Impacts of Cancer on Platelet Production, Activation and Education and Mechanisms of Cancer-Associated Thrombosis

 ,
,  and
and 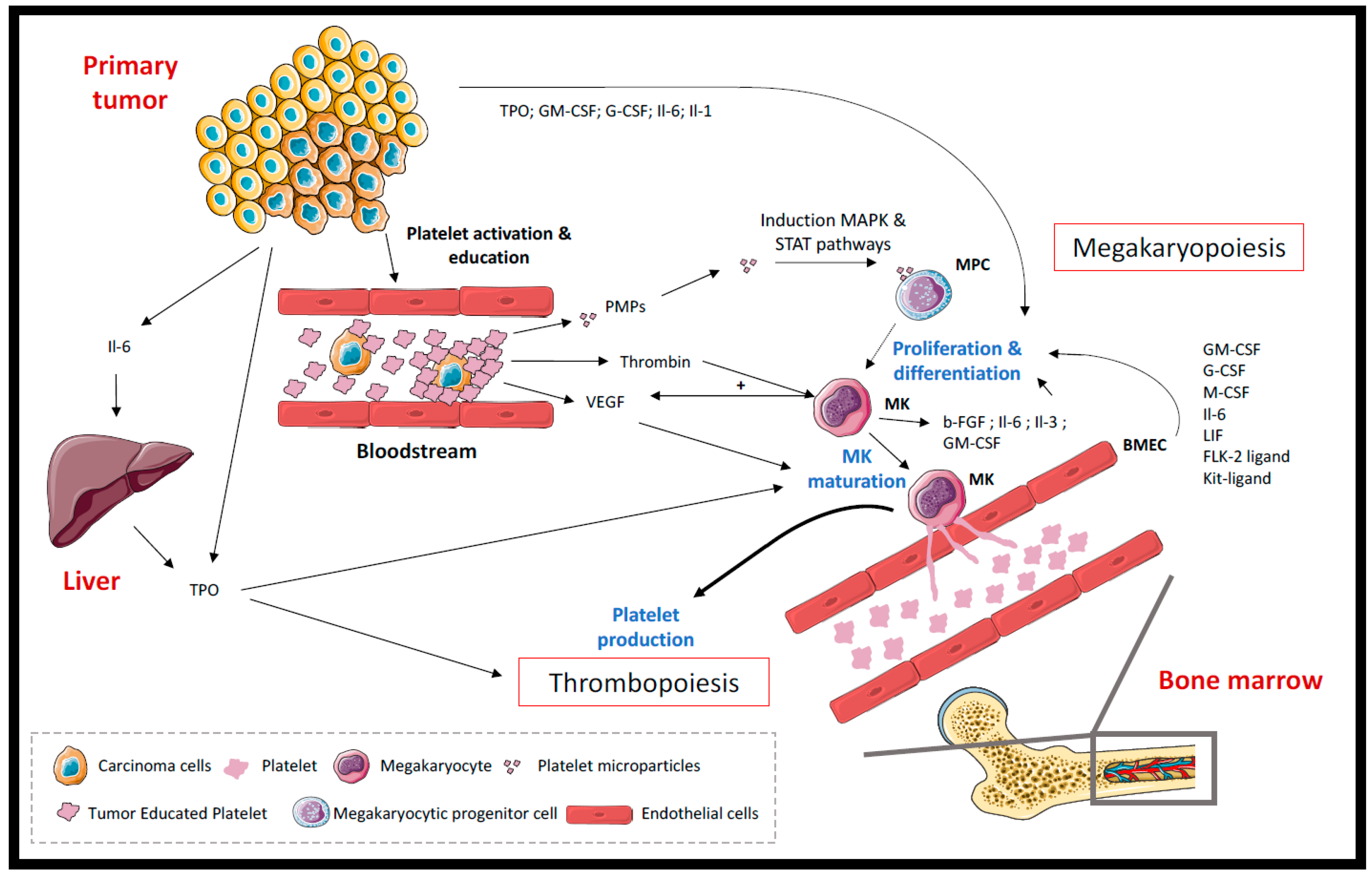{kind=link}
{kind=link}
Abstract
1. Introduction
2. Effects of Cancer on Platelets and Mechanisms Involved in Cancer-Associated Thrombosis
2.1. Thrombocytosis
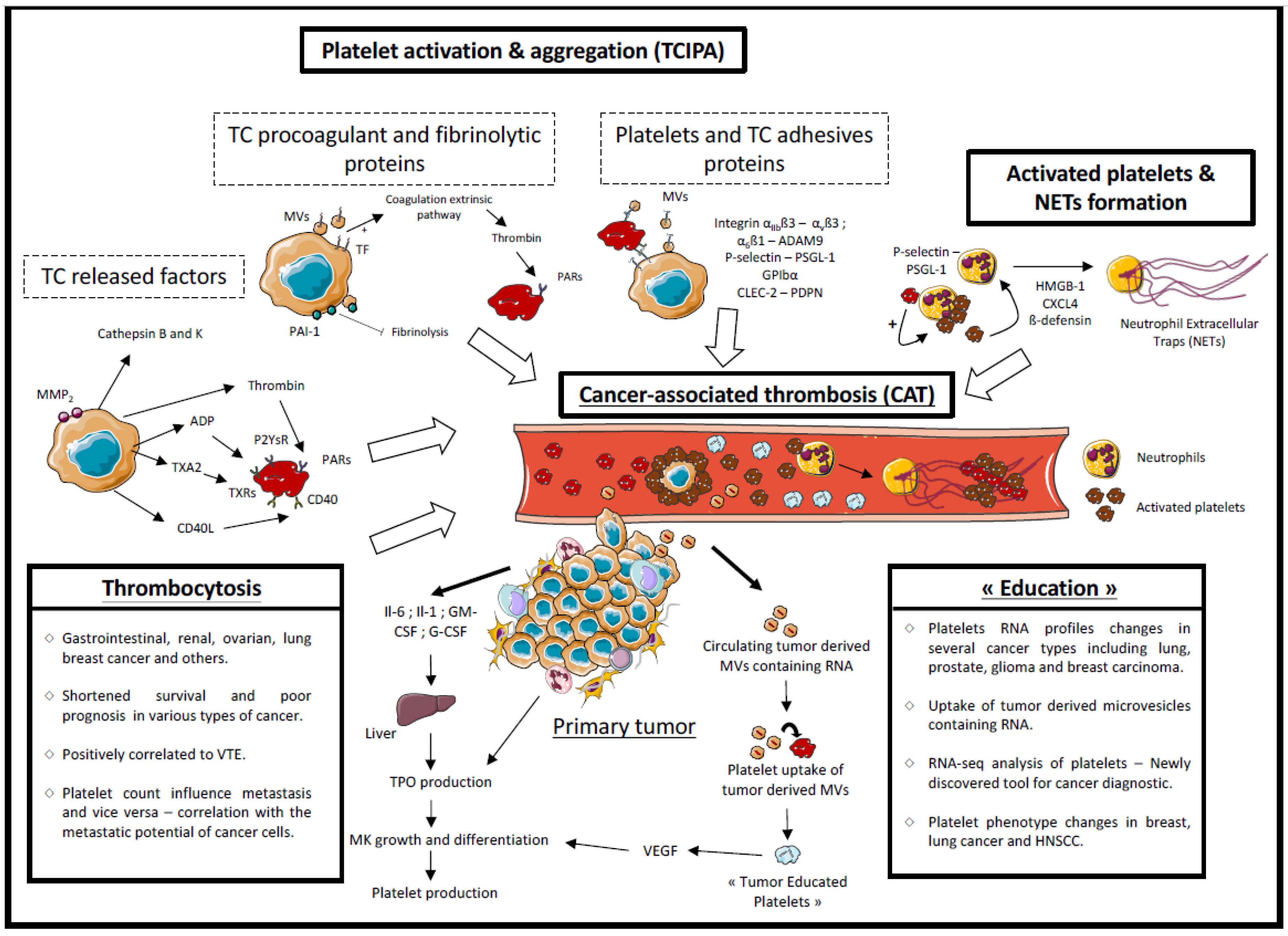
2.2. Platelets and Hemostatic System Activation
2.2.1. Released Factors
2.2.2. Tumor Cell Procoagulant Proteins
2.2.3. Microvesicles
2.2.4. Adhesive Proteins
2.2.5. Involvement of Activated Platelets in Cancer-Associated Thrombosis
2.3. Tumor Educated Platelets
2.3.1. Platelet RNA Profiles in Cancer Patients
2.3.2. Platelet Protein Profiles in Cancer Patients
2.4. Antiplatelet and Platelet Based Therapies
2.4.1. Antiplatelet Therapies
2.4.2. Platelet-Based Therapies
3. Conclusions
Funding
Conflicts of Interest
References
- Varki, A. Trousseau’s syndrome: Multiple definitions and multiple mechanisms. Blood 2007, 110, 1723–1729. [Google Scholar] [CrossRef] [PubMed]
- Mezouar, S.; Frère, C.; Darbousset, R.; Mege, D.; Crescence, L.; Dignat-George, F.; Panicot-Dubois, L.; Dubois, C. Role of platelets in cancer and cancer-associated thrombosis: Experimental and clinical evidences. Thromb. Res. 2016, 139, 65–76. [Google Scholar] [CrossRef] [PubMed]
- Palumbo, J.S.; Talmage, K.E.; Massari, J.V.; Jeunesse, C.M.L.; Flick, M.J.; Kombrinck, K.W.; Jirousková, M.; Degen, J.L. Platelets and fibrin(ogen) increase metastatic potential by impeding natural killer cell–mediated elimination of tumor cells. Blood 2005, 105, 178–185. [Google Scholar] [CrossRef] [PubMed]
- Labelle, M.; Begum, S.; Hynes, R.O. Direct Signaling between Platelets and Cancer Cells Induces an Epithelial-Mesenchymal-Like Transition and Promotes Metastasis. Cancer Cell 2011, 20, 576–590. [Google Scholar] [CrossRef] [PubMed]
- Labelle, M.; Begum, S.; Hynes, R.O. Platelets guide the formation of early metastatic niches. Proc. Natl. Acad. Sci. USA 2014, 111, 3053–3061. [Google Scholar] [CrossRef] [PubMed]
- Plantureux, L.; Crescence, L.; Dignat-George, F.; Panicot-Dubois, L.; Dubois, C. Effects of platelets on cancer progression. Thromb. Res. 2018, 164 (Suppl. 1), S40–S47. [Google Scholar] [CrossRef] [PubMed]
- Gasic, G.J.; Gasic, T.B.; Stewart, C.C. Antimetastatic effects associated with platelet reduction. Proc. Natl. Acad. Sci. USA 1968, 61, 46–52. [Google Scholar] [CrossRef] [PubMed]
- Karpatkin, S.; Pearlstein, E. Role of platelets in tumor cell metastases. Ann. Intern. Med. 1981, 95, 636–641. [Google Scholar] [CrossRef] [PubMed]
- Baranyai, Z.; Jósa, V.; Tóth, A.; Szilasi, Z.; Tihanyi, B.; Zaránd, A.; Harsanyi, L.; Szállási, Z. Paraneoplastic thrombocytosis in gastrointestinal cancer. Platelets 2016, 27, 269–275. [Google Scholar] [CrossRef] [PubMed]
- Khorana, A.A.; Francis, C.W.; Culakova, E.; Lyman, G.H. Risk factors for chemotherapy-associated venous thromboembolism in a prospective observational study. Cancer 2005, 104, 2822–2829. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, M.; Furukawa, H.; Imamura, H.; Shimizu, J.; Ishida, H.; Masutani, S.; Tatsuta, M.; Satomi, T. Poor prognosis associated with thrombocytosis in patients with gastric cancer. Ann. Surg. Oncol. 2002, 9, 287–291. [Google Scholar] [CrossRef] [PubMed]
- Gücer, F.; Moser, F.; Tamussino, K.; Reich, O.; Haas, J.; Arikan, G.; Petru, E.; Winter, R. Thrombocytosis as a prognostic factor in endometrial carcinoma. Gynecol. Oncol. 1998, 70, 210–214. [Google Scholar] [CrossRef] [PubMed]
- Chadha, A.S.; Kocak-Uzel, E.; Das, P.; Minsky, B.D.; Delclos, M.E.; Mahmood, U.; Guha, S.; Ahmad, M.; Varadhachary, G.R.; Javle, M.; et al. Paraneoplastic thrombocytosis independently predicts poor prognosis in patients with locally advanced pancreatic cancer. Acta Oncol. 2015, 54, 971–978. [Google Scholar] [CrossRef] [PubMed]
- Monreal, M.; Fernandez-Llamazares, J.; Piñol, M.; Julian, J.F.; Broggi, M.; Escola, D.; Abad, A. Platelet count and survival in patients with colorectal cancer—A preliminary study. Thromb. Haemost. 1998, 79, 916–918. [Google Scholar] [PubMed]
- Jefferson, K.; Persad, R. Poor prognosis associated with thrombocytosis in patients with renal cell carcinoma. BJU Int. 2001, 87, 715–716. [Google Scholar] [CrossRef] [PubMed]
- Tranum, B.L.; Haut, A. Thrombocytosis: Platelet kinetics in neoplasia. J. Lab. Clin. Med. 1974, 84, 615–619. [Google Scholar] [PubMed]
- Ji, Y.; Sheng, L.; Du, X.; Qiu, G.; Su, D. Elevated platelet count is a strong predictor of poor prognosis in stage I non-small cell lung cancer patients. Platelets 2015, 26, 138–142. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, A.; Takahashi, T.; Nakamura, K.; Tsuyuoka, R.; Okuno, Y.; Enomoto, T.; Fukumoto, M.; Imura, H. Thrombocytosis in patients with tumors producing colony-stimulating factor. Blood 1992, 80, 2052–2059. [Google Scholar] [PubMed]
- Stone, R.L.; Nick, A.M.; McNeish, I.A.; Balkwill, F.; Han, H.D.; Bottsford-Miller, J.; Rupairmoole, R.; Armaiz-Pena, G.N.; Pecot, C.V.; Coward, J.; et al. Paraneoplastic thrombocytosis in ovarian cancer. N. Engl. J. Med. 2012, 366, 610–618. [Google Scholar] [CrossRef] [PubMed]
- Kaser, A.; Brandacher, G.; Steurer, W.; Kaser, S.; Offner, F.A.; Zoller, H.; Theurl, I.; Widder, W.; Molnar, C.; Ludwiczek, O.; et al. Interleukin-6 stimulates thrombopoiesis through thrombopoietin: Role in inflammatory thrombocytosis. Blood 2001, 98, 2720–2725. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, Y.; Takahashi, T.; Miyazaki, H.; Matsumoto, A.; Kato, T.; Nakamura, K.; Iho, S.; Okuno, Y.; Nakao, K. Production of thrombopoietin by human carcinomas and its novel isoforms. Blood 1999, 94, 1952–1960. [Google Scholar] [PubMed]
