The Cooperative Relationship between STAT5 and Reactive Oxygen Species in Leukemia: Mechanism and Therapeutic Potential

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
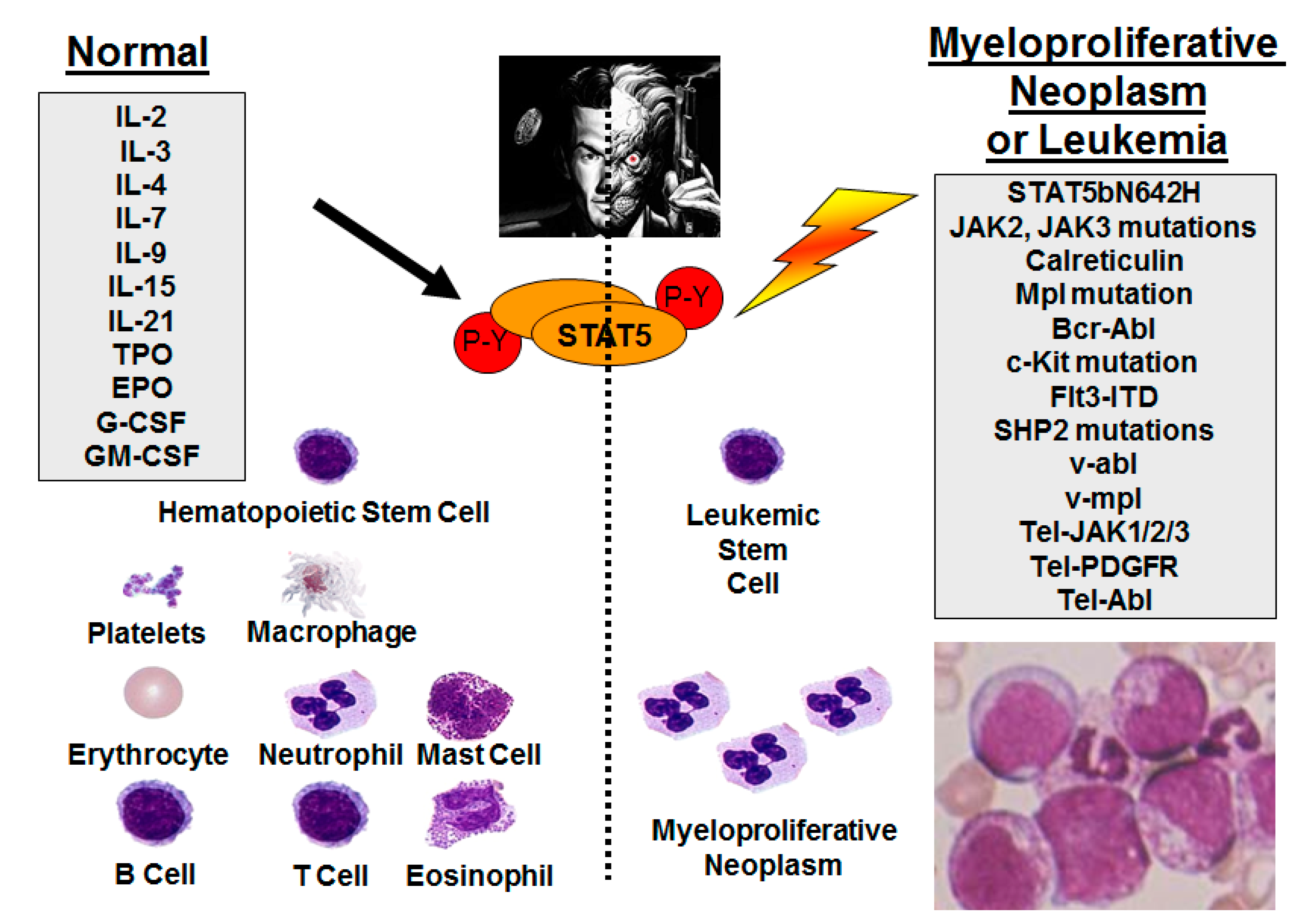
2. Persistent STAT5 Activation through Upstream Mutant Tyrosine Kinases
3. STAT5b Gene Mutations in Leukemia/Lymphoma
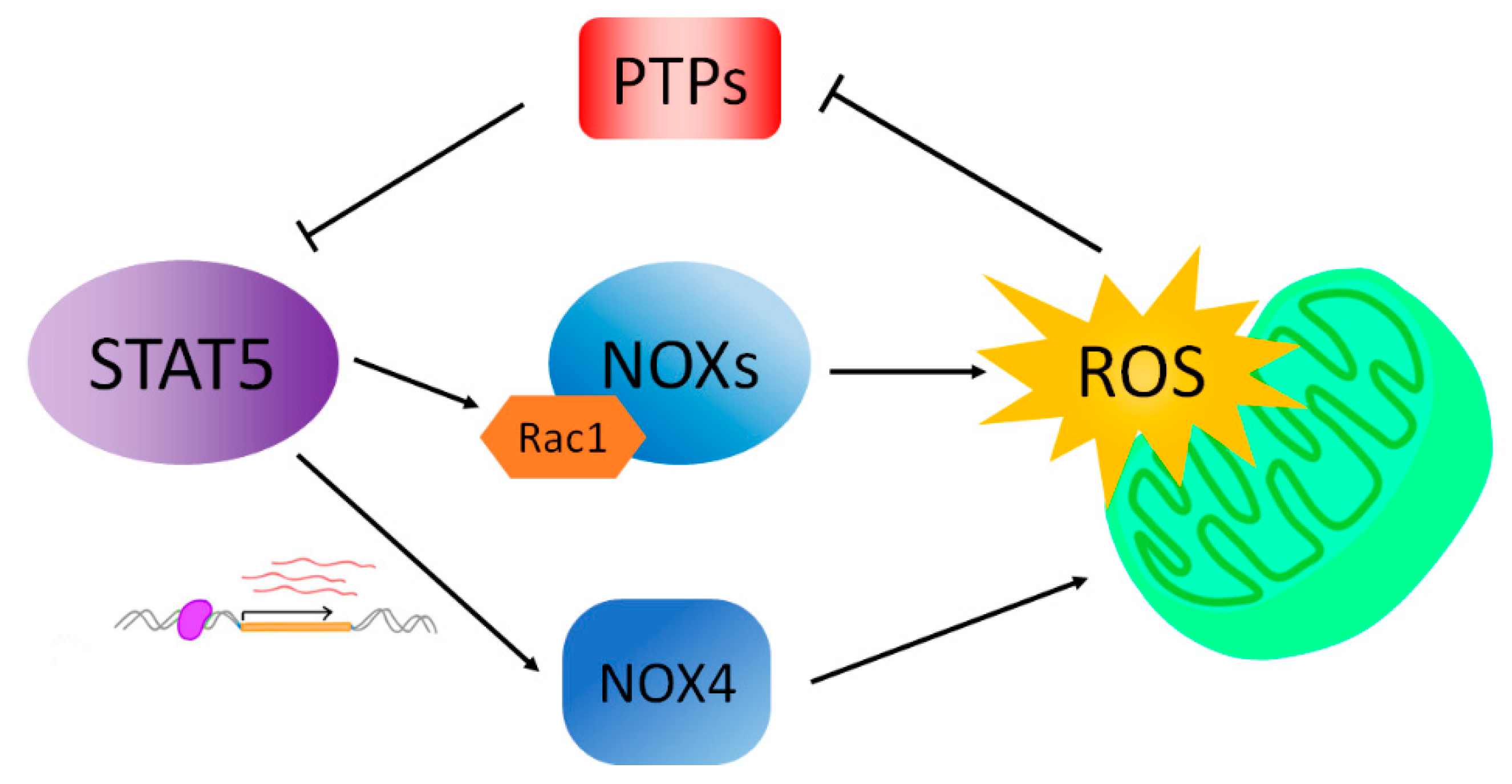
4. STAT5 Drives ROS Formation
5. ROS Promotes STAT5 Phosphorylation
6. Non-Canonical STAT5 Activation in High Risk Subsets of Acute Myeloid Leukemia
7. Emerging ROS-Reducing Cancer Therapies
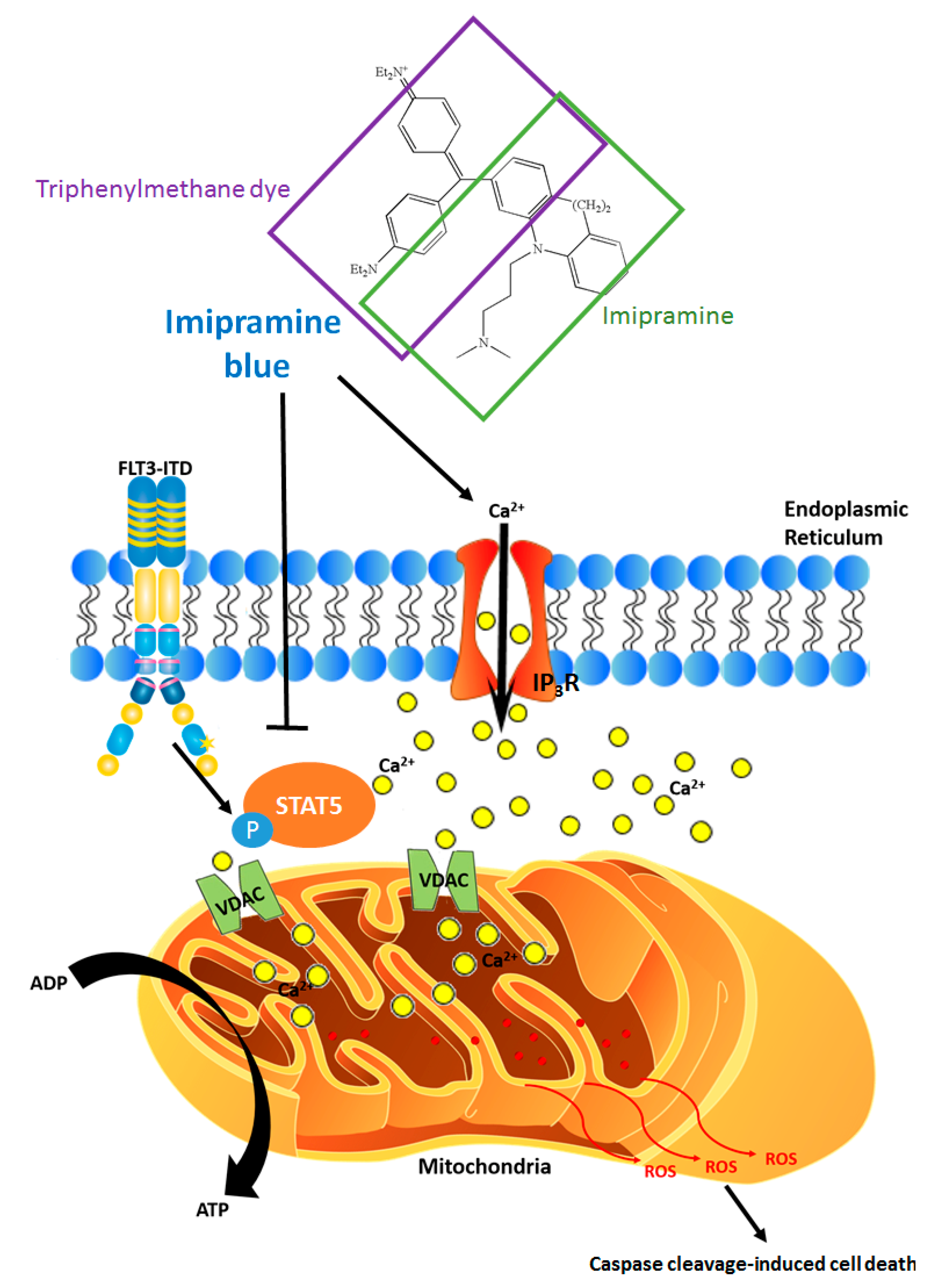
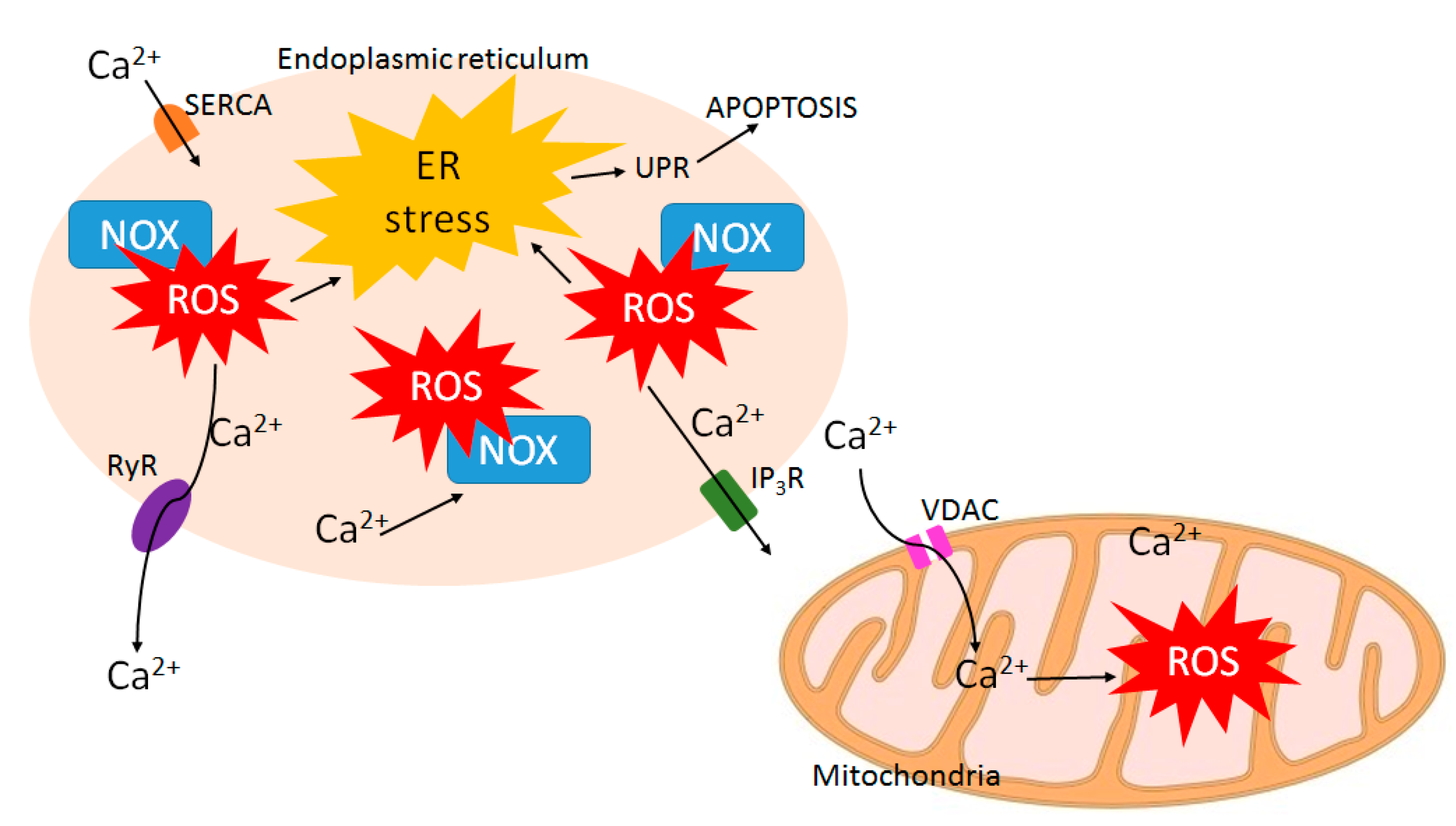
8. ROS-Calcium Connection and Potential for ROS-Increasing Cancer Therapies
9. Exploiting the ROS-STAT5 Kinome to Identify New Therapeutic Targets
10. STAT5 in Normal Hematopoiesis—Potential Side Effects of STAT5 Inhibition
11. Conclusions and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Thannickal, V.J.; Fanburg, B.L. Reactive oxygen species in cell signaling. Am. J. Physiol. Lung Cell. Mol. Physiol. 2000, 279, L1005–L1028. [Google Scholar] [CrossRef] [PubMed]
- Ray, P.D.; Huang, B.W.; Tsuji, Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell. Signal. 2012, 24, 981–990. [Google Scholar] [CrossRef] [PubMed]
- Schumacker, P.T. Reactive oxygen species in cancer cells: Live by the sword, die by the sword. Cancer Cell 2006, 10, 175–176. [Google Scholar] [CrossRef] [PubMed]
- Jayavelu, A.K.; Moloney, J.N.; Bohmer, F.D.; Cotter, T.G. NOX-driven ROS formation in cell transformation of FLT3-ITD-positive AML. Exp. Hematol. 2016, 44, 1113–1122. [Google Scholar] [CrossRef] [PubMed]
- Hole, P.S.; Zabkiewicz, J.; Munje, C.; Newton, Z.; Pearn, L.; White, P.; Marquez, N.; Hills, R.K.; Burnett, A.K.; Tonks, A.; et al. Overproduction of NOX-derived ROS in AML promotes proliferation and is associated with defective oxidative stress signaling. Blood 2013, 122, 3322–3330. [Google Scholar] [CrossRef] [PubMed]
- Touyz, R.M. Reactive oxygen species and angiotensin II signaling in vascular cells—Implications in cardiovascular disease. Braz. J. Med. Boil. Res. 2004, 37, 1263–1273. [Google Scholar] [CrossRef]
- Hensley, K.; Robinson, K.A.; Gabbita, S.P.; Salsman, S.; Floyd, R.A. Reactive oxygen species, cell signaling, and cell injury. Free Radic. Boil. Med. 2000, 28, 1456–1462. [Google Scholar] [CrossRef]
- Simon, A.R.; Rai, U.; Fanburg, B.L.; Cochran, B.H. Activation of the JAK-STAT pathway by reactive oxygen species. Am. J. Physiol. 1998, 275, C1640–C1652. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Jove, R. The STATs of cancer—New molecular targets come of age. Nat. Rev. Cancer 2004, 4, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Constantinescu, S.N.; Girardot, M.; Pecquet, C. Mining for JAK-STAT mutations in cancer. Trends Biochem. Sci. 2008, 33, 122–131. [Google Scholar] [CrossRef]
- Rani, A.; Murphy, J.J. STAT5 in cancer and immunity. J. Interferon Cytokine Res. 2016, 36, 226–237. [Google Scholar] [CrossRef] [PubMed]
- Wittig, I.; Groner, B. Signal transducer and activator of transcription 5 (STAT5), a crucial regulator of immune and cancer cells. Curr. Drug Targets Immune Endocr. Metab. Disord. 2005, 5, 449–463. [Google Scholar] [CrossRef]
- Shuai, K.; Halpern, J.; ten Hoeve, J.; Rao, X.; Sawyers, C.L. Constitutive activation of STAT5 by the Bcr-Abl oncogene in chronic myelogenous leukemia. Oncogene 1996, 13, 247–254. [Google Scholar] [PubMed]
