010, 3, 191–195. [Google Scholar] [CrossRef] [PubMed]
- Agnelli, M.G.; Bolis, M.G.; Capussotti, M.L.; Scarpa, M.R.; Tonelli, M.F.; Bonizzoni, M.E.; Moia, M.M.; Parazzini, M.F.; Rossi, M.R.; Sonaglia, M.F.; et al. A Clinical Outcome-Based Prospective Study on Venous Thromboembolism After Cancer Surgery: The @RISTOS Project. Ann. Surg. 2006, 243, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Cushman, M. Epidemiology and Risk Factors for Venous Thrombosis. Semin. Hematol. 2007, 44, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Connolly, G.; Khorana, A.A. Emerging risk stratification approaches to cancer-associated thrombosis: Risk factors, biomarkers and a risk score. Thromb. Res. 2010, 125, S1–S7. [Google Scholar] [CrossRef]
- Khorana, A.A.; Connolly, G.C. Assessing Risk of Venous Thromboembolism in the Patient With Cancer. J. Clin. Oncol. 2009, 27, 4839–4847. [Google Scholar] [CrossRef] [PubMed]
- Horsted, F.; West, J.; Grainge, M.J. Risk of Venous Thromboembolism in Patients with Cancer: A Systematic Review and Meta-Analysis. PLoS Med. 2012, 9, e1001275. [Google Scholar] [CrossRef] [PubMed]
- Haddad, T.C.; Greeno, E.W. Chemotherapy-induced thrombosis. Thrombo. Res. 2006, 118, 555–568. [Google Scholar] [CrossRef] [PubMed]
- Cronin-Fenton, D.P.; Søndergaard, F.; Pedersen, L.A.; Fryzek, J.P.; Cetin, K.; Acquavella, J.; Baron, J.A.; Sørensen, H.T. Hospitalisation for venous thromboembolism in cancer patients and the general population: A population-based cohort study in Denmark, 1997–2006. Br. J. Cancer 2010, 103, 947. [Google Scholar] [CrossRef] [PubMed]
- Dickmann, B.; Ahlbrecht, J.; Ay, C.; Dunkler, D.; Thaler, J.; Scheithauer, W.; Quehenberger, P.; Zielinski, C.; Pabinger, I. Regional lymph node metastases are a strong risk factor for venous thromboembolism: Results from the Vienna Cancer and Thrombosis Study. Haematologica 2013, 98, 1309–1314. [Google Scholar] [CrossRef] [PubMed]
- Blom, J.W.; Osanto, S.; Rosendaal, F.R. The risk of a venous thrombotic event in lung cancer patients: Higher risk for adenocarcinoma than squamous cell carcinoma. J.Thrombo. Haemost. 2004, 2, 1760–1765. [Google Scholar] [CrossRef] [PubMed]
- Alcalay, A.; Wun, T.; Khatri, V.; Chew, H.K.; Harvey, D.; Zhou, H.; White, R.H. Venous Thromboembolism in Patients With Colorectal Cancer: Incidence and Effect on Survival. J. Clin. Oncol. 2006, 24, 1112–1118. [Google Scholar] [CrossRef] [PubMed]
- Chew, H.K.; Wun, T.; Harvey, D.J.; Zhou, H.; White, R.H. Incidence of Venous Thromboembolism and the Impact on Survival in Breast Cancer Patients. J. Clin. Oncol. 2006, 25, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Ahlbrecht, J.; Dickmann, B.; Ay, C.; Dunkler, D.; Thaler, J.; Schmidinger, M.; Quehenberger, P.; Haitel, A.; Zielinski, C.; Pabinger, I. Tumor Grade Is Associated With Venous Thromboembolism in Patients With Cancer: Results From the Vienna Cancer and Thrombosis Study. J. Clin. Oncol. 2012, 30, 3870–3875. [Google Scholar] [CrossRef] [PubMed]
- Easaw, J.C.; McCall, S.; Azim, A. ClotAssist: A program to treat cancer-associated thrombosis in an outpatient pharmacy setting. J. Oncol. Pharm. Pract. 2018. [Google Scholar] [CrossRef] [PubMed]
- Khorana, A.A.; Dalal, M.; Lin, J.; Connolly, G.C. Incidence and predictors of venous thromboembolism (VTE) among ambulatory high-risk cancer patients undergoing chemotherapy in the United States. Cancer 2013, 119, 648–655. [Google Scholar] [CrossRef] [PubMed]