- Ryu, T.; Nishimura, S.; Miura, H.; Yamada, H.; Morita, H.; Miyazaki, H.; Kitamura, S.; Miura, Y.; Saito, T. Thrombopoietin-producing hepatocellular carcinoma. Intern. Med. 2003, 42, 730–734. [Google Scholar] [CrossRef] [PubMed]
- Rafii, S.; Shapiro, F.; Pettengell, R.; Ferris, B.; Nachman, R.L.; Moore, M.A.; Asch, A.S. Human bone marrow microvascular endothelial cells support long-term proliferation and differentiation of myeloid and megakaryocytic progenitors. Blood 1995, 86, 3353–3363. [Google Scholar] [PubMed]
- Rafii, S.; Mohle, R.; Shapiro, F.; Frey, B.M.; Moore, M.A.S. Regulation of Hematopoiesis by Microvascular Endothelium. Leuk. Lymphoma 1997, 27, 375–386. [Google Scholar] [CrossRef] [PubMed]
- Wickenhauser, C.; Lorenzen, J.; Thiele, J.; Hillienhof, A.; Jungheim, K.; Schmitz, B.; Hansmann, M.L.; Fischer, R. Secretion of cytokines (interleukins-1 alpha, -3, and -6 and granulocyte-macrophage colony-stimulating factor) by normal human bone marrow megakaryocytes. Blood 1995, 85, 685–691. [Google Scholar] [PubMed]
- Baj-Krzyworzeka, M.; Majka, M.; Pratico, D.; Ratajczak, J.; Vilaire, G.; Kijowski, J.; Reca, R.; Janowska-Wieczorek, A.; Ratajczak, M.Z. Platelet-derived microparticles stimulate proliferation, survival, adhesion, and chemotaxis of hematopoietic cells. Exp. Hematol. 2002, 30, 450–459. [Google Scholar] [CrossRef]
- Möhle, R.; Green, D.; Moore, M.A.; Nachman, R.L.; Rafii, S. Constitutive production and thrombin-induced release of vascular endothelial growth factor by human megakaryocytes and platelets. Proc. Natl. Acad. Sci. USA 1997, 94, 663–668. [Google Scholar] [CrossRef] [PubMed]
- Holmes, C.E.; Levis, J.E.; Schneider, D.J.; Bambace, N.M.; Sharma, D.; Lal, I.; Wood, M.E.; Muss, H.B. Platelet phenotype changes associated with breast cancer and its treatment. Platelets 2016, 27, 703–711. [Google Scholar] [CrossRef] [PubMed]
- Casella, I.; Feccia, T.; Chelucci, C.; Samoggia, P.; Castelli, G.; Guerriero, R.; Parolini, I.; Petrucci, E.; Pelosi, E.; Morsilli, O.; et al. Autocrine-paracrine VEGF loops potentiate the maturation of megakaryocytic precursors through Flt1 receptor. Blood 2003, 101, 1316–1323. [Google Scholar] [CrossRef] [PubMed]
- Broxmeyer, H.E.; Cooper, S.; Li, Z.H.; Lu, L.; Song, H.Y.; Kwon, B.S.; Warren, R.E.; Donner, D.B. Myeloid progenitor cell regulatory effects of vascular endothelial cell growth factor. Int. J. Hematol. 1995, 62, 203–215. [Google Scholar] [CrossRef]
- Avraham, H.; Banu, N.; Scadden, D.T.; Abraham, J.; Groopman, J.E. Modulation of megakaryocytopoiesis by human basic fibroblast growth factor. Blood 1994, 83, 2126–2132. [Google Scholar] [PubMed]
- Ay, C.; Pabinger, I. Predictive potential of haemostatic biomarkers for venous thromboembolism in cancer patients. Thromb. Res. 2012, 129, S6–S9. [Google Scholar] [CrossRef]
- Starlinger, P.; Moll, H.P.; Assinger, A.; Nemeth, C.; Hoetzenecker, K.; Gruenberger, B.; Gruenberger, T.; Kuehrer, I.; Schoppmann, S.F.; Gnant, M.; et al. Thrombospondin-1: A unique marker to identify in vitro platelet activation when monitoring in vivo processes. J. Thromb. Haemost. 2010, 8, 1809–1819. [Google Scholar] [CrossRef] [PubMed]
- Suh, E.J.; Kabir, M.H.; Kang, U.-B.; Lee, J.W.; Yu, J.; Noh, D.-Y.; Lee, C. Comparative profiling of plasma proteome from breast cancer patients reveals thrombospondin-1 and BRWD3 as serological biomarkers. Exp. Mol. Med. 2012, 44, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Jochems, C.; Talaie, T.; Anderson, A.; Jales, A.; Tsang, K.Y.; Madan, R.A.; Gulley, J.L.; Schlom, J. Elevated serum soluble CD40 ligand in cancer patients may play an immunosuppressive role. Blood 2012, 120, 3030–3038. [Google Scholar] [CrossRef] [PubMed]
- Benedetti Panici, P.; Scambia, G.; Massidda, B.; Chessa, P.; Tarquini, A.; Mancuso, S. Elevated plasma levels of beta-thromboglobulin in breast cancer. Oncology 1986, 43, 208–211. [Google Scholar] [CrossRef] [PubMed]
- Poruk, K.E.; Firpo, M.A.; Huerter, L.M.; Scaife, C.L.; Emerson, L.L.; Boucher, K.M.; Jones, K.A.; Mulvihill, S.J. Serum platelet factor 4 is an independent predictor of survival and venous thromboembolism in patients with pancreatic adenocarcinoma. Cancer Epidemiol. Biomark. Prev. 2010, 19, 2605–2610. [Google Scholar] [CrossRef] [PubMed]
- Mantur, M.; Kemona, H.; Kozłowski, R.; Kemona-Chetnik, I. Effect of tumor stage and nephrectomy on CD62P expression and sP-selectin concentration in renal cancer. Neoplasma 2003, 50, 262–265. [Google Scholar] [PubMed]
- Riedl, J.; Hell, L.; Kaider, A.; Koder, S.; Marosi, C.; Zielinski, C.; Panzer, S.; Pabinger, I.; Ay, C. Association of platelet activation markers with cancer-associated venous thromboembolism. Platelets 2016, 27, 80–85. [Google Scholar] [CrossRef] [PubMed]
- Falanga, A.; Panova-Noeva, M.; Russo, L. Procoagulant mechanisms in tumour cells. Best Pract. Res. Clin. Haematol. 2009, 22, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Karpatkin, S.; Pearlstein, E.; Ambrogio, C.; Coller, B.S. Role of adhesive proteins in platelet tumor interaction in vitro and metastasis formation in vivo. J. Clin. Investig. 1988, 81, 1012–1019. [Google Scholar] [CrossRef] [PubMed]
