1. Introduction
Triple-negative breast cancer (TNBC) accounts for 15% of breast cancer cases and it is associated with an advanced stage at the time of diagnosis and early occurrence of distant metastases. It also has the poorest survival outcome of all breast cancer subtypes [
1]. The lack of expression of the HER2 (receptor tyrosine-protein kinase erbB-2), the estrogen receptor (ER), and the progesterone receptor (PR) characterizes the TNBC cells [
1,
2]. Due to the lack of these proteins and the possibility of using targeted therapy against these receptors, chemotherapy is currently the only systemic treatment method available for TNBC patients [
1,
3]. Therefore, TNBC is the most challenging breast cancer subtype in terms of treatment. It is believed that receptor tyrosine kinases (RTKs) particularly the hepatocyte growth factor receptor (MET) and epidermal growth factor receptor (EGFR) are promising therapeutic targets due to their elevated expression in multiple TNBC subtypes [
2].
It was shown that EGFR is overexpressed in approximately 50% of TNBC cases and its high expression correlates with a poor prognosis [
4,
5,
6,
7]. EGFR is a tyrosine kinase receptor involved in cell growth and survival [
8,
9]. It belongs to the membrane anchored receptor tyrosine kinase ERBB/HER family consisting of EGFR, HER2, HER3, and HER4 [
10]. Ligand activation of EGFR results in homo- and hetero-dimerization with other family members. This dimerization enables EGFR auto-phosphorylation, which results in the recruitment of signaling proteins to the receptor [
8,
9]. Aberrant activation of EGFR in cancer cells as a result of gene copy number amplification, protein overproduction, or point mutations leads to unregulated proliferation, malignant transformation, invasion, metastasis, and resistance to apoptosis [
5,
10]. EGFR overexpression correlates with a loss of estrogen responsiveness and a poor prognosis [
11]. However, therapies directed only against EGFR based on inhibitors or monoclonal antibodies were not effective in TNBC [
3].
Receptor tyrosine kinase MET of which the ligand is a hepatocyte growth factor is also highly expressed in TNBC [
12]. It is overexpressed in 20% to 30% of breast cancer cases and appears to be associated with a worse prognosis [
13]. MET can be activated through ligand-dependent and ligand-independent mechanisms [
9]. Ligand-dependent activation of this receptor occurs in the mammary gland where stromal cells such as fibroblasts produce HGF [
14]. Ligand-independent activation of MET occurs through MET mutation, constitutive dimerization of the receptor as a result of overexpression, crosstalk with other membrane receptors including the EGFR, or the loss of negative regulators [
9,
15,
16,
17]. Excessive MET activation promotes the growth, survival, and migration of cancer cells [
17]. MET signaling, which is similar to EGFR, activates a number of downstream effectors such as AKT, extracellular signal-related kinase (ERK), phosphoinositide 3-kinase, RAS, and SRC [
16,
18]. MET expression correlates positively with EGFR expression in basal-type breast cancers [
9]. Of particular interest is the fact that MET gene amplification and overexpression leads to resistance to anti-EGFR therapies in non-small cell lung cancer [
19] and in brain tumors through receptor co-activation with EGFR [
20].
In TNBC, even in the presence of EGFR inhibitors, cancer cells exhibit perpetual phosphorylation of this receptor, which correlates with the resistance to tyrosine kinase inhibitors (TKI) [
2,
21]. This resistance may be related to MET–EGFR crosstalk, which was reported to be involved in therapeutic resistance to EGFR inhibitors in colon and lung cancers [
19,
22]. EGFR and MET interact both physically and functionally in a way independent of both receptors’ kinase activities and this interplay can promote resistance to EGFR TKI via the constitutive phosphorylation of MET [
9]. In this scenario, we report on the influence of a mix of selected EGFR and MET inhibitors on TNBC cell viability, cytoskeleton organization, and invasion in vitro. The aim of our study is to support new potential therapeutic strategies for triple negative breast cancer treatment.