- Piovella, F.; Wang, C.J.; Lu, H.; Lee, K.; Lee, L.H.; Lee, W.; Turpie, A.; Gallus, A.; Planes, A.; Passera, R.; et al. Deep-vein thrombosis rates after major orthopedic surgery in Asia. An epidemiological study based on postoperative screening with centrally adjudicated bilateral venography. J. Thromb. Haemost. 2005, 3, 2664–2670. [Google Scholar] [CrossRef] [PubMed]
- Hakkim, A.; Fuchs, A.T.; Martinez, E.N.; Hess, S.; Prinz, H.; Zychlinsky, A.; Waldmann, H. Activation of the Raf-MEK-ERK pathway is required for neutrophil extracellular trap formation. Nat. Chem. Biol. 2010, 7, 75. [Google Scholar] [CrossRef] [PubMed]
- Khorana, A.A.; Kuderer, N.M.; Culakova, E.; Lyman, G.H.; Francis, C.W. Development and validation of a predictive model for chemotherapy-associated thrombosis. Blood 2008, 111, 4902–4907. [Google Scholar] [CrossRef] [PubMed]
- Rogers, M.A.M.; Levine, D.A.; Blumberg, N.; Flanders, S.A.; Chopra, V.; Langa, K.M. Triggers of Hospitalization for Venous Thromboembolism. Circulation 2012, 125, 2092–2099. [Google Scholar] [CrossRef] [PubMed]
- Doll, D.C.; List, A.F.; Greco, F.; Hainsworth, J.D.; Hande, K.R.; Johnson, D.H. Acute vascular ischemic events after cisplatin-based combination chemotherapy for germ-cell tumors of the testis. Ann. Intern. Med. 1986, 105, 48–51. [Google Scholar] [CrossRef] [PubMed]
- Numico, G.; Garrone, O.; Dongiovanni, V.; Silvestris, N.; Colantonio, I.; Costanzo, G.D.; Granetto, C.; Occelli, M.; Fea, E.; Heouaine, A.; et al. Prospective evaluation of major vascular events in patients with nonsmall cell lung carcinoma treated with cisplatin and gemcitabine. Cancer 2005, 103, 994–999. [Google Scholar] [CrossRef] [PubMed]
- Czaykowski, P.M.; Moore, M.J.; Tannock, I.F. High Risk of Vascular Events in Patients With Urothelial Transitional Cell Carcinoma Treated with Cisplatin Based Chemotherapy. J. Urol. 1998, 160, 2021–2024. [Google Scholar] [CrossRef]
- Cunningham, D.; Starling, N.; Rao, S.; Iveson, T.; Nicolson, M.; Coxon, F.; Middleton, G.; Daniel, F.; Oates, J.; Norman, A.R. Capecitabine and Oxaliplatin for Advanced Esophagogastric Cancer. N. Engl. J. Med. 2008, 358, 36–46. [Google Scholar] [CrossRef] [PubMed]
- John, A.H. Epidemiology of venous thromboembolism. Nat. Rev. Cardiol. 2015, 12, 464–474. [Google Scholar]
- Nalluri, S.; Chu, D.; Keresztes, R.; Zhu, X.; Wu, S. Risk of venous thromboembolism with the angiogenesis inhibitor bevacizumab in cancer patients: A meta-analysis. JAMA 2008, 300, 2277–2285. [Google Scholar] [CrossRef] [PubMed]
- Schutz, F.A.B.; Je, Y.; Azzi, G.R.; Nguyen, P.L.; Choueiri, T.K. Bevacizumab increases the risk of arterial ischemia: A large study in cancer patients with a focus on different subgroup outcomes. Ann. Oncol. 2011, 22, 1404–1412. [Google Scholar] [CrossRef] [PubMed]
- Scappaticci, F.A.; Skillings, J.R.; Holden, S.N.; Gerber, H.-P.; Miller, K.; Kabbinavar, F.; Bergsland, E.; Ngai, J.; Holmgren, E.; Wang, J.; et al. Arterial Thromboembolic Events in Patients with Metastatic Carcinoma Treated with Chemotherapy and Bevacizumab. JNCI 2007, 99, 1232–1239. [Google Scholar] [CrossRef] [PubMed]
- DeLoughery, T.G. Hemostasis and Thrombosis, 3rd ed.; DeLoughery, T.G., Ed.; Springer International Publishing: Cham, Switzerland, 2015. [Google Scholar]
- Lee, A.Y.Y.; Levine, M.N.; Butler, G.; Webb, C.; Costantini, L.; Gu, C.; Julian, J.A. Incidence, risk factors, and outcomes of catheter-related thrombosis in adult patients with cancer. J. Clin. Oncol. 2006, 24, 1404. [Google Scholar] [CrossRef] [PubMed]