- Haemmerle, M.; Bottsford-Miller, J.; Pradeep, S.; Taylor, M.L.; Choi, H.-J.; Hansen, J.M.; Dalton, H.J.; Stone, R.L.; Cho, M.S.; Nick, A.M.; et al. FAK regulates platelet extravasation and tumor growth after antiangiogenic therapy withdrawal. J. Clin. Investig. 2016, 126, 1885–1896. [Google Scholar] [CrossRef] [PubMed]
- Camez, A.; Dupuy, E.; Bellucci, S.; Calvo, F.; Bryckaert, M.C.; Tobelem, G. Human platelet-tumor cell interactions vary with the tumor cell lines. Invasion Metastasis 1986, 6, 321–334. [Google Scholar] [PubMed]
- Bastida, E.; Escolar, G.; Almirall, L.; Ordinas, A. Platelet activation induced by a human neuroblastoma tumor cell line is reduced by prior administration of ticlopidine. Thromb. Haemost. 1986, 55, 333–337. [Google Scholar] [CrossRef] [PubMed]
- Boukerche, H.; Berthier-Vergnes, O.; Penin, F.; Tabone, E.; Lizard, G.; Bailly, M.; McGregor, J.L. Human melanoma cell lines differ in their capacity to release ADP and aggregate platelets. Br. J. Haematol. 1994, 87, 763–772. [Google Scholar] [CrossRef] [PubMed]
- Alonso-Escolano, D.; Strongin, A.Y.; Chung, A.W.; Deryugina, E.I.; Radomski, M.W. Membrane type-1 matrix metalloproteinase stimulates tumour cell-induced platelet aggregation: Role of receptor glycoproteins. Br. J. Pharmacol. 2004, 141, 241–252. [Google Scholar] [CrossRef] [PubMed]
- Heinmöller, E.; Weinel, R.J.; Heidtmann, H.H.; Salge, U.; Seitz, R.; Schmitz, I.; Müller, K.M.; Zirngibl, H. Studies on tumor-cell-induced platelet aggregation in human lung cancer cell lines. J. Cancer Res. Clin. Oncol. 1996, 122, 735–744. [Google Scholar] [CrossRef] [PubMed]
- Sakai, H.; Suzuki, T.; Takahashi, Y.; Ukai, M.; Tauchi, K.; Fujii, T.; Horikawa, N.; Minamimura, T.; Tabuchi, Y.; Morii, M.; et al. Upregulation of thromboxane synthase in human colorectal carcinoma and the cancer cell proliferation by thromboxane A2. FEBS Lett. 2006, 580, 3368–3374. [Google Scholar] [CrossRef] [PubMed]
- Cathcart, M.-C.; Gately, K.; Cummins, R.; Kay, E.; O’Byrne, K.J.; Pidgeon, G.P. Examination of thromboxane synthase as a prognostic factor and therapeutic target in non-small cell lung cancer. Mol. Cancer 2011, 10, 25. [Google Scholar] [CrossRef] [PubMed]
- Kajita, S.; Ruebel, K.H.; Casey, M.B.; Nakamura, N.; Lloyd, R.V. Role of COX-2, thromboxane A2 synthase, and prostaglandin I2 synthase in papillary thyroid carcinoma growth. Mod. Pathol. 2005, 18, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Moussa, O.; Yordy, J.S.; Abol-Enein, H.; Sinha, D.; Bissada, N.K.; Halushka, P.V.; Ghoneim, M.A.; Watson, D.K. Prognostic and functional significance of thromboxane synthase gene overexpression in invasive bladder cancer. Cancer Res. 2005, 65, 11581–11587. [Google Scholar] [CrossRef] [PubMed]
- Nie, D.; Che, M.; Zacharek, A.; Qiao, Y.; Li, L.; Li, X.; Lamberti, M.; Tang, K.; Cai, Y.; Guo, Y.; et al. Differential expression of thromboxane synthase in prostate carcinoma: Role in tumor cell motility. Am. J. Pathol. 2004, 164, 429–439. [Google Scholar] [CrossRef]
- de Leval, X.; Benoit, V.; Delarge, J.; Julémont, F.; Masereel, B.; Pirotte, B.; Merville, M.P.; David, J.L.; Dogné, J.M. Pharmacological evaluation of the novel thromboxane modulator BM-567 (II/II). Effects of BM-567 on osteogenic sarcoma-cell-induced platelet aggregation. Prostaglandins Leukot. Essent. Fatty Acids 2003, 68, 55–59. [Google Scholar] [CrossRef]
- Nie, D.; Lamberti, M.; Zacharek, A.; Li, L.; Szekeres, K.; Tang, K.; Chen, Y.; Honn, K.V. Thromboxane A(2) regulation of endothelial cell migration, angiogenesis, and tumor metastasis. Biochem. Biophys. Res. Commun. 2000, 267, 245–251. [Google Scholar] [CrossRef] [PubMed]
- Stout, R.D.; Suttles, J. The many roles of CD40 in cell-mediated inflammatory responses. Immunol. Today 1996, 17, 487–492. [Google Scholar] [CrossRef]
- Henn, V.; Slupsky, J.R.; Gräfe, M.; Anagnostopoulos, I.; Förster, R.; Müller-Berghaus, G.; Kroczek, R.A. CD40 ligand on activated platelets triggers an inflammatory reaction of endothelial cells. Nature 1998, 391, 591–594. [Google Scholar] [CrossRef] [PubMed]
- Braesch-Andersen, S.; Paulie, S.; Koho, H.; Nika, H.; Aspenström, P.; Perlmann, P. Biochemical characteristics and partial amino acid sequence of the receptor-like human B cell and carcinoma antigen CDw40. J. Immunol. 1989, 142, 562–567. [Google Scholar] [PubMed]
- Ingersoll, S.B.; Langer, F.; Walker, J.M.; Meyer, T.; Robson, T.; Amaya, M.; Desai, H.; Francis, J.L.; Amirkhosravi, A. Deficiencies in the CD40 and CD154 receptor-ligand system reduce experimental lung metastasis. Clin. Exp. Metastasis 2009, 26, 829–837. [Google Scholar] [CrossRef] [PubMed]
- Young, L.S.; Dawson, C.W.; Brown, K.W.; Rickinson, A.B. Identification of a human epithelial cell surface protein sharing an epitope with the C3d/Epstein-Barr virus receptor molecule of B lymphocytes. Int. J. Cancer 1989, 43, 786–794. [Google Scholar] [CrossRef] [PubMed]
- Bussolati, B.; Russo, S.; Deambrosis, I.; Cantaluppi, V.; Volpe, A.; Ferrando, U.; Camussi, G. Expression of CD154 on renal cell carcinomas and effect on cell proliferation, motility and platelet-activating factor synthesis. Int. J. Cancer 2002, 100, 654–661. [Google Scholar] [CrossRef] [PubMed]
- Henn, V.; Steinbach, S.; Büchner, K.; Presek, P.; Kroczek, R.A. The inflammatory action of CD40 ligand (CD154) expressed on activated human platelets is temporally limited by coexpressed CD40. Blood 2001, 98, 1047–1054. [Google Scholar] [CrossRef] [PubMed]