- Huang, M.; Dorsey, J.F.; Epling-Burnette, P.K.; Nimmanapalli, R.; Landowski, T.H.; Mora, L.B.; Niu, G.; Sinibaldi, D.; Bai, F.; Kraker, A.; et al. Inhibition of Bcr-Abl kinase activity by PD180970 blocks constitutive activation of STAT5 and growth of cml cells. Oncogene 2002, 21, 8804–8816. [Google Scholar] [CrossRef] [PubMed]
- Spiekermann, K.; Bagrintseva, K.; Schwab, R.; Schmieja, K.; Hiddemann, W. Overexpression and constitutive activation of FLT3 induces STAT5 activation in primary acute myeloid leukemia blast cells. Clin. Cancer Res. 2003, 9, 2140–2150. [Google Scholar] [PubMed]
- Harir, N.; Boudot, C.; Friedbichler, K.; Sonneck, K.; Kondo, R.; Martin-Lanneree, S.; Kenner, L.; Kerenyi, M.; Yahiaoui, S.; Gouilleux-Gruart, V.; et al. Oncogenic kit controls neoplastic mast cell growth through a STAT5/PI3-kinase signaling cascade. Blood 2008, 112, 2463–2473. [Google Scholar] [CrossRef] [PubMed]
- Wilbanks, A.M.; Mahajan, S.; Frank, D.A.; Druker, B.J.; Gilliland, D.G.; Carroll, M. Tel/PDGFbetar fusion protein activates STAT1 and STAT5: A common mechanism for transformation by tyrosine kinase fusion proteins. Exp. Hematol. 2000, 28, 584–593. [Google Scholar] [CrossRef]
- Quintas-Cardama, A.; Verstovsek, S. Molecular pathways: JAK/STAT pathway: Mutations, inhibitors, and resistance. Clin. Cancer Res. 2013, 19, 1933–1940. [Google Scholar] [CrossRef] [PubMed]
- Silva, C.M. Role of STATs as downstream signal transducers in Src family kinase-mediated tumorigenesis. Oncogene 2004, 23, 8017–8023. [Google Scholar] [CrossRef] [PubMed]
- Ozawa, Y.; Williams, A.H.; Estes, M.L.; Matsushita, N.; Boschelli, F.; Jove, R.; List, A.F. Src family kinases promote AML cell survival through activation of signal transducers and activators of transcription (STAT). Leuk. Res. 2008, 32, 893–903. [Google Scholar] [CrossRef] [PubMed]
- Rajala, H.L.; Eldfors, S.; Kuusanmaki, H.; van Adrichem, A.J.; Olson, T.; Lagstrom, S.; Andersson, E.I.; Jerez, A.; Clemente, M.J.; Yan, Y.; et al. Discovery of somatic STAT5b mutations in large granular lymphocytic leukemia. Blood 2013, 121, 4541–4550. [Google Scholar] [CrossRef] [PubMed]
- Rajala, H.L.; Porkka, K.; Maciejewski, J.P.; Loughran, T.P., Jr.; Mustjoki, S. Uncovering the pathogenesis of large granular lymphocytic leukemia-novel STAT3 and STAT5b mutations. Ann. Med. 2014, 46, 114–122. [Google Scholar] [CrossRef] [PubMed]
- Kucuk, C.; Jiang, B.; Hu, X.; Zhang, W.; Chan, J.K.; Xiao, W.; Lack, N.; Alkan, C.; Williams, J.C.; Avery, K.N.; et al. Activating mutations of STAT5b and STAT3 in lymphomas derived from γδ-T or NK cells. Nat. Commun. 2015, 6, 6025. [Google Scholar] [CrossRef] [PubMed]
- Roberti, A.; Dobay, M.P.; Bisig, B.; Vallois, D.; Boechat, C.; Lanitis, E.; Bouchindhomme, B.; Parrens, M.C.; Bossard, C.; Quintanilla-Martinez, L.; et al. Type II enteropathy-associated T-cell lymphoma features a unique genomic profile with highly recurrent SETD2 alterations. Nat. Commun. 2016, 7, 12602. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Wen, L.; Wu, L.; Wang, Q.; Yao, H.; Wang, Q.; Ma, L.; Chen, S. Rare occurrence of a STAT5b N642H mutation in adult T-cell acute lymphoblastic leukemia. Cancer Genet. 2015, 208, 52–53. [Google Scholar] [CrossRef] [PubMed]