- Ruf, W.; Edgington, T.S. Structural biology of tissue factor, the initiator of thrombogenesis in vivo. FASEB J. 1994, 8, 385–390. [Google Scholar] [CrossRef] [PubMed]
- Khorana, A.A.; Ahrendt, S.A.; Ryan, C.K.; Francis, C.W.; Hruban, R.H.; Hu, Y.C.; Hostetter, G.; Harvey, J.; Taubman, M.B. Tissue Factor Expression, Angiogenesis, and Thrombosis in Pancreatic Cancer. Clin. Cancer Res. 2007, 13, 2870–2875. [Google Scholar] [CrossRef] [PubMed]
- Uno, K.; Homma, S.; Satoh, T.; Nakanishi, K.; Abe, D.; Matsumoto, K.; Oki, A.; Tsunoda, H.; Yamaguchi, I.; Nagasawa, T.; et al. Tissue factor expression as a possible determinant of thromboembolism in ovarian cancer. Br. J. Cancer 2007, 96, 290–295. [Google Scholar] [CrossRef] [PubMed]
- Kakkar, A.K.; Lemoine, N.R.; Scully, M.F.; Tebbutt, S.; Williamson, R.C.N. Tissue factor expression correlates with histological grade in human pancreatic cancer. Br. J. Surg. 1995, 82, 1101–1104. [Google Scholar] [CrossRef] [PubMed]
- Nitori, N.; Ino, Y.; Nakanishi, Y.; Yamada, T.; Honda, K.; Yanagihara, K.; Kosuge, T.; Kanai, Y.; Kitajima, M.; Hirohashi, S. Prognostic Significance of Tissue Factor in Pancreatic Ductal Adenocarcinoma. Clin. Cancer Res. 2005, 11, 2531–2539. [Google Scholar] [CrossRef] [PubMed]
- Zwicker, J.I.; Liebman, H.A.; Neuberg, D.; Lacroix, R.; Bauer, K.A.; Furie, B.C.; Furie, B. Tumor-Derived Tissue Factor-Bearing Microparticles are Associated with Venous Thromboembolic Events in Malignancy. Clin. Cancer Res. 2009, 15, 6830–6840. [Google Scholar] [CrossRef] [PubMed]
- Dvorak, H.; Quay, S.; Orenstein, N.; Dvorak, A.; Hahn, P.; Bitzer, A.; Carvalho, A. Tumor shedding and coagulation. Science 1981, 212, 923–924. [Google Scholar] [CrossRef] [PubMed]
- Thomas, G.M.; Panicot-Dubois, L.; Lacroix, R.; Dignat-George, F.; Lombardo, D.; Dubois, C. Cancer cell–derived microparticles bearing P-selectin glycoprotein ligand 1 accelerate thrombus formation in vivo. J. Exp. Med. 2009, 206, 1913–1927. [Google Scholar] [CrossRef] [PubMed]
- Geddings, J.E.; Hisada, Y.; Boulaftali, Y.; Getz, T.M.; Whelihan, M.; Fuentes, R.; Dee, R.; Cooley, B.C.; Key, N.S.; Wolberg, A.S.; et al. Tissue Factor-positive Tumor Microvesicles Activate Platelets and Enhance Thrombosis in Mice. JTH 2016, 14, 153–166. [Google Scholar] [CrossRef] [PubMed]
- Tesselaar, M.E.; Romijn, F.P.; Van Der Linden, I.K.; Prins, F.A.; Bertina, R.M.; Osanto, S. Microparticle-associated tissue factor activity: A link between cancer and thrombosis? J. Thromb. Haemost. 2007, 5, 520–527. [Google Scholar] [CrossRef] [PubMed]
- Gardiner, C.; Harrison, P.; Belting, M.; Böing, A.; Campello, E.; Carter, B.S.; Collier, M.E.; Coumans, F.; Ettelaie, C.; van Es, N.; et al. Extracellular vesicles, tissue factor, cancer and thrombosis–discussion themes of the ISEV 2014 Educational Day. J. Extracell. Vesicles 2015, 4. [Google Scholar] [CrossRef] [PubMed]
- Stark, K.; Schubert, I.; Joshi, U.; Kilani, B.; Hoseinpour, P.; Thakur, M.; Grünauer, P.; Pfeiler, S.; Schmidergall, T.; Stockhausen, S.; et al. Distinct Pathogenesis of Pancreatic Cancer Microvesicle–Associated Venous Thrombosis Identifies New Antithrombotic Targets In Vivo. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 772–786. [Google Scholar] [CrossRef] [PubMed]
- Geddings, J.E.; Mackman, N. Tumor-derived tissue factor–positive microparticles and venous thrombosis in cancer patients. Blood 2013, 122, 1873–1880. [Google Scholar] [CrossRef] [PubMed]