- McNicol, A.; Israels, S.J. Beyond hemostasis: The role of platelets in inflammation, malignancy and infection. Cardiovasc. Hematol. Disord. Drug Targets 2008, 8, 99–117. [Google Scholar] [CrossRef] [PubMed]
- Inwald, D.P.; McDowall, A.; Peters, M.J.; Callard, R.E.; Klein, N.J. CD40 is constitutively expressed on platelets and provides a novel mechanism for platelet activation. Circ. Res. 2003, 92, 1041–1048. [Google Scholar] [CrossRef] [PubMed]
- Tong, A.W.; Papayoti, M.H.; Netto, G.; Armstrong, D.T.; Ordonez, G.; Lawson, J.M.; Stone, M.J. Growth-inhibitory effects of CD40 ligand (CD154) and its endogenous expression in human breast cancer. Clin. Cancer Res. 2001, 7, 691–703. [Google Scholar] [PubMed]
- Yamada, M.; Shiroko, T.; Kawaguchi, Y.; Sugiyama, Y.; Egilmez, N.K.; Chen, F.A.; Bankert, R.B. CD40-CD40 ligand (CD154) engagement is required but not sufficient for modulating MHC class I, ICAM-1 and Fas expression and proliferation of human non-small cell lung tumors. Int. J. Cancer 2001, 92, 589–599. [Google Scholar] [CrossRef] [PubMed]
- van den Oord, J.J.; Maes, A.; Stas, M.; Nuyts, J.; Battocchio, S.; Kasran, A.; Garmyn, M.; De Wever, I.; De Wolf-Peeters, C. CD40 is a prognostic marker in primary cutaneous malignant melanoma. Am. J. Pathol. 1996, 149, 1953–1961. [Google Scholar] [PubMed]
- Santilli, F.; Basili, S.; Ferroni, P.; Davì, G. CD40/CD40L system and vascular disease. Intern. Emerg. Med. 2007, 2, 256–268. [Google Scholar] [CrossRef] [PubMed]
- Jurasz, P.; Sawicki, G.; Duszyk, M.; Sawicka, J.; Miranda, C.; Mayers, I.; Radomski, M.W. Matrix metalloproteinase 2 in tumor cell-induced platelet aggregation: Regulation by nitric oxide. Cancer Res. 2001, 61, 376–382. [Google Scholar] [PubMed]
- Medina, C.; Jurasz, P.; Santos-Martinez, M.J.; Jeong, S.S.; Mitsky, T.; Chen, R.; Radomski, M.W. Platelet aggregation-induced by caco-2 cells: Regulation by matrix metalloproteinase-2 and adenosine diphosphate. J. Pharmacol. Exp. Ther. 2006, 317, 739–745. [Google Scholar] [CrossRef] [PubMed]
- Honn, K.V.; Cavanaugh, P.; Evens, C.; Taylor, J.D.; Sloane, B.F. Tumor cell-platelet aggregation: Induced by cathepsin B-like proteinase and inhibited by prostacyclin. Science 1982, 217, 540–542. [Google Scholar] [CrossRef] [PubMed]
- Andrade, S.S.; Gouvea, I.E.; Silva, M.C.C.; Castro, E.D.; de Paula, C.A.A.; Okamoto, D.; Oliveira, L.; Peres, G.B.; Ottaiano, T.; Facina, G.; et al. Cathepsin K induces platelet dysfunction and affects cell signaling in breast cancer—Molecularly distinct behavior of cathepsin K in breast cancer. BMC Cancer 2016, 16, 173. [Google Scholar] [CrossRef] [PubMed]
- Bode, M.; Mackman, N. Regulation of tissue factor gene expression in monocytes and endothelial cells: Thromboxane A2 as a new player. Vascul. Pharmacol. 2014, 62, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Jiang, P.; Capkova, K.; Xue, D.; Ye, L.; Sinha, S.C.; Mackman, N.; Janda, K.D.; Liu, C. Tissue factor-activated coagulation cascade in the tumor microenvironment is critical for tumor progression and an effective target for therapy. Cancer Res. 2011, 71, 6492–6502. [Google Scholar] [CrossRef] [PubMed]
- Ruf, W.; Yokota, N.; Schaffner, F. Tissue factor in cancer progression and angiogenesis. Thromb. Res. 2010, 125, S36–S38. [Google Scholar] [CrossRef]
- Böhm, L.; Serafin, A.; Akudugu, J.; Fernandez, P.; van der Merwe, A.; Aziz, N.A. uPA/PAI-1 ratios distinguish benign prostatic hyperplasia and prostate cancer. J. Cancer Res. Clin. Oncol. 2013, 139, 1221–1228. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Ling, D.; Tan, J.; Zhang, J.; Li, L. Expression of urokinase plasminogen activator and plasminogen activator inhibitor type-1 in ovarian cancer and its clinical significance. Oncol. Rep. 2013, 29, 637–645. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Peng, H.; Liu, W.; Sun, Y.; Su, N.; Tang, W.; Zhang, X.; Wang, J.; Cui, L.; Hu, P.; et al. Silencing of plasminogen activator inhibitor-1 suppresses colorectal cancer progression and liver metastasis. Surgery 2015, 158, 1704–1713. [Google Scholar] [CrossRef] [PubMed]
- Ferroni, P.; Roselli, M.; Portarena, I.; Formica, V.; Riondino, S.; LA Farina, F.; Costarelli, L.; Melino, A.; Massimiani, G.; Cavaliere, F.; et al. Plasma plasminogen activator inhibitor-1 (PAI-1) levels in breast cancer—Relationship with clinical outcome. Anticancer Res. 2014, 34, 1153–1161. [Google Scholar] [PubMed]
- Rickles, F.R.; Falanga, A. Molecular basis for the relationship between thrombosis and cancer. Thromb. Res. 2001, 102, V215–V224. [Google Scholar] [CrossRef]
- Mezouar, S.; Mege, D.; Darbousset, R.; Farge, D.; Debourdeau, P.; Dignat-George, F.; Panicot-Dubois, L.; Dubois, C. Involvement of platelet-derived microparticles in tumor progression and thrombosis. Semin. Oncol. 2014, 41, 346–358. [Google Scholar] [CrossRef] [PubMed]
- Chargaff, E.; West, R. The biological significance of the thromboplastic protein of blood. J. Biol. Chem. 1946, 166, 189–197. [Google Scholar] [PubMed]
- Nomura, S.; Niki, M.; Nisizawa, T.; Tamaki, T.; Shimizu, M. Microparticles as Biomarkers of Blood Coagulation in Cancer. Biomark. Cancer 2015, 7, 51–56. [Google Scholar] [CrossRef] [PubMed]
- Geddings, J.E.; Mackman, N. Tumor-derived tissue factor–positive microparticles and venous thrombosis in cancer patients. Blood 2013, 122, 1873–1880. [Google Scholar] [CrossRef] [PubMed]