- Bandapalli, O.R.; Schuessele, S.; Kunz, J.B.; Rausch, T.; Stutz, A.M.; Tal, N.; Geron, I.; Gershman, N.; Izraeli, S.; Eilers, J.; et al. The activating STAT5B N642H mutation is a common abnormality in pediatric T-cell acute lymphoblastic leukemia and confers a higher risk of relapse. Haematologica 2014, 99, e188–e192. [Google Scholar] [CrossRef] [PubMed]
- McKinney, M.; Moffitt, A.B.; Gaulard, P.; Travert, M.; De Leval, L.; Nicolae, A.; Raffeld, M.; Jaffe, E.S.; Pittaluga, S.; Xi, L.; et al. The genetic basis of hepatosplenic T-cell lymphoma. Cancer Discov. 2017, 7, 369–379. [Google Scholar] [CrossRef] [PubMed]
- Pham, H.T.T.; Maurer, B.; Prchal-Murphy, M.; Grausenburger, R.; Grundschober, E.; Javaheri, T.; Nivarthi, H.; Boersma, A.; Kolbe, T.; Elabd, M. STAT5B N642H is a driver mutation for T cell neoplasia. J. Clin. Investig. 2018, 128, 387–401. [Google Scholar] [CrossRef] [PubMed]
- Bourgeais, J.; Gouilleux-Gruart, V.; Gouilleux, F. Oxidative metabolism in cancer: A STAT affair? JAKSTAT 2013, 2, e25764. [Google Scholar] [CrossRef] [PubMed]
- Warsch, W.; Grundschober, E.; Berger, A.; Gille, L.; Cerny-Reiterer, S.; Tigan, A.S.; Hoelbl-Kovacic, A.; Valent, P.; Moriggl, R.; Sexl, V. STAT5 triggers BCR-ABL1 mutation by mediating ROS production in chronic myeloid leukaemia. Oncotarget 2012, 3, 1669–1687. [Google Scholar] [CrossRef] [PubMed]
- Sallmyr, A.; Fan, J.; Rassool, F.V. Genomic instability in myeloid malignancies: Increased reactive oxygen species (ROS), DNA double strand breaks (DSBs) and error-prone repair. Cancer Lett. 2008, 270, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Bedard, K.; Krause, K.H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef] [PubMed]
- Jayavelu, A.K.; Muller, J.P.; Bauer, R.; Bohmer, S.A.; Lassig, J.; Cerny-Reiterer, S.; Sperr, W.R.; Valent, P.; Maurer, B.; Moriggl, R.; et al. NOX4-driven ROS formation mediates PTP inactivation and cell transformation in FLT3ITD-positive AML cells. Leukemia 2016, 30, 473–483. [Google Scholar] [CrossRef] [PubMed]
- Woolley, J.F.; Naughton, R.; Stanicka, J.; Gough, D.R.; Bhatt, L.; Dickinson, B.C.; Chang, C.J.; Cotter, T.G. H2O2 production downstream of FLT3 is mediated by p22phox in the endoplasmic reticulum and is required for STAT5 signalling. PLoS ONE 2012, 7, e34050. [Google Scholar] [CrossRef] [PubMed]
- Bourgeais, J.; Ishac, N.; Medrzycki, M.; Brachet-Botineau, M.; Desbourdes, L.; Gouilleux-Gruart, V.; Pecnard, E.; Rouleux-Bonnin, F.; Gyan, E.; Domenech, J.; et al. Oncogenic STAT5 signaling promotes oxidative stress in chronic myeloid leukemia cells by repressing antioxidant defenses. Oncotarget 2017, 8, 41876–41889. [Google Scholar] [CrossRef] [PubMed]
- Casetti, L.; Martin-Lanneree, S.; Najjar, I.; Plo, I.; Auge, S.; Roy, L.; Chomel, J.C.; Lauret, E.; Turhan, A.G.; Dusanter-Fourt, I. Differential contributions of STAT5a and STAT5b to stress protection and tyrosine kinase inhibitor resistance of chronic myeloid leukemia stem/progenitor cells. Cancer Res. 2013, 73, 2052–2058. [Google Scholar] [CrossRef] [PubMed]
- Iiyama, M.; Kakihana, K.; Kurosu, T.; Miura, O. Reactive oxygen species generated by hematopoietic cytokines play roles in activation of receptor-mediated signaling and in cell cycle progression. Cell. Signal. 2006, 18, 174–182. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Sedwick, D.; Wang, Z. Genetic alterations of protein tyrosine phosphatases in human cancers. Oncogene 2015, 34, 3885–3894. [Google Scholar] [CrossRef] [PubMed]