- Shindo, K.; Aishima, S.; Ohuchida, K.; Fujiwara, K.; Fujino, M.; Mizuuchi, Y.; Hattori, M.; Mizumoto, K.; Tanaka, M.; Oda, Y. Podoplanin expression in cancer-associated fibroblasts enhances tumor progression of invasive ductal carcinoma of the pancreas. Mol. Cancer 2013, 12, 168. [Google Scholar] [CrossRef] [PubMed]
- Kitano, H.; Kageyama, S.-I.; Hewitt, S.M.; Hayashi, R.; Doki, Y.; Ozaki, Y.; Fujino, S.; Takikita, M.; Kubo, H.; Fukuoka, J. Podoplanin Expression in Cancerous Stroma Induces Lymphangiogenesis and Predicts Lymphatic Spread and Patient Survival. Arch. Pathol. Lab. Med. 2010, 134, 1520–1527. [Google Scholar] [PubMed]
- Suzuki-Inoue, K.; Kato, Y.; Inoue, O.; Kaneko, M.K.; Mishima, K.; Yatomi, Y.; Yamazaki, Y.; Narimatsu, H.; Ozaki, Y. Involvement of the Snake Toxin Receptor CLEC-2, in Podoplanin-mediated Platelet Activation, by Cancer Cells. J. Biol. Chem. 2007, 282, 25993–26001. [Google Scholar] [CrossRef] [PubMed]
- Gagliano, N.; Celesti, G.; Tacchini, L.; Pluchino, S.; Sforza, C.; Rasile, M.; Valerio, V.; Laghi, L.; Conte, V.; Procacci, P. Epithelial-to-mesenchymal transition in pancreatic ductal adenocarcinoma: Characterization in a 3D-cell culture model. World J. Gastroenterol. 2016, 22, 4466–4483. [Google Scholar] [CrossRef] [PubMed]
- Payne, H.; Ponomaryov, T.; Watson, S.P.; Brill, A. Mice with a deficiency in CLEC-2 are protected against deep vein thrombosis. Blood 2017, 129, 2013–2020. [Google Scholar] [CrossRef] [PubMed]
- Riedl, J.; Preusser, M.; Nazari, P.M.S.; Posch, F.; Panzer, S.; Marosi, C.; Birner, P.; Thaler, J.; Brostjan, C.; Lötsch, D.; et al. Podoplanin expression in primary brain tumors induces platelet aggregation and increases risk of venous thromboembolism. Blood 2017, 129, 1831–1839. [Google Scholar] [CrossRef] [PubMed]
- Zwicker, J.I. Risking thromboembolism: Podoplanin and glioma. Blood 2017, 129, 1742–1743. [Google Scholar] [CrossRef] [PubMed]
- Mege, D.; Laurence, P.D.; Mehdi, O.; Stéphane, R.; Igor, S.; Bernard, S.; Françoise, D.G.; Christophe, D. The origin and concentration of circulating microparticles differ according to cancer type and evolution: A prospective single-center study. Int. J. Cancer 2016, 138, 939–948. [Google Scholar] [CrossRef] [PubMed]
- Lupu-Meiri, M.; Geras-Raaka, E.; Lupu, R.; Shapira, H.; Sandbank, J.; Segal, L.; Gershengorn, M.C.; Oron, Y. Knock-down of plasminogen-activator inhibitor-1 enhances expression of E-cadherin and promotes epithelial differentiation of human pancreatic adenocarcinoma cells. J. Cell. Physiol. 2012, 227, 3621–3628. [Google Scholar] [CrossRef] [PubMed]
- Westrick, R.; Eitzman, D. Plasminogen activator inhibitor-1 in vascular thrombosis. Curr. Drug Targets 2007, 8, 966–1002. [Google Scholar] [CrossRef] [PubMed]
- Andrén-Sandberg, Å.; Lecander, I.; Martinsson, G.; Åstedt, B. Peaks in plasma plasminogen activator inhibitor-1 concentration may explain thrombotic events in cases of pancreatic carcinoma. Cancer 1992, 69, 2884–2887. [Google Scholar] [CrossRef]
- Chen, N.; Ren, M.; Li, R.; Deng, X.; Li, Y.; Yan, K.; Xiao, L.; Yang, Y.; Wang, L.; Luo, M.; et al. Bevacizumab promotes venous thromboembolism through the induction of PAI-1 in a mouse xenograft model of human lung carcinoma. Mol. Cancer 2015, 14, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Gordon, S.G.; Franks, J.J.; Lewis, B. Cancer procoagulant A: A factor X activating procoagulant from malignant tissue. Thromb. Res. 1975, 6, 127–137. [Google Scholar] [CrossRef]
- Gordon, S.S. A proteolytic procoagulant associated with malignant transformation. J. Histochem. Cytochem. 1981, 29, 457–463. [Google Scholar] [CrossRef] [PubMed]