- Hron, G.; Kollars, M.; Weber, H.; Sagaster, V.; Quehenberger, P.; Eichinger, S.; Kyrle, P.A.; Weltermann, A. Tissue factor-positive microparticles: Cellular origin and association with coagulation activation in patients with colorectal cancer. Thromb. Haemost. 2007, 97, 119–123. [Google Scholar] [CrossRef] [PubMed]
- Baran, J.; Baj-Krzyworzeka, M.; Weglarczyk, K.; Szatanek, R.; Zembala, M.; Barbasz, J.; Czupryna, A.; Szczepanik, A.; Zembala, M. Circulating tumour-derived microvesicles in plasma of gastric cancer patients. Cancer Immunol. Immunother. 2010, 59, 841–850. [Google Scholar] [CrossRef] [PubMed]
- Yates, K.R.; Welsh, J.; Echrish, H.H.; Greenman, J.; Maraveyas, A.; Madden, L.A. Pancreatic cancer cell and microparticle procoagulant surface characterization: Involvement of membrane-expressed tissue factor, phosphatidylserine and phosphatidylethanolamine. Blood Coagul. Fibrinolysis 2011, 22, 680–687. [Google Scholar] [CrossRef] [PubMed]
- Fleitas, T.; Martínez-Sales, V.; Vila, V.; Reganon, E.; Mesado, D.; Martín, M.; Gómez-Codina, J.; Montalar, J.; Reynés, G. Circulating Endothelial Cells and Microparticles as Prognostic Markers in Advanced Non-Small Cell Lung Cancer. PLoS ONE 2012, 7, e47365. [Google Scholar] [CrossRef] [PubMed]
- Graves, L.E.; Ariztia, E.V.; Navari, J.R.; Matzel, H.J.; Stack, M.S.; Fishman, D.A. Proinvasive Properties of Ovarian Cancer Ascites-Derived Membrane Vesicles. Cancer Res. 2004, 64, 7045–7049. [Google Scholar] [CrossRef] [PubMed]
- Liebhardt, S.; Ditsch, N.; Nieuwland, R.; Rank, A.; Jeschke, U.; Von Koch, F.; Friese, K.; Toth, B. CEA-, Her2/neu-, BCRP- and Hsp27-positive microparticles in breast cancer patients. Anticancer Res. 2010, 30, 1707–1712. [Google Scholar] [PubMed]
- Mege, D.; Panicot-Dubois, L.; Ouaissi, M.; Robert, S.; Sielezneff, I.; Sastre, B.; Dignat-George, F.; Dubois, C. The origin and concentration of circulating microparticles differ according to cancer type and evolution: A prospective single-center study. Int. J. Cancer 2016, 138, 939–948. [Google Scholar] [CrossRef] [PubMed]
- Thomas, G.M.; Brill, A.; Mezouar, S.; Crescence, L.; Gallant, M.; Dubois, C.; Wagner, D.D. Tissue factor expressed by circulating cancer cell-derived microparticles drastically increases the incidence of deep vein thrombosis in mice. J. Thromb. Haemost. 2015, 13, 1310–1319. [Google Scholar] [CrossRef] [PubMed]
- Hisada, Y.; Ay, C.; Auriemma, A.C.; Cooley, B.C.; Mackman, N. Human pancreatic tumors grown in mice release tissue factor-positive microvesicles that increase venous clot size. J. Thromb. Haemost. 2017, 15, 2208–2217. [Google Scholar] [CrossRef] [PubMed]
- Demers, M.; Wagner, D.D. NETosis: A new factor in tumor progression and cancer-associated thrombosis. Semin. Thromb. Hemost. 2014, 40, 277–283. [Google Scholar] [CrossRef] [PubMed]
- Collier, M.E.W.; Mah, P.-M.; Xiao, Y.; Maraveyas, A.; Ettelaie, C. Microparticle-associated tissue factor is recycled by endothelial cells resulting in enhanced surface tissue factor activity. Thromb. Haemost. 2013, 110, 966–976. [Google Scholar] [CrossRef] [PubMed]
- Leal, A.C.; Mizurini, D.M.; Gomes, T.; Rochael, N.C.; Saraiva, E.M.; Dias, M.S.; Werneck, C.C.; Sielski, M.S.; Vicente, C.P.; Monteiro, R.Q. Tumor-Derived Exosomes Induce the Formation of Neutrophil Extracellular Traps: Implications For The Establishment of Cancer-Associated Thrombosis. Sci. Rep. 2017, 7, 6438. [Google Scholar] [CrossRef] [PubMed]
- Engebraaten, O.; Trikha, M.; Juell, S.; Garman-Vik, S.; Fodstad, Ø. Inhibition of in vivo tumour growth by the blocking of host alpha(v)beta3 and alphaII(b)beta3 integrins. Anticancer Res. 2009, 29, 131–137. [Google Scholar] [PubMed]
- Weber, M.R.; Zuka, M.; Lorger, M.; Tschan, M.; Torbett, B.E.; Zijlstra, A.; Quigley, J.P.; Staflin, K.; Eliceiri, B.P.; Krueger, J.S.; et al. Activated tumor cell integrin αvβ3 cooperates with platelets to promote extravasation and metastasis from the blood stream. Thromb. Res. 2016, 140 (Suppl. 1), S27–S36. [Google Scholar] [CrossRef]
- Mammadova-Bach, E.; Zigrino, P.; Brucker, C.; Bourdon, C.; Freund, M.; De Arcangelis, A.; Abrams, S.I.; Orend, G.; Gachet, C.; Mangin, P.H. Platelet integrin α6β1 controls lung metastasis through direct binding to cancer cell-derived ADAM9. JCI Insight 2016, 1, e88245. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.J.; Borsig, L.; Varki, N.M.; Varki, A. P-selectin deficiency attenuates tumor growth and metastasis. PNAS 1998, 95, 9325–9330. [Google Scholar] [CrossRef] [PubMed]
- Qi, C.; Wei, B.; Zhou, W.; Yang, Y.; Li, B.; Guo, S.; Li, J.; Ye, J.; Li, J.; Zhang, Q.; et al. P-selectin-mediated platelet adhesion promotes tumor growth. Oncotarget 2015, 6, 6584–6596. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Geng, J.-G. P-selectin mediates adhesion of leukocytes, platelets, and cancer cells in inflammation, thrombosis, and cancer growth and metastasis. Arch. Immunol. Ther. Exp. 2006, 54, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Wehmeier, A.; Tschöpe, D.; Esser, J.; Menzel, C.; Nieuwenhuis, H.K.; Schneider, W. Circulating activated platelets in myeloproliferative disorders. Thromb. Res. 1991, 61, 271–278. [Google Scholar] [CrossRef]
- Bastida, E.; Almirall, L.; Ordinas, A. Tumor-cell-induced platelet aggregation is a glycoprotein-dependent and lipoxygenase-associated process. Int. J. Cancer 1987, 39, 760–763. [Google Scholar] [CrossRef] [PubMed]
- Grossi, I.M.; Fitzgerald, L.A.; Kendall, A.; Taylor, J.D.; Sloane, B.F.; Honn, K.V. Inhibition of Human Tumor Cell Induced Platelet Aggregation by Antibodies to Platelet Glycoproteins lb and llb/llla. Proc. Soc. Exp. Biol. Med. 1987, 186, 378–383. [Google Scholar] [CrossRef] [PubMed]