- Chiarugi, P.; Cirri, P. Redox regulation of protein tyrosine phosphatases during receptor tyrosine kinase signal transduction. Trends Biochem. Sci. 2003, 28, 509–514. [Google Scholar] [CrossRef]
- Ostman, A.; Bohmer, F.D. Regulation of receptor tyrosine kinase signaling by protein tyrosine phosphatases. Trends Cell Boil. 2001, 11, 258–266. [Google Scholar] [CrossRef]
- Duhe, R.J. Redox regulation of Janus kinase: The elephant in the room. JAK-STAT 2013, 2, e26141. [Google Scholar] [CrossRef] [PubMed]
- Duhe, R.J.; Evans, G.A.; Erwin, R.A.; Kirken, R.A.; Cox, G.W.; Farrar, W.L. Nitric oxide and thiol redox regulation of janus kinase activity. Proc. Natl. Acad. Sci. USA 1998, 95, 126–131. [Google Scholar] [CrossRef] [PubMed]
- Klco, J.M.; Miller, C.A.; Griffith, M.; Petti, A.; Spencer, D.H.; Ketkar-Kulkarni, S.; Wartman, L.D.; Christopher, M.; Lamprecht, T.L.; Helton, N.M.; et al. Association between mutation clearance after induction therapy and outcomes in acute myeloid leukemia. JAMA 2015, 314, 811–822. [Google Scholar] [CrossRef] [PubMed]
- Grimwade, D.; Hills, R.K.; Moorman, A.V.; Walker, H.; Chatters, S.; Goldstone, A.H.; Wheatley, K.; Harrison, C.J.; Burnett, A.K. Refinement of cytogenetic classification in acute myeloid leukemia: Determination of prognostic significance of rare recurring chromosomal abnormalities among 5876 younger adult patients treated in the united kingdom medical research council trials. Blood 2010, 116, 354–365. [Google Scholar] [CrossRef] [PubMed]
- Harrison, C.J.; Hills, R.K.; Moorman, A.V.; Grimwade, D.J.; Hann, I.; Webb, D.K.; Wheatley, K.; de Graaf, S.S.; van den Berg, E.; Burnett, A.K.; et al. Cytogenetics of childhood acute myeloid leukemia: United kingdom medical research council treatment trials AML 10 and 12. J. Clin. Oncol. 2010, 28, 2674–2681. [Google Scholar] [CrossRef] [PubMed]
- Tyner, J.W.; Erickson, H.; Deininger, M.W.; Willis, S.G.; Eide, C.A.; Levine, R.L.; Heinrich, M.C.; Gattermann, N.; Gilliland, D.G.; Druker, B.J.; et al. High-throughput sequencing screen reveals novel, transforming ras mutations in myeloid leukemia patients. Blood 2009, 113, 1749–1755. [Google Scholar] [CrossRef] [PubMed]
- Heuser, M.; Sly, L.M.; Argiropoulos, B.; Kuchenbauer, F.; Lai, C.; Weng, A.; Leung, M.; Lin, G.; Brookes, C.; Fung, S.; et al. Modelling the functional heterogeneity of leukemia stem cells: Role of STAT5 in leukemia stem cell self-renewal. Blood 2009, 114, 3983–3993. [Google Scholar] [CrossRef] [PubMed]
- Sallmyr, A.; Fan, J.; Datta, K.; Kim, K.T.; Grosu, D.; Shapiro, P.; Small, D.; Rassool, F. Internal tandem duplication of FLT3 (FLT3/ITD) induces increased ros production, DNA damage, and misrepair: Implications for poor prognosis in AML. Blood 2008, 111, 3173–3182. [Google Scholar] [CrossRef] [PubMed]
- Bunting, K.D. STAT5 signaling in normal and pathologic hematopoiesis. Front. Biosci. 2007, 12, 2807–2820. [Google Scholar] [CrossRef] [PubMed]
- Schepers, H.; van, G.D.; Wierenga, A.T.; Eggen, B.J.; Schuringa, J.J.; Vellenga, E. STAT5 is required for long-term maintenance of normal and leukemic human stem/progenitor cells. Blood 2007, 110, 2880–2888. [Google Scholar] [CrossRef] [PubMed]