- Mielicki, W.P.; Tenderenda, M.; Rutkowski, P.; Chojnowski, K. Activation of blood coagulation and the activity of cancer procoagulant (EC 3.4.22.26) in breast cancer patients. Cancer Lett. 1999, 146, 61–66. [Google Scholar] [CrossRef]
- Francis, J.L.; El-Baruni, K.; Roath, O.S.; Taylor, I. Factor X-activating activity in normal and malignant colorectal tissue. Thromb. Res. 1988, 52, 207–217. [Google Scholar] [CrossRef]
- Raasi, S.; Mielicki, W.P.; Gordon, S.G.; Korte, W. Properties of proteins in cancer procoagulant preparations that are detected by anti-tissue factor antibodies. Arch. Biochem. Biophys. 2004, 428, 131–135. [Google Scholar] [CrossRef] [PubMed]
- Grignani, G.; Jamieson, G.A. Platelets in tumor metastasis: Generation of adenosine diphosphate by tumor cells is specific but unrelated to metastatic potential. Blood 1988, 71, 844. [Google Scholar] [PubMed]
- Wojtukiewicz, M.Z.; Rucinska, M.; Zimnoch, L.; Jaromin, J.; Piotrowski, Z.; Rózanska-Kudelska, M.; Kisiel, W.; Kudryk, B.J. Expression of Prothrombin Fragment 1+2 in Cancer Tissue as an Indicator of Local Activation of Blood Coagulation. Thromb. Res. 2000, 97, 335–342. [Google Scholar] [CrossRef]
- Haas, S.L.; Jesnowski, R.; Steiner, M.; Hummel, F.; Ringel, J.; Burstein, C.; Nizze, H.; Liebe, S.; Löhr, J.M. Expression of tissue factor in pancreatic adenocarcinoma is associated with activation of coagulation. WJG 2006, 12, 4843–4849. [Google Scholar] [PubMed]
- Falanga, A.; Panova-Noeva, M.; Russo, L. Procoagulant mechanisms in tumour cells. Best Pract. Res. Clin. Haematol. 2009, 22, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Bevilacqua, M.P.; Pober, J.S.; Majeau, G.R.; Fiers, W.; Cotran, R.S.; Gimbrone, M.A. Recombinant Tumor Necrosis Factor Induces Procoagulant Activity in Cultured Human Vascular Endothelium: Characterization and Comparison with the Actions of Interleukin 1. Proc. Natl. Acad. Sci. USA 1986, 83, 4533–4537. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.; Esmon, C.; Esmon, N. Tumor necrosis factor leads to the internalization and degradation of thrombomodulin from the surface of bovine aortic endothelial cells in culture. Blood 1989, 73, 159–165. [Google Scholar] [PubMed]
- Nawroth, P.P.; Stern, D.M. Endothelial Cell Procoagulant Properties and the Host Response. Semin. Thromb. Hemost. 1987, 13, 391–397. [Google Scholar] [CrossRef] [PubMed]
- Kanno, K.; Hirata, Y.; Imai, T.; Iwashina, M.; Marumo, F. Regulation of inducible nitric oxide synthase gene by interleukin-1 beta in rat vascular endothelial cells. Am. J. Physiol. Heart Circ. Physiol. 1994, 267, H2318–H2324. [Google Scholar] [CrossRef] [PubMed]
- Johnson, A.; Phelps, D.T.; Ferro, T.J. Tumor necrosis factor-alpha decreases pulmonary artery endothelial nitrovasodilator via protein kinase C. Am. J. Physiol. Lung Cell. Mol. Physiol. 1994, 267, L318–L325. [Google Scholar] [CrossRef] [PubMed]
- Clauss, M. Vascular permeability factor: A tumor-derived polypeptide that induces endothelial cell and monocyte procoagulant activity, and promotes monocyte migration. J. Exp. Med. 1990, 172, 1535–1545. [Google Scholar] [CrossRef] [PubMed]
- Falanga, A.; Marchetti, M.; Evangelista, V.; Manarini, S.; Oldani, E.; Giovanelli, S.; Galbusera, M.; Cerletti, C.; Barbui, T. Neutrophil Activation and Hemostatic Changes in Healthy Donors Receiving Granulocyte Colony-Stimulating Factor. Blood 1999, 93, 2506–2514. [Google Scholar] [PubMed]