- Jain, S.; Zuka, M.; Liu, J.; Russell, S.; Dent, J.; Guerrero, J.A.; Forsyth, J.; Maruszak, B.; Gartner, T.K.; Felding-Habermann, B.; et al. Platelet glycoprotein Ibα supports experimental lung metastasis. PNAS 2007, 104, 9024–9028. [Google Scholar] [CrossRef] [PubMed]
- Oleksowicz, L.; Mrowiec, Z.; Schwartz, E.; Khorshidi, M.; Dutcher, J.P.; Puszkin, E. Characterization of tumor-induced platelet aggregation: The role of immunorelated GPIb and GPIIb IIIa expression by MCF-7 breast cancer cells. Thromb. Res. 1995, 79, 261–274. [Google Scholar] [CrossRef]
- Erpenbeck, L.; Nieswandt, B.; Schön, M.; Pozgajova, M.; Schön, M.P. Inhibition of Platelet GPIbα and Promotion of Melanoma Metastasis. J. Investig. Dermatol. 2010, 130, 576–586. [Google Scholar] [CrossRef] [PubMed]
- Fujita, N.; Takagi, S. The impact of Aggrus/podoplanin on platelet aggregation and tumour metastasis. J. Biochem. 2012, 152, 407–413. [Google Scholar] [CrossRef] [PubMed]
- Kato, Y.; Kaneko, M.; Sata, M.; Fujita, N.; Tsuruo, T.; Osawa, M. Enhanced Expression of Aggrus (T1alpha/Podoplanin), a Platelet-Aggregation-Inducing Factor in Lung Squamous Cell Carcinoma. TBI 2005, 26, 195–200. [Google Scholar] [CrossRef] [PubMed]
- Takagi, S.; Oh-hara, T.; Sato, S.; Gong, B.; Takami, M.; Fujita, N. Expression of Aggrus/podoplanin in bladder cancer and its role in pulmonary metastasis. Int. J. Cancer 2014, 134, 2605–2614. [Google Scholar] [CrossRef] [PubMed]
- Kato, Y.; Fujita, N.; Kunita, A.; Sato, S.; Kaneko, M.; Osawa, M.; Tsuruo, T. Molecular identification of Aggrus/T1alpha as a platelet aggregation-inducing factor expressed in colorectal tumors. J. Biol. Chem. 2003, 278, 51599–51605. [Google Scholar] [CrossRef] [PubMed]
- Takagi, S.; Sato, S.; Oh-hara, T.; Takami, M.; Koike, S.; Mishima, Y.; Hatake, K.; Fujita, N. Platelets Promote Tumor Growth and Metastasis via Direct Interaction between Aggrus/Podoplanin and CLEC-2. PLoS ONE 2013, 8, e73609. [Google Scholar] [CrossRef] [PubMed]
- Vanschoonbeek, K.; Feijge, M. a. H.; Kampen, R.J.W.V.; Kenis, H.; Hemker, H.C.; Giesen, P.L.A.; Heemskerk, J.W.M. Initiating and potentiating role of platelets in tissue factor-induced thrombin generation in the presence of plasma: Subject-dependent variation in thrombogram characteristics. J. Thromb. Haemost. 2004, 2, 476–484. [Google Scholar] [CrossRef] [PubMed]
- Wahrenbrock, M.; Borsig, L.; Le, D.; Varki, N.; Varki, A. Selectin-mucin interactions as a probable molecular explanation for the association of Trousseau syndrome with mucinous adenocarcinomas. J. Clin. Investig. 2003, 112, 853–862. [Google Scholar] [CrossRef] [PubMed]
- Cedervall, J.; Hamidi, A.; Olsson, A.-K. Platelets, NETs and cancer. Thromb. Res. 2018, 164 (Suppl. 1), S148–S152. [Google Scholar] [CrossRef]
- Caudrillier, A.; Kessenbrock, K.; Gilliss, B.M.; Nguyen, J.X.; Marques, M.B.; Monestier, M.; Toy, P.; Werb, Z.; Looney, M.R. Platelets induce neutrophil extracellular traps in transfusion-related acute lung injury. J. Clin. Investig. 2012, 122, 2661–2671. [Google Scholar] [CrossRef] [PubMed]
- Etulain, J.; Fondevila, C.; Negrotto, S.; Schattner, M. Platelet-mediated angiogenesis is independent of VEGF and fully inhibited by aspirin. Br. J. Pharmacol. 2013, 170, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Rossaint, J.; Herter, J.M.; Aken, H.V.; Napirei, M.; Döring, Y.; Weber, C.; Soehnlein, O.; Zarbock, A. Synchronized integrin engagement and chemokine activation is crucial in neutrophil extracellular trap–mediated sterile inflammation. Blood 2014, 123, 2573–2584. [Google Scholar] [CrossRef] [PubMed]
- von Brühl, M.-L.; Stark, K.; Steinhart, A.; Chandraratne, S.; Konrad, I.; Lorenz, M.; Khandoga, A.; Tirniceriu, A.; Coletti, R.; Köllnberger, M.; et al. Monocytes, neutrophils, and platelets cooperate to initiate and propagate venous thrombosis in mice in vivo. J. Exp. Med. 2012, 209, 819–835. [Google Scholar] [CrossRef] [PubMed]
- Maugeri, N.; Campana, L.; Gavina, M.; Covino, C.; Metrio, M.D.; Panciroli, C.; Maiuri, L.; Maseri, A.; D’Angelo, A.; Bianchi, M.E.; et al. Activated platelets present high mobility group box 1 to neutrophils, inducing autophagy and promoting the extrusion of neutrophil extracellular traps. J. Thromb. Haemost. 2014, 12, 2074–2088. [Google Scholar] [CrossRef] [PubMed]
- Kraemer, B.F.; Campbell, R.A.; Schwertz, H.; Cody, M.J.; Franks, Z.; Tolley, N.D.; Kahr, W.H.A.; Lindemann, S.; Seizer, P.; Yost, C.C.; et al. Novel Anti-bacterial Activities of β-defensin 1 in Human Platelets: Suppression of Pathogen Growth and Signaling of Neutrophil Extracellular Trap Formation. PLOS Pathog. 2011, 7, e1002355. [Google Scholar] [CrossRef] [PubMed]
- Carestia, A.; Kaufman, T.; Rivadeneyra, L.; Landoni, V.I.; Pozner, R.G.; Negrotto, S.; D’Atri, L.P.; Gómez, R.M.; Schattner, M. Mediators and molecular pathways involved in the regulation of neutrophil extracellular trap formation mediated by activated platelets. J. Leuk. Biol. 2016, 99, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Amirkhosravi, A.; Amaya, M.; Desai, H.; Francis, J.L. Platelet-CD40 ligand interaction with melanoma cell and monocyte CD40 enhances cellular procoagulant activity. Blood Coagul. Fibrinolysis 2002, 13, 505–512. [Google Scholar] [CrossRef] [PubMed]