- Warsch, W.; Kollmann, K.; Eckelhart, E.; Fajmann, S.; Cerny-Reiterer, S.; Holbl, A.; Gleixner, K.V.; Dworzak, M.; Mayerhofer, M.; Hoermann, G.; et al. High STAT5 levels mediate imatinib resistance and indicate disease progression in chronic myeloid leukemia. Blood 2011, 117, 3409–3420. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Bi, C.; Janakakumara, J.V.; Liu, S.C.; Chng, W.J.; Tay, K.G.; Poon, L.F.; Xie, Z.; Palaniyandi, S.; Yu, H.; et al. Enhanced activation of STAT pathways and overexpression of survivin confer resistance to FLT3 inhibitors and could be therapeutic targets in AML. Blood 2009, 113, 4052–4062. [Google Scholar] [CrossRef] [PubMed]
- Weisberg, E.; Boulton, C.; Kelly, L.M.; Manley, P.; Fabbro, D.; Meyer, T.; Gilliland, D.G.; Griffin, J.D. Inhibition of mutant FLT3 receptors in leukemia cells by the small molecule tyrosine kinase inhibitor pkc412. Cancer Cell 2002, 1, 433–443. [Google Scholar] [CrossRef]
- Starr, P. Midostaurin the first targeted therapy to improve survival in AML: Potentially practice-changing. Am. Health Drug Benefits 2016, 9, 1–21. [Google Scholar] [PubMed]
- Choudhary, C.; Brandts, C.; Schwable, J.; Tickenbrock, L.; Sargin, B.; Ueker, A.; Bohmer, F.D.; Berdel, W.E.; Muller-Tidow, C.; Serve, H. Activation mechanisms of STAT5 by oncogenic FLT3-ITD. Blood 2007, 110, 370–374. [Google Scholar] [CrossRef] [PubMed]
- Rocnik, J.L.; Okabe, R.; Yu, J.C.; Giese, N.; Schenkein, D.P.; Gilliland, D.G. Roles of tyrosine 589 and 591 in STAT5 activation and transformation mediated by FLT3-ITD. Blood 2006, 108, 1339–1345. [Google Scholar] [CrossRef] [PubMed]
- Schmidt-Arras, D.; Bohmer, S.A.; Koch, S.; Muller, J.P.; Blei, L.; Cornils, H.; Bauer, R.; Korasikha, S.; Thiede, C.; Bohmer, F.D. Anchoring of FLT3 in the endoplasmic reticulum alters signaling quality. Blood 2009, 113, 3568–3576. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, C.; Olsen, J.V.; Brandts, C.; Cox, J.; Reddy, P.N.; Bohmer, F.D.; Gerke, V.; Schmidt-Arras, D.E.; Berdel, W.E.; Muller-Tidow, C.; et al. Mislocalized activation of oncogenic RTKs switches downstream signaling outcomes. Mol. Cell 2009, 36, 326–339. [Google Scholar] [CrossRef] [PubMed]
- Obata, Y.; Toyoshima, S.; Wakamatsu, E.; Suzuki, S.; Ogawa, S.; Esumi, H.; Abe, R. Oncogenic kit signals on endolysosomes and endoplasmic reticulum are essential for neoplastic mast cell proliferation. Nat. Commun. 2014, 5, 5715. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Li, G.; Tse, W.; Bunting, K.D. Conditional deletion of STAT5 in adult mouse hematopoietic stem cells causes loss of quiescence and permits efficient nonablative stem cell replacement. Blood 2009, 113, 4856–4865. [Google Scholar] [CrossRef] [PubMed]
- Couldrey, C.; Bradley, H.L.; Bunting, K.D. A STAT5 modifier locus on murine chromosome 7 modulates engraftment of hematopoietic stem cells during steady-state hematopoiesis. Blood 2005, 105, 1476–1483. [Google Scholar] [CrossRef] [PubMed]
- Bradley, H.L.; Couldrey, C.; Bunting, K.D. Hematopoietic-repopulating defects from STAT5-deficient bone marrow are not fully accounted for by loss of thrombopoietin responsiveness. Blood 2004, 103, 2965–2972. [Google Scholar] [CrossRef] [PubMed]
- Bunting, K.D.; Bradley, H.L.; Hawley, T.S.; Moriggl, R.; Sorrentino, B.P.; Ihle, J.N. Reduced lymphomyeloid repopulating activity from adult bone marrow and fetal liver of mice lacking expression of STAT5. Blood 2002, 99, 479–487. [Google Scholar] [CrossRef] [PubMed]