- Kaneko, T.; Fujii, S.; Matsumoto, A.; Goto, D.; Makita, N.; Hamada, J.; Moriuchi, T.; Kitabatake, A. Induction of Tissue Factor Expression in Endothelial Cells by Basic Fibroblast Growth Factor and its Modulation by Fenofibric acid. Thromb. J. 2003, 1, 6. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Giavazzi, R.; Foppolo, M.; Dossi, R.; Remuzzi, A. Rolling and adhesion of human tumor cells on vascular endothelium under physiological flow conditions. J. Clin. Investig. 1993, 92, 3038–3044. [Google Scholar] [CrossRef] [PubMed]
- Abdol Razak, N.; Elaskalani, O.; Metharom, P. Pancreatic Cancer-Induced Neutrophil Extracellular Traps: A Potential Contributor to Cancer-Associated Thrombosis. Int. J. Mol. Sci. 2017, 18, 487. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil Extracellular Traps Kill Bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef] [PubMed]
- von Brühl, M.-L.; Stark, K.; Steinhart, A.; Chandraratne, S.; Konrad, I.; Lorenz, M.; Khandoga, A.; Tirniceriu, A.; Coletti, R.; Köllnberger, M.; et al. Monocytes, neutrophils, and platelets cooperate to initiate and propagate venous thrombosis in mice in vivo. J. Exp. Med. 2012, 209, 819–835. [Google Scholar] [CrossRef] [PubMed]
- Brill, A.; Fuchs, T.A.; Savchenko, A.S.; Thomas, G.M.; Martinod, K.; De Meyer, S.F.; Bhandari, A.A.; Wagner, D.D. Neutrophil extracellular traps promote deep vein thrombosis in mice. J. Thromb. Haemost. 2012, 10, 136–144. [Google Scholar] [CrossRef] [PubMed]
- Leal, A.C.; Mizurini, D.M.; Gomes, T.; Rochael, N.C.; Saraiva, E.M.; Dias, M.S.; Werneck, C.C.; Sielski, M.S.; Vicente, C.P.; Monteiro, R.Q. Tumor-Derived Exosomes Induce the Formation of Neutrophil Extracellular Traps: Implications For The Establishment of Cancer-Associated Thrombosis. Sci. Rep. 2017, 7, 6438. [Google Scholar] [CrossRef] [PubMed]
- Lam, F.; Cruz, M.; Parikh, K.; Rumbaut, R. Histones stimulate von Willebrand factor release in vitro and in vivo. Haematologica 2016, 101, e277–e279. [Google Scholar] [CrossRef] [PubMed]
- McDonald, B.; Davis, R.P.; Kim, S.-J.; Tse, M.; Esmon, C.T.; Kolaczkowska, E.; Jenne, C.N. Platelets and neutrophil extracellular traps collaborate to promote intravascular coagulation during sepsis in mice. Blood 2017, 129, 1357–1367. [Google Scholar] [CrossRef] [PubMed]
- Mauracher, L.M.; Posch, F.; Martinod, K.; Grilz, E.; Däullary, T.; Hell, L.; Brostjan, C.; Zielinski, C.; Ay, C.; Wagner, D.D.; et al. Citrullinated histone H3, a biomarker of neutrophil extracellular trap formation, predicts the risk of venous thromboembolism in cancer patients. J. Thromb. Haemost. 2018, 16, 508–518. [Google Scholar] [CrossRef] [PubMed]
- Kaur, S.; Kumar, S.; Momi, N.; Sasson, A.R.; Batra, S.K. Mucins in pancreatic cancer and its microenvironment. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 607–620. [Google Scholar] [CrossRef] [PubMed]
- Hollingsworth, M.; Swanson, J. Mucins in cancer: Protection and control of the cell surface. Nat. Rev. Cancer 2004, 4, 45. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.J.; Borsig, L.; Han, H.-L.; Varki, N.M.; Varki, A. Distinct Selectin Ligands on Colon Carcinoma Mucins Can Mediate Pathological Interactions among Platelets, Leukocytes, and Endothelium. Am. J. Pathol. 1999, 155, 461–472. [Google Scholar] [CrossRef]
- Wahrenbrock, M.; Borsig, L.; Le, D.; Varki, N.; Varki, A. Selectin-mucin interactions as a probable molecular explanation for the association of Trousseau syndrome with mucinous adenocarcinomas. J. Clin. Investig. 2003, 112, 853–862. [Google Scholar] [CrossRef] [PubMed]