3. Discussion
To better characterize and understand different kinds of breast cancers, they were classified based on the presence of molecular markers. Luminal A and B subtypes exhibit expression of estrogen and/or progesterone receptors while the HER2 subtype demonstrates enrichment in HER2 expression with a lack of response to hormones. Triple-negative breast cancer is deficient in both hormone receptors and HER2 expression and shows high similarity to basal-like breast cancer (BLBC). Nevertheless, around 5% to 15% cases of BLBC are not triple-receptor-negative [
1,
12]. Despite constant furthering of our knowledge concerning the molecular background of this tumor, existing therapeutic strategies still fail to fully elicit a successful outcome. Currently used targeted therapies against breast cancer include the use of tamoxifen, fulvestrant, and aromatase inhibitors for ER-positive breast cancers and Herceptin and HER2 inhibitors for the HER2-positive breast cancer treatment [
29]. In the case of TNBC, patients show initial responsiveness to conventional chemotherapy. However, they frequently experience a relapse that eventually leads to worse survival outcome compared to those with other breast cancer subtypes [
12]. The lack of effective treatment following relapses and high metastasis rates also contribute to the bad prognosis of patients with TNBC [
30].
A suggested new potential therapy directed against TNBC is the combined targeting of the growth factors receptors MET and EGFR. (1) Both are overexpressed simultaneously in this subtype [
17]. (2) They activate numerous, partly overlapping, signaling pathways that promote cell growth, invasion, and cell survival with the predominant ones being Ras/ERK and PI3K/AKT pathways. (3) In a monotherapy context, they are frequently associated with therapeutic resistance to TKIs [
2]. Moreover, in cancer development, their functional crosstalk has been reported with EGFR activating MET or, conversely, with MET expression regulating EGFR tyrosine phosphorylation and cell growth, e.g., in the presence of gefitinib (EGFR inhibitor) in SUM229 breast cancer cells [
9]. Mueller et al. suggested that MET may mediate EGFR phosphorylation through co-association, which will lead to conformational changes in both receptors and regulation of the kinase activity of MET. This association of EGFR and MET and their mutual regulation suggest that inhibiting the kinase activity of these molecules individually may be insufficient. Thus, shifting from monotherapy to combination therapy, targeting both primary and rescue pathways, could be a promising approach. In non-small cell lung cancer cells, MET overexpression increased EGFR downstream signaling and the combined use of dual EGFR/HER2 and MET tyrosine kinase inhibitors resulted in a maximal growth inhibition [
31]. Importantly, in view of therapy development, the MET and EGFR tyrosine kinases are actionable therapeutic targets due to their high expression in TNBC and several selective kinase inhibitors of which both are FDA approved for other cancers or in clinical trials [
2].
In this study, we showed that two selected MET and EGFR inhibitors, foretinib and lapatinib, act effectively in the two TNBC cell lines. In a context of permanent ligand-based activation (as is occurring in the tumor), the inhibitors join forces to reduce several malignant properties of the TNBC cells including survival, AKT activation, cell growth, invadopodia formation, matrix proteolysis, and, importantly, migration and invasion. This is, to our knowledge, the first report showing that a combination of EGFR and MET inhibitors reduce not only TNBC cell viability but also cell invasion, which is connected to the devastating ability of these cells to form metastasis. The simultaneous usage of inhibitors of these two receptors led to not only better treatment efficiency but also and equally importantly prevented crosstalk between examined receptors.