- Calverley, D.C.; Phang, T.L.; Choudhury, Q.G.; Gao, B.; Oton, A.B.; Weyant, M.J.; Geraci, M.W. Significant downregulation of platelet gene expression in metastatic lung cancer. Clin. Transl. Sci. 2010, 3, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, R.J.A.; Balaj, L.; Hulleman, E.; van Rijn, S.; Pegtel, D.M.; Walraven, M.; Widmark, A.; Gerritsen, W.R.; Verheul, H.M.; Vandertop, W.P.; et al. Blood platelets contain tumor-derived RNA biomarkers. Blood 2011, 118, 3680–3683. [Google Scholar] [CrossRef] [PubMed]
- Best, M.G.; Sol, N.; Kooi, I.; Tannous, J.; Westerman, B.A.; Rustenburg, F.; Schellen, P.; Verschueren, H.; Post, E.; Koster, J.; et al. RNA-Seq of Tumor-Educated Platelets Enables Blood-Based Pan-Cancer, Multiclass, and Molecular Pathway Cancer Diagnostics. Cancer Cell 2015, 28, 666–676. [Google Scholar] [CrossRef] [PubMed]
- Denis, M.M.; Tolley, N.D.; Bunting, M.; Schwertz, H.; Jiang, H.; Lindemann, S.; Yost, C.C.; Rubner, F.J.; Albertine, K.H.; Swoboda, K.J.; et al. Escaping the Nuclear Confines: Signal-Dependent Pre-mRNA Splicing in Anucleate Platelets. Cell 2005, 122, 379–391. [Google Scholar] [CrossRef] [PubMed]
- Alhasan, A.A.; Izuogu, O.G.; Al-Balool, H.H.; Steyn, J.S.; Evans, A.; Colzani, M.; Ghevaert, C.; Mountford, J.C.; Marenah, L.; Elliott, D.J.; et al. Circular RNA enrichment in platelets is a signature of transcriptome degradation. Blood 2016, 127, e1–e11. [Google Scholar] [CrossRef] [PubMed]
- Rowley, J.W.; Oler, A.J.; Tolley, N.D.; Hunter, B.N.; Low, E.N.; Nix, D.A.; Yost, C.C.; Zimmerman, G.A.; Weyrich, A.S. Genome-wide RNA-seq analysis of human and mouse platelet transcriptomes. Blood 2011, 118, e101–e111. [Google Scholar] [CrossRef] [PubMed]
- Schubert, S.; Weyrich, A.S.; Rowley, J.W. A tour through the transcriptional landscape of platelets. Blood 2014, 124, 493–502. [Google Scholar] [CrossRef] [PubMed]
- Landry, P.; Plante, I.; Ouellet, D.L.; Perron, M.P.; Rousseau, G.; Provost, P. Existence of a microRNA pathway in anucleate platelets. Nat. Struct. Mol. Biol. 2009, 16, 961–966. [Google Scholar] [CrossRef] [PubMed]
- Bray, P.F.; McKenzie, S.E.; Edelstein, L.C.; Nagalla, S.; Delgrosso, K.; Ertel, A.; Kupper, J.; Jing, Y.; Londin, E.; Loher, P.; et al. The complex transcriptional landscape of the anucleate human platelet. BMC Genom. 2013, 14, 1. [Google Scholar] [CrossRef] [PubMed]
- Plé, H.; Landry, P.; Benham, A.; Coarfa, C.; Gunaratne, P.H.; Provost, P. The repertoire and features of human platelet microRNAs. PLoS ONE 2012, 7, e50746. [Google Scholar] [CrossRef] [PubMed]
- Agam, G.; Bessler, H.; Djaldetti, M. In vitro DNA and RNA synthesis by human platelets. Biochim. Biophys. Acta 1976, 425, 41–48. [Google Scholar] [CrossRef]
- Booyse, F.M.; Rafelson, M.E. Stable messenger RNA in the synthesis of contractile protein in human platelets. Biochim. Biophys. Acta 1967, 145, 188–190. [Google Scholar] [CrossRef]
- Brown, G.T.; McIntyre, T.M. Lipopolysaccharide signaling without a nucleus: Kinase cascades stimulate platelet shedding of proinflammatory IL-1β-rich microparticles. J. Immunol. 2011, 186, 5489–5496. [Google Scholar] [CrossRef] [PubMed]
- Schwertz, H.; Tolley, N.D.; Foulks, J.M.; Denis, M.M.; Risenmay, B.W.; Buerke, M.; Tilley, R.E.; Rondina, M.T.; Harris, E.M.; Kraiss, L.W.; et al. Signal-dependent splicing of tissue factor pre-mRNA modulates the thrombogenicity of human platelets. J. Exp. Med. 2006, 203, 2433–2440. [Google Scholar] [CrossRef] [PubMed]
- Rosenwald, I.B.; Pechet, L.; Han, A.; Lu, L.; Pihan, G.; Woda, B.; Chen, J.J.; Szymanski, I. Expression of translation initiation factors elF-4E and elF-2alpha and a potential physiologic role of continuous protein synthesis in human platelets. Thromb. Haemost. 2001, 85, 142–151. [Google Scholar] [PubMed]
- Kishore, S.; Khanna, A.; Zhang, Z.; Hui, J.; Balwierz, P.J.; Stefan, M.; Beach, C.; Nicholls, R.D.; Zavolan, M.; Stamm, S. The snoRNA MBII-52 (SNORD 115) is processed into smaller RNAs and regulates alternative splicing. Hum. Mol. Genet. 2010, 19, 1153–1164. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, R.J.A.; Karachaliou, N.; Berenguer, J.; Gimenez-Capitan, A.; Schellen, P.; Teixido, C.; Tannous, J.; Kuiper, J.L.; Drees, E.; Grabowska, M.; et al. Rearranged EML4-ALK fusion transcripts sequester in circulating blood platelets and enable blood-based crizotinib response monitoring in non-small-cell lung cancer. Oncotarget 2015, 7, 1066–1075. [Google Scholar] [CrossRef] [PubMed]
- Best, M.G.; Sol, N.; In’t Veld, S.G.J.G.; Vancura, A.; Muller, M.; Niemeijer, A.-L.N.; Fejes, A.V.; Tjon Kon Fat, L.-A.; Huis In ’t Veld, A.E.; Leurs, C.; et al. Swarm Intelligence-Enhanced Detection of Non-Small-Cell Lung Cancer Using Tumor-Educated Platelets. Cancer Cell 2017, 32, 238–252. [Google Scholar] [CrossRef] [PubMed]
- Bian, D.; Shi, W.; Shao, Y.; Li, P.; Song, G. Long non-coding RNA GAS5 inhibits tumorigenesis via miR-137 in melanoma. Am. J. Transl. Res. 2017, 9, 1509–1520. [Google Scholar] [PubMed]
- Luo, G.; Liu, D.; Huang, C.; Wang, M.; Xiao, X.; Zeng, F.; Wang, L.; Jiang, G. LncRNA GAS5 Inhibits Cellular Proliferation by Targeting P27Kip1. Mol. Cancer Res. 2017, 15, 789–799. [Google Scholar] [CrossRef] [PubMed]
- Luo, C.-L.; Xu, Z.-G.; Chen, H.; Ji, J.; Wang, Y.-H.; Hu, W.; Wang, K.; Zhang, W.-W.; Yuan, C.-H.; Wang, F.-B. LncRNAs and EGFRvIII sequestered in TEPs enable blood-based NSCLC diagnosis. Cancer Manag. Res. 2018, 10, 1449–1459. Available online: https://www.dovepress.com/lncrnas-and-egfrviii-sequestered-in-teps-enable-blood-based-nsclc-diag-peer-reviewed-article-CMAR (accessed on 5 September 2018).