- Miaczynska, M. Effects of membrane trafficking on signaling by receptor tyrosine kinases. Cold Spring Harb. Perspect. Boil. 2013, 5, a009035. [Google Scholar] [CrossRef] [PubMed]
- Monteith, G.R.; Davis, F.M.; Roberts-Thomson, S.J. Calcium channels and pumps in cancer: Changes and consequences. J. Biol. Chem. 2012, 287, 31666–31673. [Google Scholar] [CrossRef] [PubMed]
- Rimessi, A.; Patergnani, S.; Bonora, M.; Wieckowski, M.R.; Pinton, P. Mitochondrial Ca(2+) remodeling is a prime factor in oncogenic behavior. Front. Oncol. 2015, 5, 143. [Google Scholar] [CrossRef] [PubMed]
- Munson, J.M.; Fried, L.; Rowson, S.A.; Bonner, M.Y.; Karumbaiah, L.; Diaz, B.; Courtneidge, S.A.; Knaus, U.G.; Brat, D.J.; Arbiser, J.L.; et al. Anti-invasive adjuvant therapy with imipramine blue enhances chemotherapeutic efficacy against glioma. Sci. Transl. Med. 2012, 4, 127ra136. [Google Scholar] [CrossRef] [PubMed]
- Perry, B.N.; Govindarajan, B.; Bhandarkar, S.S.; Knaus, U.G.; Valo, M.; Sturk, C.; Carrillo, C.O.; Sohn, A.; Cerimele, F.; Dumont, D.; et al. Pharmacologic blockade of angiopoietin-2 is efficacious against model hemangiomas in mice. J. Investig. Dermatol. 2006, 126, 2316–2322. [Google Scholar] [CrossRef] [PubMed]
- Colston, J.; Horobin, R.W.; Rashid-Doubell, F.; Pediani, J.; Johal, K.K. Why fluorescent probes for endoplasmic reticulum are selective: An experimental and QSAR-modelling study. Biotech. Histochem. 2003, 78, 323–332. [Google Scholar] [CrossRef] [PubMed]
- Kazmi, F.; Hensley, T.; Pope, C.; Funk, R.S.; Loewen, G.J.; Buckley, D.B.; Parkinson, A. Lysosomal sequestration (trapping) of lipophilic amine (cationic amphiphilic) drugs in immortalized human hepatocytes (Fa2N-4 cells). Drug Metab. Dispos. Boil. Fate Chem. 2013, 41, 897–905. [Google Scholar] [CrossRef] [PubMed]
- Quintero, J.L.; Arenas, M.I.; Garcia, D.E. The antidepressant imipramine inhibits m current by activating a phosphatidylinositol 4,5-bisphosphate (PIP2)-dependent pathway in rat sympathetic neurones. Br. J. Pharmacol. 2005, 145, 837–843. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, H.; Nishida, A.; Saito, H.; Shimizu, M.; Yamawaki, S. Imipramine stimulates phospholipase C activity in rat brain. Neurochem. Int. 1994, 25, 567–571. [Google Scholar] [CrossRef]
- Metts, J.; Bradley, H.L.; Wang, Z.; Shah, N.P.; Kapur, R.; Arbiser, J.L.; Bunting, K.D. Imipramine blue sensitively and selectively targets FLT3-ITD positive acute myeloid leukemia cells. Sci. Rep. 2017, 7, 4447. [Google Scholar] [CrossRef] [PubMed]
- Stewart, T.A.; Yapa, K.T.; Monteith, G.R. Altered calcium signaling in cancer cells. Biochim. Biophys. Acta 2015, 1848, 2502–2511. [Google Scholar] [CrossRef] [PubMed]
- Skupin, A.; Thurley, K. Calcium signaling: From single channels to pathways. Adv. Exp. Med. Boil. 2012, 740, 531–551. [Google Scholar]
- Gorlach, A.; Bertram, K.; Hudecova, S.; Krizanova, O. Calcium and ROS: A mutual interplay. Redox Boil. 2015, 6, 260–271. [Google Scholar] [CrossRef] [PubMed]
- Gordeeva, A.V.; Zvyagilskaya, R.A.; Labas, Y.A. Cross-talk between reactive oxygen species and calcium in living cells. Biochem. Biokhimiia 2003, 68, 1077–1080. [Google Scholar] [CrossRef]
- Brookes, P.S.; Yoon, Y.; Robotham, J.L.; Anders, M.W.; Sheu, S.S. Calcium, ATP, and ROS: A mitochondrial love-hate triangle. Am. J. Physiol. Cell Physiol. 2004, 287, C817–C833. [Google Scholar] [CrossRef] [PubMed]