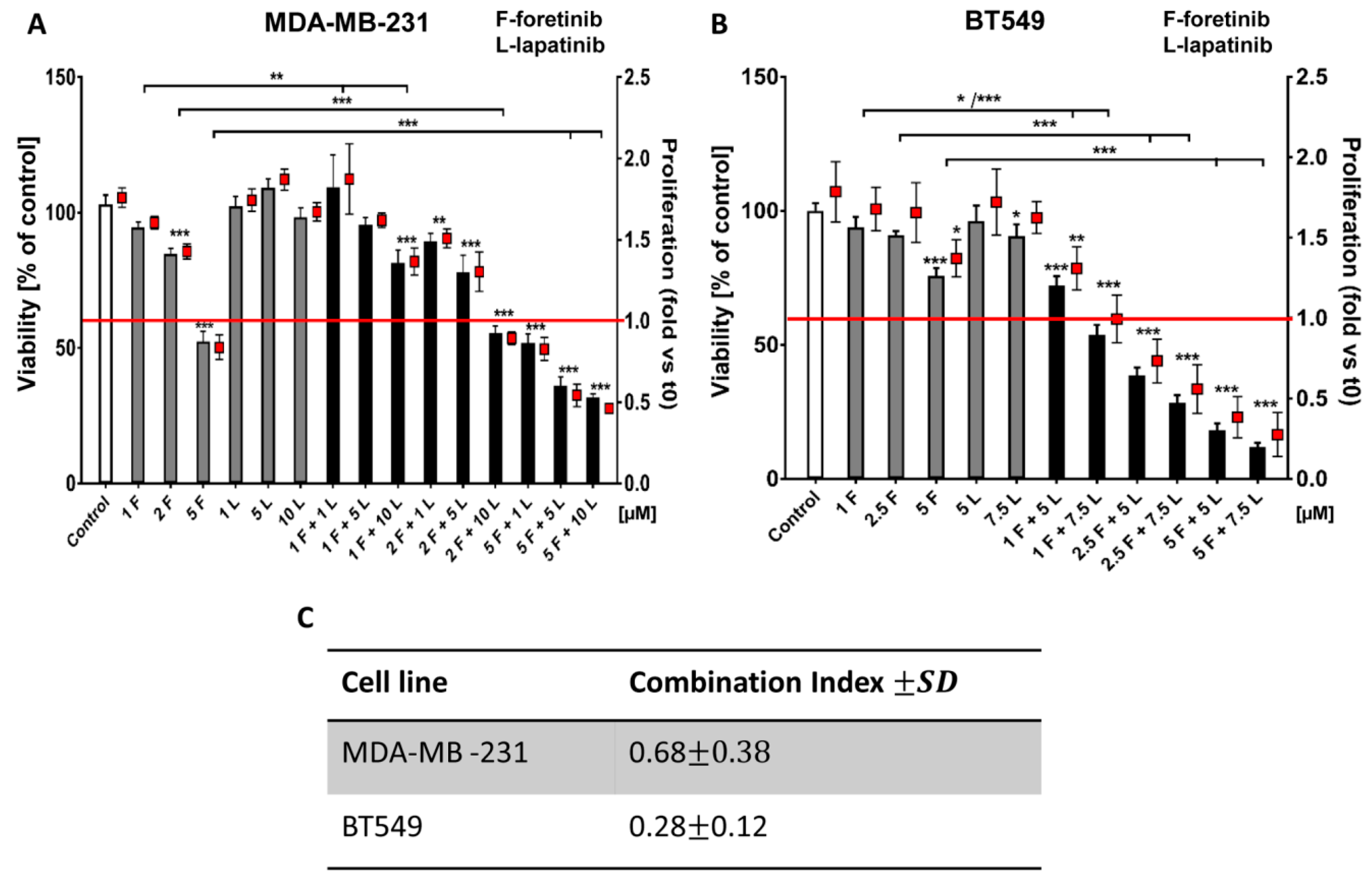
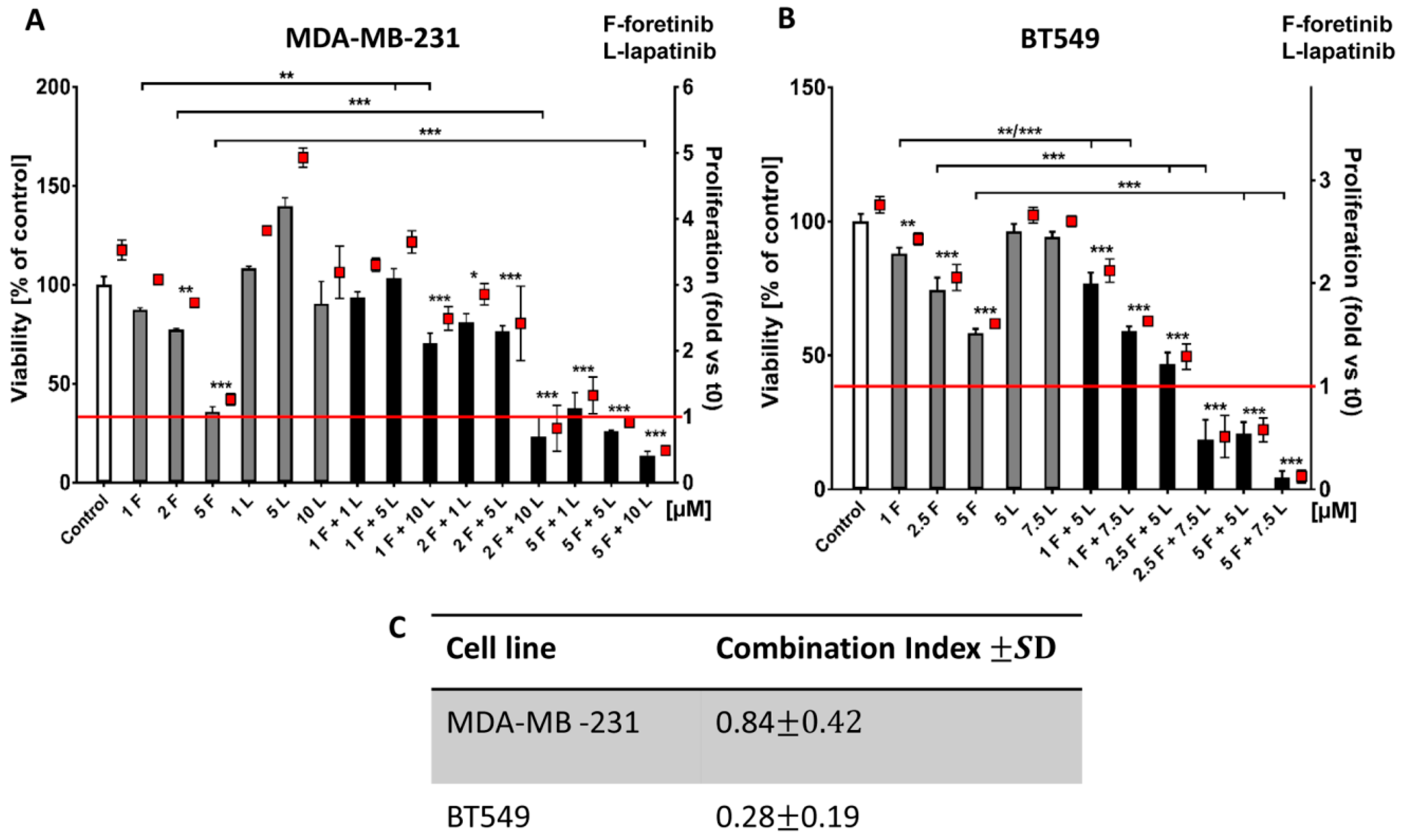
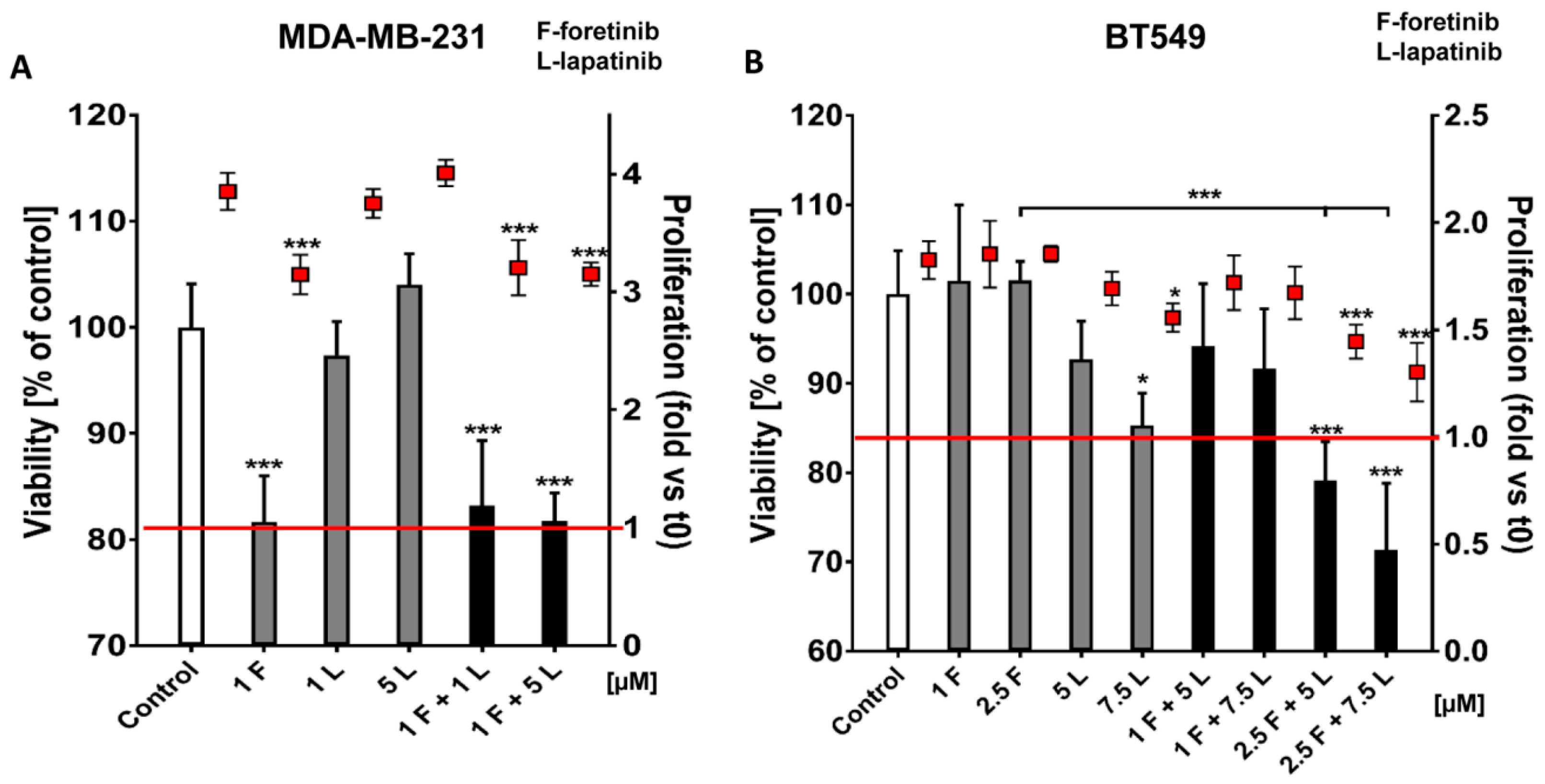
Going into detail, we noticed that the combination of drugs inhibited cell viability in much stronger ways than the inhibitors when used independently. The average combination index was 0.68 for MDA-MB-231 cells and 0.28 for BT549 cells, which demonstrates the synergistic effect of foretinib and lapatinib (
Figure 1). These results suggested that used inhibitors work collectively to decrease cell viability and proliferation in TNBC cells. Similar results were obtained by Kim et al. [