- Kaudewitz, D.; Skroblin, P.; Bender, L.H.; Barwari, T.; Willeit, P.; Pechlaner, R.; Sunderland, N.P.; Willeit, K.; Morton, A.C.; Armstrong, P.C.; et al. Association of MicroRNAs and YRNAs With Platelet FunctionNovelty and Significance. Circ. Res. 2016, 118, 420–432. [Google Scholar] [CrossRef] [PubMed]
- Gidlöf, O.; van der Brug, M.; Ohman, J.; Gilje, P.; Olde, B.; Wahlestedt, C.; Erlinge, D. Platelets activated during myocardial infarction release functional miRNA, which can be taken up by endothelial cells and regulate ICAM1 expression. Blood 2013, 121, 3908–3917. [Google Scholar] [CrossRef] [PubMed]
- Londin, E.R.; Hatzimichael, E.; Loher, P.; Edelstein, L.; Shaw, C.; Delgrosso, K.; Fortina, P.; Bray, P.F.; McKenzie, S.E.; Rigoutsos, I. The human platelet: Strong transcriptome correlations among individuals associate weakly with the platelet proteome. Biol. Direct 2014, 9, 3. [Google Scholar] [CrossRef] [PubMed]
- McRedmond, J.P.; Park, S.D.; Reilly, D.F.; Coppinger, J.A.; Maguire, P.B.; Shields, D.C.; Fitzgerald, D.J. Integration of proteomics and genomics in platelets: A profile of platelet proteins and platelet-specific genes. Mol. Cell Proteomics 2004, 3, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Burkhart, J.M.; Vaudel, M.; Gambaryan, S.; Radau, S.; Walter, U.; Martens, L.; Geiger, J.; Sickmann, A.; Zahedi, R.P. The first comprehensive and quantitative analysis of human platelet protein composition allows the comparative analysis of structural and functional pathways. Blood 2012, 120, e73–e82. [Google Scholar] [CrossRef] [PubMed]
- Rowley, J.W.; Weyrich, A.S. Coordinate expression of transcripts and proteins in platelets. Blood 2013, 121, 5255–5256. [Google Scholar] [CrossRef] [PubMed]
- Hung, W.S.; Hung, C.L.; Fan, J.T.; Huang, D.Y.; Yeh, C.F.; Cheng, J.C.; Tseng, C.P. The endocytic adaptor protein Disabled-2 is required for cellular uptake of fibrinogen. Biochim. Biophys. Acta Mol. Cell Res. 2012, 1823, 1778–1788. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, M.; Joshi, S.; Zhang, J.; Moncman, C.L.; Yadav, S.; Bouchard, B.A.; Storrie, B.; Whiteheart, S.W. Cellubrevin/vesicle-associated membrane protein-3—Mediated endocytosis and trafficking regulate platelet functions. Blood 2017, 130, 2872–2883. [Google Scholar] [CrossRef] [PubMed]
- Klement, G.L.; Yip, T.-T.; Cassiola, F.; Kikuchi, L.; Cervi, D.; Podust, V.; Italiano, J.E.; Wheatley, E.; Abou-Slaybi, A.; Bender, E.; et al. Platelets actively sequester angiogenesis regulators. Blood 2009, 113, 2835–2842. [Google Scholar] [CrossRef] [PubMed]
- Rolfes, V.; Idel, C.; Pries, R.; Plötze-Martin, K.; Habermann, J.; Gemoll, T.; Bohnet, S.; Latz, E.; Ribbat-Idel, J.; Franklin, B.S.; et al. PD-L1 is expressed on human platelets and is affected by immune checkpoint therapy. Oncotarget 2018, 9, 27460–27470. [Google Scholar] [CrossRef] [PubMed]
- Rothwell, P.M.; Wilson, M.; Price, J.F.; Belch, J.F.; Meade, T.W.; Mehta, Z. Effect of daily aspirin on risk of cancer metastasis: A study of incident cancers during randomised controlled trials. Lancet 2012, 379, 1591–1601. [Google Scholar] [CrossRef]
- Guillem-Llobat, P.; Dovizio, M.; Bruno, A.; Ricciotti, E.; Cufino, V.; Sacco, A.; Grande, R.; Alberti, S.; Arena, V.; Cirillo, M.; et al. Aspirin prevents colorectal cancer metastasis in mice by splitting the crosstalk between platelets and tumor cells. Oncotarget 2016, 7, 32462–32477. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Nishihara, R.; Wu, K.; Wang, M.; Ogino, S.; Willett, W.C.; Spiegelman, D.; Fuchs, C.S.; Giovannucci, E.L.; Chan, A.T. Population-wide Impact of Long-term Use of Aspirin and the Risk for Cancer. JAMA Oncol. 2016, 2, 762–769. [Google Scholar] [CrossRef] [PubMed]
- Santilli, F.; Boccatonda, A.; Davì, G. Aspirin, platelets, and cancer: The point of view of the internist. Eur. J. Intern. Med. 2016, 34, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Gebremeskel, S.; LeVatte, T.; Liwski, R.S.; Johnston, B.; Bezuhly, M. The reversible P2Y12 inhibitor ticagrelor inhibits metastasis and improves survival in mouse models of cancer. Int. J. Cancer 2015, 136, 234–240. [Google Scholar] [CrossRef] [PubMed]
- Mezouar, S.; Darbousset, R.; Dignat-George, F.; Panicot-Dubois, L.; Dubois, C. Inhibition of platelet activation prevents the P-selectin and integrin-dependent accumulation of cancer cell microparticles and reduces tumor growth and metastasis in vivo. Int. J. Cancer 2015, 136, 462–475. [Google Scholar] [CrossRef] [PubMed]
- Denslow, A.; Świtalska, M.; Jarosz, J.; Papiernik, D.; Porshneva, K.; Nowak, M.; Wietrzyk, J. Clopidogrel in a combined therapy with anticancer drugs—Effect on tumor growth, metastasis, and treatment toxicity: Studies in animal models. PLoS ONE 2017, 12, e0188740. [Google Scholar] [CrossRef] [PubMed]
- Serebruany, V.L. Platelet Inhibition with Prasugrel and Increased Cancer Risks: Potential Causes and Implications. Am. J. Med. 2009, 122, 407–408. [Google Scholar] [CrossRef] [PubMed]
- Leader, A.; Zelikson-Saporta, R.; Pereg, D.; Spectre, G.; Rozovski, U.; Raanani, P.; Hermoni, D.; Lishner, M. The Effect of Combined Aspirin and Clopidogrel Treatment on Cancer Incidence. Am. J. Med. 2017, 130, 826–832. [Google Scholar] [CrossRef] [PubMed]
- Leader, A.; Zelikson-Saporta, R.; Rozovski, U.; Pereg, D.; Raanani, P.; Spectre, G.; Lishner, M.; Hermoni, D. Clopidogrel Treatment Is Associated with a Decrease in Cancer Incidence. Blood 2015, 126, 1124. [Google Scholar]
- Huo, Y.; Schober, A.; Forlow, S.B.; Smith, D.F.; Hyman, M.C.; Jung, S.; Littman, D.R.; Weber, C.; Ley, K. Circulating activated platelets exacerbate atherosclerosis in mice deficient in apolipoprotein E. Nat. Med. 2003, 9, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Sachais, B.S.; Turrentine, T.; Dawicki McKenna, J.M.; Rux, A.H.; Rader, D.; Kowalska, M.A. Elimination of platelet factor 4 (PF4) from platelets reduces atherosclerosis in C57Bl/6 and apoE-/- mice. Thromb. Haemost. 2007, 98, 1108–1113. [Google Scholar] [PubMed]
- Swaim, A.F.; Field, D.J.; Fox-Talbot, K.; Baldwin, W.M.; Morrell, C.N. Platelets Contribute to Allograft Rejection through Glutamate Receptor Signaling. J. Immunol. 2010, 185, 6999–7006. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Zhang, X.; Mannon, R.B.; Kirk, A.D. Platelet-derived or soluble CD154 induces vascularized allograft rejection independent of cell-bound CD154. J. Clin. Investig. 2006, 116, 769–774. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Ren, M.; Chen, N.; Luo, M.; Deng, X.; Xia, J.; Yu, G.; Liu, J.; He, B.; Zhang, X.; et al. Presence of intratumoral platelets is associated with tumor vessel structure and metastasis. BMC Cancer 2014, 14, 167. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.-T.; Nishikawa, T.; Kaneda, Y. Platelet-cytokine Complex Suppresses Tumour Growth by Exploiting Intratumoural Thrombin-dependent Platelet Aggregation. Sci. Rep. 2016, 6, 25077. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Sharkey, C.C.; Wun, B.; Liesveld, J.; King, M.R. Genetic engineering of platelets to neutralize circulating tumor cells. J. Control. Release 2016, 228, 38. [Google Scholar] [CrossRef] [PubMed]


© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Plantureux, L.; Mège, D.; Crescence, L.; Dignat-George, F.; Dubois, C.; Panicot-Dubois, L. Impacts of Cancer on Platelet Production, Activation and Education and Mechanisms of Cancer-Associated Thrombosis. Cancers 2018, 10, 441. https://doi.org/10.3390/cancers10110441
Plantureux L, Mège D, Crescence L, Dignat-George F, Dubois C, Panicot-Dubois L. Impacts of Cancer on Platelet Production, Activation and Education and Mechanisms of Cancer-Associated Thrombosis. Cancers. 2018; 10(11):441. https://doi.org/10.3390/cancers10110441
Chicago/Turabian StylePlantureux, Léa, Diane Mège, Lydie Crescence, Françoise Dignat-George, Christophe Dubois, and Laurence Panicot-Dubois. 2018. "Impacts of Cancer on Platelet Production, Activation and Education and Mechanisms of Cancer-Associated Thrombosis" Cancers 10, no. 11: 441. https://doi.org/10.3390/cancers10110441
APA StylePlantureux, L., Mège, D., Crescence, L., Dignat-George, F., Dubois, C., & Panicot-Dubois, L. (2018). Impacts of Cancer on Platelet Production, Activation and Education and Mechanisms of Cancer-Associated Thrombosis. Cancers, 10(11), 441. https://doi.org/10.3390/cancers10110441