- Koch, G.L. The endoplasmic reticulum and calcium storage. Bioessays News Rev. Mol. Cell. Dev. Boil. 1990, 12, 527–531. [Google Scholar] [CrossRef] [PubMed]
- Verfaillie, T.; Garg, A.D.; Agostinis, P. Targeting ER stress induced apoptosis and inflammation in cancer. Cancer Lett. 2013, 332, 249–264. [Google Scholar] [CrossRef] [PubMed]
- Schepers, H.; Wierenga, A.T.; Vellenga, E.; Schuringa, J.J. STAT5-mediated self-renewal of normal hematopoietic and leukemic stem cells. JAKSTAT 2012, 1, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Pesakhov, S.; Nachliely, M.; Barvish, Z.; Aqaqe, N.; Schwartzman, B.; Voronov, E.; Sharoni, Y.; Studzinski, G.P.; Fishman, D.; Danilenko, M. Cancer-selective cytotoxic Ca2+ overload in acute myeloid leukemia cells and attenuation of disease progression in mice by synergistically acting polyphenols curcumin and carnosic acid. Oncotarget 2016, 7, 31847–31861. [Google Scholar] [CrossRef] [PubMed]
- Aboukhatwa, M.A.; Undieh, A.S. Antidepressant stimulation of CDP-diacylglycerol synthesis does not require monoamine reuptake inhibition. BMC Neurosci. 2010, 11, 10. [Google Scholar] [CrossRef] [PubMed]
- Tyeryar, K.R.; Vongtau, H.O.; Undieh, A.S. Diverse antidepressants increase CDP-diacylglycerol production and phosphatidylinositide resynthesis in depression-relevant regions of the rat brain. BMC Neurosci. 2008, 9, 12. [Google Scholar] [CrossRef] [PubMed]
- Schenk, T.; Chen, W.C.; Gollner, S.; Howell, L.; Jin, L.; Hebestreit, K.; Klein, H.U.; Popescu, A.C.; Burnett, A.; Mills, K.; et al. Inhibition of the lsd1 (KDM1A) demethylase reactivates the all-trans-retinoic acid differentiation pathway in acute myeloid leukemia. Nat. Med. 2012, 18, 605–611. [Google Scholar] [CrossRef] [PubMed]
- Amit, B.H.; Gil-Ad, I.; Taler, M.; Bar, M.; Zolokov, A.; Weizman, A. Proapoptotic and chemosensitizing effects of selective serotonin reuptake inhibitors on T cell lymphoma/leukemia (Jurkat) in vitro. Eur. Neuropsychopharmacol. J. Eur. Coll. Neuropsychopharmacol. 2009, 19, 726–734. [Google Scholar] [CrossRef] [PubMed]
- Walker, A.J.; Card, T.; Bates, T.E.; Muir, K. Tricyclic antidepressants and the incidence of certain cancers: A study using the GPRD. Br. J. Cancer 2011, 104, 193–197. [Google Scholar] [CrossRef] [PubMed]
- Si, J.; Collins, S.J. Activated Ca2+/calmodulin-dependent protein kinase IIgamma is a critical regulator of myeloid leukemia cell proliferation. Cancer Res. 2008, 68, 3733–3742. [Google Scholar] [CrossRef] [PubMed]
- Metzelder, S.K.; Michel, C.; von Bonin, M.; Rehberger, M.; Hessmann, E.; Inselmann, S.; Solovey, M.; Wang, Y.; Sohlbach, K.; Brendel, C.; et al. NFATc1 as a therapeutic target in FLT3-ITD-positive AML. Leukemia 2015, 29, 1470–1477. [Google Scholar] [CrossRef] [PubMed]
- Schardt, J.A.; Weber, D.; Eyholzer, M.; Mueller, B.U.; Pabst, T. Activation of the unfolded protein response is associated with favorable prognosis in acute myeloid leukemia. Clin. Cancer Res. 2009, 15, 3834–3841. [Google Scholar] [CrossRef] [PubMed]
- Roti, G.; Carlton, A.; Ross, K.N.; Markstein, M.; Pajcini, K.; Su, A.H.; Perrimon, N.; Pear, W.S.; Kung, A.L.; Blacklow, S.C.; et al. Complementary genomic screens identify SERCA as a therapeutic target in NOTCH1 mutated cancer. Cancer Cell 2013, 23, 390–405. [Google Scholar] [CrossRef] [PubMed]