17], Yi et al. [
5], and Sohn et al. [
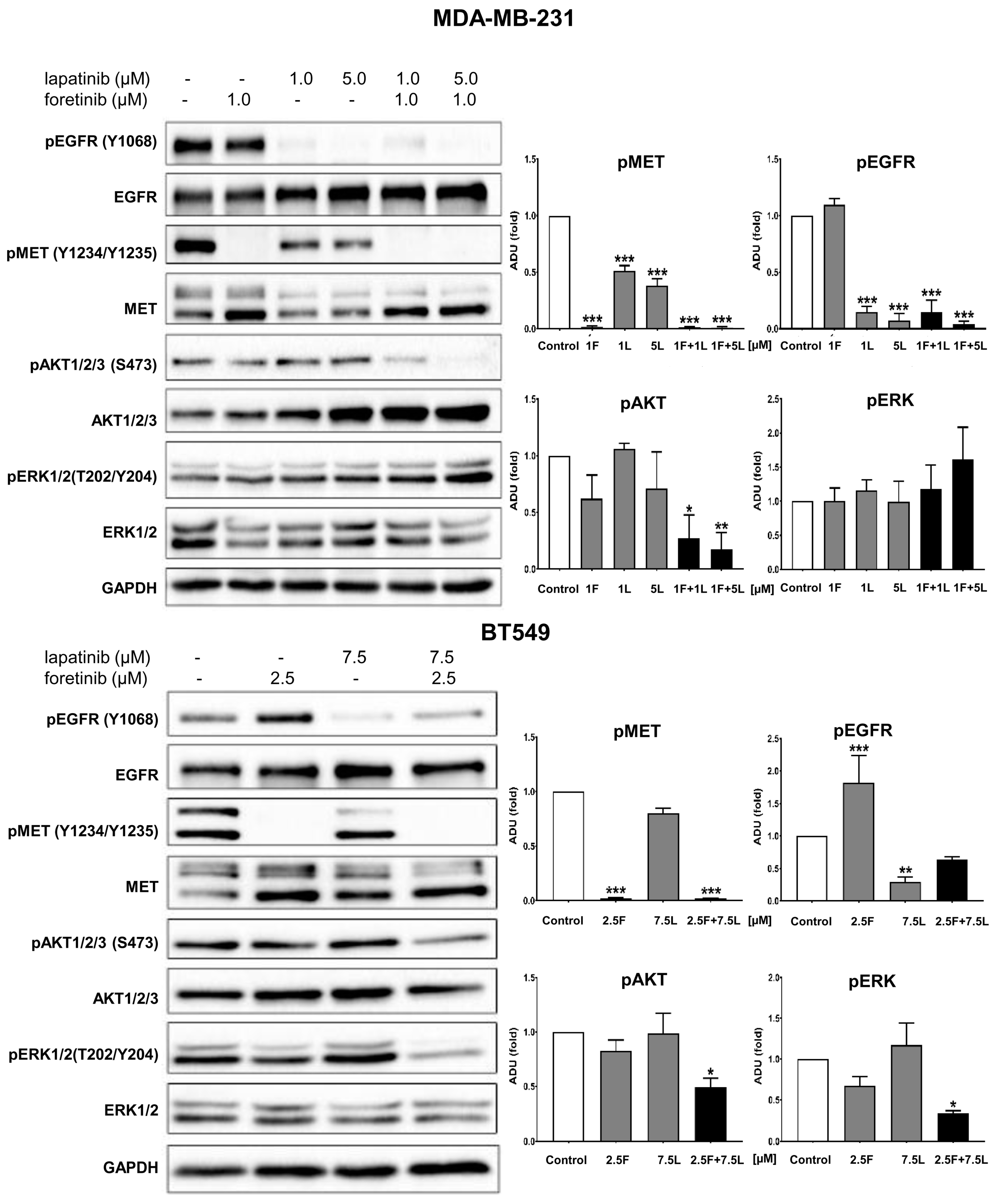
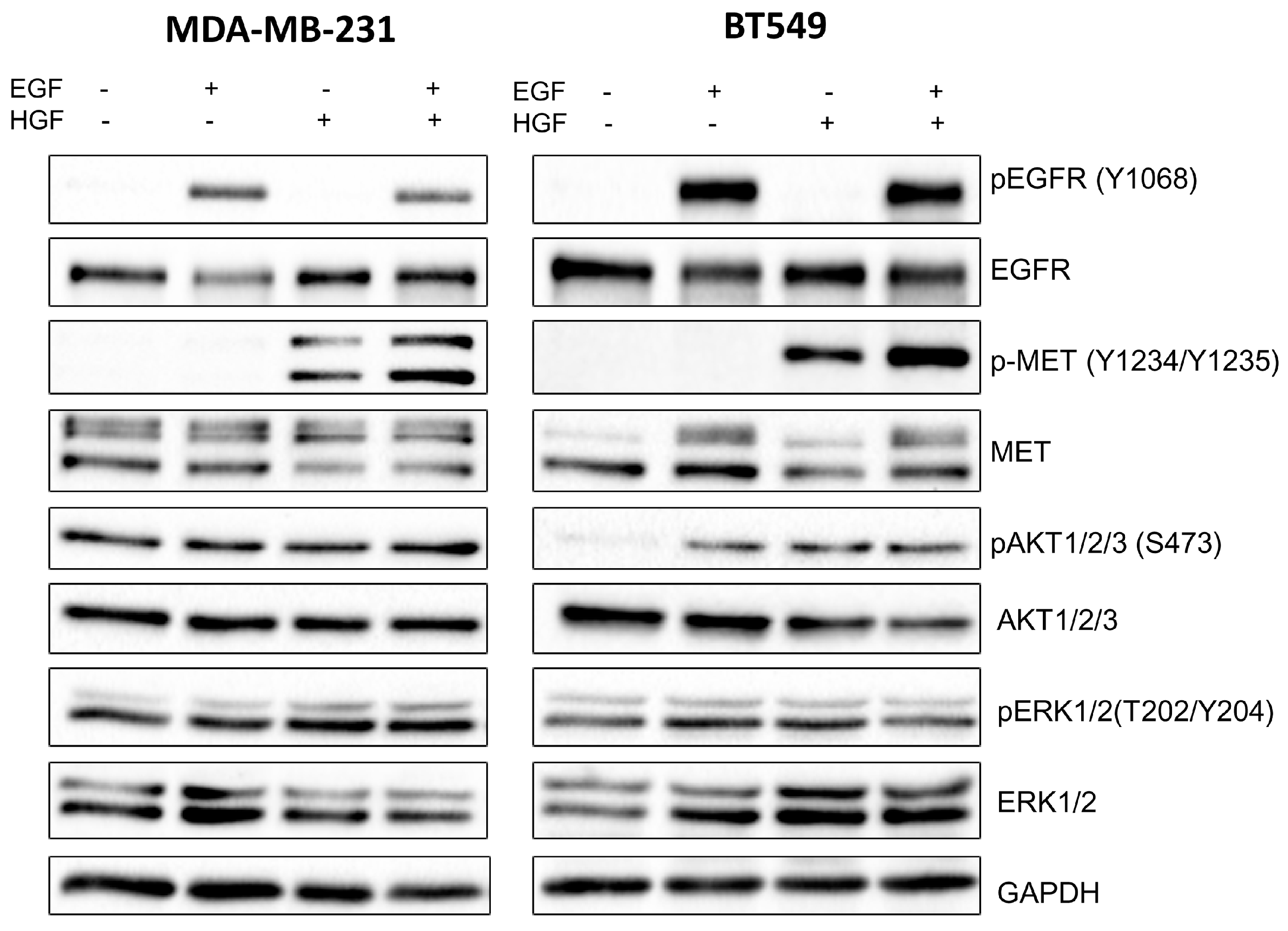
3]. Our study also indicated that the pAKT level was decreased in both cell lines only when drugs were used in combination (
Figure 2), which suggests that only a combination of EGFR and MET inhibitors is able to inhibit the downstream signaling pathways of these receptors. This result is in line with several recent studies, which have revealed that co-expression of RTKs can promote signaling