- Shao, B.; Wahrenbrock, M.G.; Yao, L.; David, T.; Coughlin, S.R.; Xia, L.; Varki, A.; McEver, R.P. Carcinoma mucins trigger reciprocal activation of platelets and neutrophils in a murine model of Trousseau syndrome. Blood 2011, 118, 4015–4023. [Google Scholar] [CrossRef] [PubMed]
- Sambrano, G.R.; Huang, W.; Faruqi, T.; Mahrus, S.; Craik, C.; Coughlin, S.R. Cathepsin G Activates Protease-activated Receptor-4 in Human Platelets. J. Biol. Chem. 2000, 275, 6819–6823. [Google Scholar] [CrossRef] [PubMed]
- Koong, A.C.; Mehta, V.K.; Le, Q.T.; Fisher, G.A.; Terris, D.J.; Brown, J.M.; Bastidas, A.J.; Vierra, M. Pancreatic tumors show high levels of hypoxia. Int. J. Radiat. Oncol. Biol. Phys. 2000, 48, 919–922. [Google Scholar] [CrossRef]
- Muz, B.; de la Puente, P.; Azab, F.; Azab, A.K. The role of hypoxia in cancer progression, angiogenesis, metastasis, and resistance to therapy. Hypoxia 2015, 3, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Caplan, M.S.; Adler, L.; Kelly, A.; Hsueh, W. Hypoxia increases stimulus-induced PAF production and release from human umbilical vein endothelial cells. Biochim. Biophys. Acta 1992, 1128, 205–210. [Google Scholar] [CrossRef]
- Takahashi, T.; Hato, F.; Yamane, T.; Fukumasu, H.; Suzuki, K.; Ogita, S.; Nishizawa, Y.; Kitagawa, S. Activation of Human Neutrophil by Cytokine-Activated Endothelial Cells. Circ. Res. 2001, 88, 422. [Google Scholar] [CrossRef] [PubMed]
- Kubes, P.; Ibbotson, G.; Russell, J.; Wallace, J.L.; Granger, D.N. Role of platelet-activating factor in ischemia/reperfusion-induced leukocyte adherence. Am. J. Physiol.-Gastrointest. Liver Physiol. 1990, 259, G300–G305. [Google Scholar] [CrossRef] [PubMed]
- Di Virgilio, F.; Adinolfi, E. Extracellular purines, purinergic receptors and tumor growth. Oncogene 2016, 36, 293. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, C.; Huebener, P.; Schwabe, R.F. Damage-associated molecular patterns in cancer: A double-edged sword. Oncogene 2016, 35, 5931–5941. [Google Scholar] [CrossRef] [PubMed]
- Rojas, A.; Delgado-López, F.; Perez-Castro, R.; Gonzalez, I.; Romero, J.; Rojas, I.; Araya, P.; Añazco, C.; Morales, E.; Llanos, J. HMGB1 enhances the protumoral activities of M2 macrophages by a RAGE-dependent mechanism. Tumour Biol. 2016, 37, 3321–3329. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Wang, H.; Zhang, M.; Liu, J.; Lv, B.; Chen, F. HMGB1: A novel protein that induced platelets active and aggregation via Toll-like receptor-4, NF-κB and cGMP dependent mechanisms. Diagn. Pathol. 2015, 10, 134. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, T.A.; Bhandari, A.A.; Wagner, D.D. Histones induce rapid and profound thrombocytopenia in mice. Blood 2011, 118, 3708–3714. [Google Scholar] [CrossRef] [PubMed]
- Tadie, J.-M.; Bae, H.-B.; Jiang, S.; Park, D.W.; Bell, C.P.; Yang, H.; Pittet, J.-F.; Tracey, K.; Thannickal, V.J.; Abraham, E.; et al. HMGB1 promotes neutrophil extracellular trap formation through interactions with Toll-like receptor 4. Am. J. Physiol. 2013, 304, L342–L349. [Google Scholar] [CrossRef] [PubMed]
- Sipes, J.N.; Suratt, P.M.; Teates, C.D.; Barada, F.A.; Davis, J.S.; Tegtmeyer, C.J. A Prospective Study of Plasma DNA in the Diagnosis of Pulmonary Embolism. Am. Rev. Respir. Dis. 1978, 118, 475–478. [Google Scholar] [PubMed]
- Lechner, D.; Kollars, M.; Gleiss, A.; Kyrle, P.A.; Weltermann, A. Chemotherapy-induced thrombin generation via procoagulant endothelial microparticles is independent of tissue factor activity. J. Thromb. Haemost. 2007, 5, 2445–2452. [Google Scholar] [CrossRef] [PubMed]