- Pereira, D.M.; Valentao, P.; Correia-da-Silva, G.; Teixeira, N.; Andrade, P.B. Translating endoplasmic reticulum biology into the clinic: A role for ER-targeted natural products? Nat. Prod. Rep. 2015, 32, 705–722. [Google Scholar] [CrossRef] [PubMed]
- Colvin, C.L.; Tankanow, R.M. Pimozide: Use in tourette’s syndrome. Drug Intell. Clin. Pharm. 1985, 19, 421–424. [Google Scholar] [PubMed]
- Santi, C.M.; Cayabyab, F.S.; Sutton, K.G.; McRory, J.E.; Mezeyova, J.; Hamming, K.S.; Parker, D.; Stea, A.; Snutch, T.P. Differential inhibition of T-type calcium channels by neuroleptics. J. Neurosci. 2002, 22, 396–403. [Google Scholar] [CrossRef] [PubMed]
- Panner, A.; Cribbs, L.L.; Zainelli, G.M.; Origitano, T.C.; Singh, S.; Wurster, R.D. Variation of T-type calcium channel protein expression affects cell division of cultured tumor cells. Cell Calcium 2005, 37, 105–119. [Google Scholar] [CrossRef] [PubMed]
- Nelson, E.A.; Walker, S.R.; Weisberg, E.; Bar-Natan, M.; Barrett, R.; Gashin, L.B.; Terrell, S.; Klitgaard, J.L.; Santo, L.; Addorio, M.R.; et al. The STAT5 inhibitor pimozide decreases survival of chronic myelogenous leukemia cells resistant to kinase inhibitors. Blood 2011, 117, 3421–3429. [Google Scholar] [CrossRef] [PubMed]
- Nelson, E.A.; Walker, S.R.; Xiang, M.; Weisberg, E.; Bar-Natan, M.; Barrett, R.; Liu, S.; Kharbanda, S.; Christie, A.L.; Nicolais, M.; et al. The STAT5 inhibitor pimozide displays efficacy in models of acute myelogenous leukemia driven by FLT3 mutations. Genes Cancer 2012, 3, 503–511. [Google Scholar] [CrossRef] [PubMed]
- Cumaraswamy, A.A.; Lewis, A.M.; Geletu, M.; Todic, A.; Diaz, D.B.; Cheng, X.R.; Brown, C.E.; Laister, R.C.; Muench, D.; Kerman, K.; et al. Nanomolar-potency small molecule inhibitor of STAT5 protein. ACS Med. Chem. Lett. 2014, 5, 1202–1206. [Google Scholar] [CrossRef] [PubMed]
- Elumalai, N.; Berg, A.; Rubner, S.; Blechschmidt, L.; Song, C.; Natarajan, K.; Matysik, J.; Berg, T. Rational development of Stafib-2: A selective, nanomolar inhibitor of the transcription factor STAT5b. Sci. Rep. 2017, 7, 819. [Google Scholar] [CrossRef] [PubMed]
- Wingelhofer, B.; Maurer, B.; Heyes, E.C.; Cumaraswamy, A.A.; Berger-Becvar, A.; de Araujo, E.D.; Orlova, A.; Freund, P.; Ruge, F.; Park, J.; et al. Pharmacologic inhibition of STAT5 in acute myeloid leukemia. Leukemia 2018, 32, 1135–1146. [Google Scholar] [CrossRef] [PubMed]




© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mi, T.; Wang, Z.; Bunting, K.D. The Cooperative Relationship between STAT5 and Reactive Oxygen Species in Leukemia: Mechanism and Therapeutic Potential. Cancers 2018, 10, 359. https://doi.org/10.3390/cancers10100359
Mi T, Wang Z, Bunting KD. The Cooperative Relationship between STAT5 and Reactive Oxygen Species in Leukemia: Mechanism and Therapeutic Potential. Cancers. 2018; 10(10):359. https://doi.org/10.3390/cancers10100359
Chicago/Turabian StyleMi, Tian, Zhengqi Wang, and Kevin D. Bunting. 2018. "The Cooperative Relationship between STAT5 and Reactive Oxygen Species in Leukemia: Mechanism and Therapeutic Potential" Cancers 10, no. 10: 359. https://doi.org/10.3390/cancers10100359
APA StyleMi, T., Wang, Z., & Bunting, K. D. (2018). The Cooperative Relationship between STAT5 and Reactive Oxygen Species in Leukemia: Mechanism and Therapeutic Potential. Cancers, 10(10), 359. https://doi.org/10.3390/cancers10100359