- Palumbo, A.; Rajkumar, S.V.; Dimopoulos, M.A.; Richardson, P.G.; Miguel, J.S.; Barlogie, B.; Harousseau, J.; Zonder, J.A.; Cavo, M.; Zangari, M.; et al. Prevention of thalidomide- and lenalidomide-associated thrombosis in myeloma. Leukemia 2007, 22, 414. [Google Scholar] [CrossRef] [PubMed]
- Key, N.; Makris, M.; O’Shaughnessy, D.; Lillicrap, D. Practical Hemostasis and Thrombosis, 2nd ed.; Wiley-Blackwell: Oxford, UK, 2009. [Google Scholar]
- Pihusch, R.; Danzl, G.; Scholz, M.; Harich, D.; Pihusch, M.; Lohse, P.; Hiller, E. Impact of thrombophilic gene mutations on thrombosis risk in patients with gastrointestinal carcinoma. Cancer 2002, 94, 3120–3126. [Google Scholar] [CrossRef] [PubMed]
- Lindahl, A.K.; Odegaard, O.R.; Sandset, P.M.; Harbitz, T.B. Coagulation inhibition and activation in pancreatic cancer. Changes during progress of disease. Cancer 1992, 70, 2067–2072. [Google Scholar] [CrossRef]
- Lee, A.Y.; Peterson, E.A. Treatment of cancer-associated thrombosis. Blood 2013, 122, 2310–2317. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.F.; Li, A.; Garcia, D. Managing thrombosis in cancer patients. Res. Pract. Thromb. Haemost. 2018, 2, 429–438. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.Y.; Levine, M.N.; Baker, R.I.; Bowden, C.; Kakkar, A.K.; Prins, M.; Rickles, F.R.; Julian, J.A.; Haley, S.; Kovacs, M.J.; et al. Low-molecular-weight heparin versus a coumarin for the prevention of recurrent venous thromboembolism in patients with cancer. N. Engl. J. Med. 2003, 349, 146–153. [Google Scholar] [CrossRef] [PubMed]
- Lyman, G.H.; Bohlke, K.; Falanga, A. Venous thromboembolism prophylaxis and treatment in patients with cancer: American Society of Clinical Oncology clinical practice guideline update. J. Oncol. Pract. 2015, 11, e442–e444. [Google Scholar] [CrossRef] [PubMed]
- Kearon, C.; Akl, E.A.; Ornelas, J.; Blaivas, A.; Jimenez, D.; Bounameaux, H.; Huisman, M.; King, C.S.; Morris, T.A.; Sood, N.; et al. Antithrombotic Therapy for VTE Disease: CHEST Guideline and Expert Panel Report. Chest 2016, 149, 315–352. [Google Scholar] [CrossRef] [PubMed]
- Raskob, G.E.; van Es, N.; Verhamme, P.; Carrier, M.; Di Nisio, M.; Garcia, D.; Grosso, M.A.; Kakkar, A.K.; Kovacs, M.J.; Mercuri, M.F.; et al. Edoxaban for the Treatment of Cancer-Associated Venous Thromboembolism. N. Engl. J. Med. 2018, 378, 615–624. [Google Scholar] [CrossRef] [PubMed]


© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abdol Razak, N.B.; Jones, G.; Bhandari, M.; Berndt, M.C.; Metharom, P. Cancer-Associated Thrombosis: An Overview of Mechanisms, Risk Factors, and Treatment. Cancers 2018, 10, 380. https://doi.org/10.3390/cancers10100380
Abdol Razak NB, Jones G, Bhandari M, Berndt MC, Metharom P. Cancer-Associated Thrombosis: An Overview of Mechanisms, Risk Factors, and Treatment. Cancers. 2018; 10(10):380. https://doi.org/10.3390/cancers10100380
Chicago/Turabian StyleAbdol Razak, Norbaini Binti, Gabrielle Jones, Mayank Bhandari, Michael C. Berndt, and Pat Metharom. 2018. "Cancer-Associated Thrombosis: An Overview of Mechanisms, Risk Factors, and Treatment" Cancers 10, no. 10: 380. https://doi.org/10.3390/cancers10100380
APA StyleAbdol Razak, N. B., Jones, G., Bhandari, M., Berndt, M. C., & Metharom, P. (2018). Cancer-Associated Thrombosis: An Overview of Mechanisms, Risk Factors, and Treatment. Cancers, 10(10), 380. https://doi.org/10.3390/cancers10100380