crosstalk and maintain active Ras/ERK and PI3K/AKT signaling in the presence of kinase inhibitors to ensure cell survival. It was demonstrated that RTKs are co-expressed in 41 cancer cell lines [
30] while ligands of RTKs may be able to overcome the drug sensitivity in “kinase-addicted” malignant cells. For example, the expression of HGF reduced sensitivity of several HER2-dependent breast cancer cell lines to lapatinib [
2]. It was also shown that failure to induce inhibition of AKT can be a major cause of resistance to EGFR inhibitors [
3]. In our research, the reduction in pEGFR level was connected to decreased pERK1/2 level only in one cell line: BT549. The fact that it is not decreased in the second cell line may be due to several reasons. First, in breast cancer cells, growth factors other than EGF and HGF may trigger the activation of the MAPK signaling pathway, which includes ERK1/2. Among these growth factors are insulin-like growth factor [
2] or nerve growth factor [
3]. Thus, even if EGFR is inhibited, other receptors of growth factors can lead to this pathway activation. Second, there may be mutations present in the genes encoding proteins present upstream of ERK1/2 in the MAPK signaling pathway, i.e., Raf or Ras. This may make these proteins constitutively active and, thus, lead to phosphorylation of ERK1/2 in the absence of an active EGFR receptor [
4]. Third, it was shown that TNBC cell lines are characterized by a profile of gene expression similar to those of k-Ras or EGFR-mutant cancers and exhibit upregulation of pMEK and pERK [
5,
6]. All these reasons can cause the pERK level to remain unchanged despite the decreased level of pEGFR.
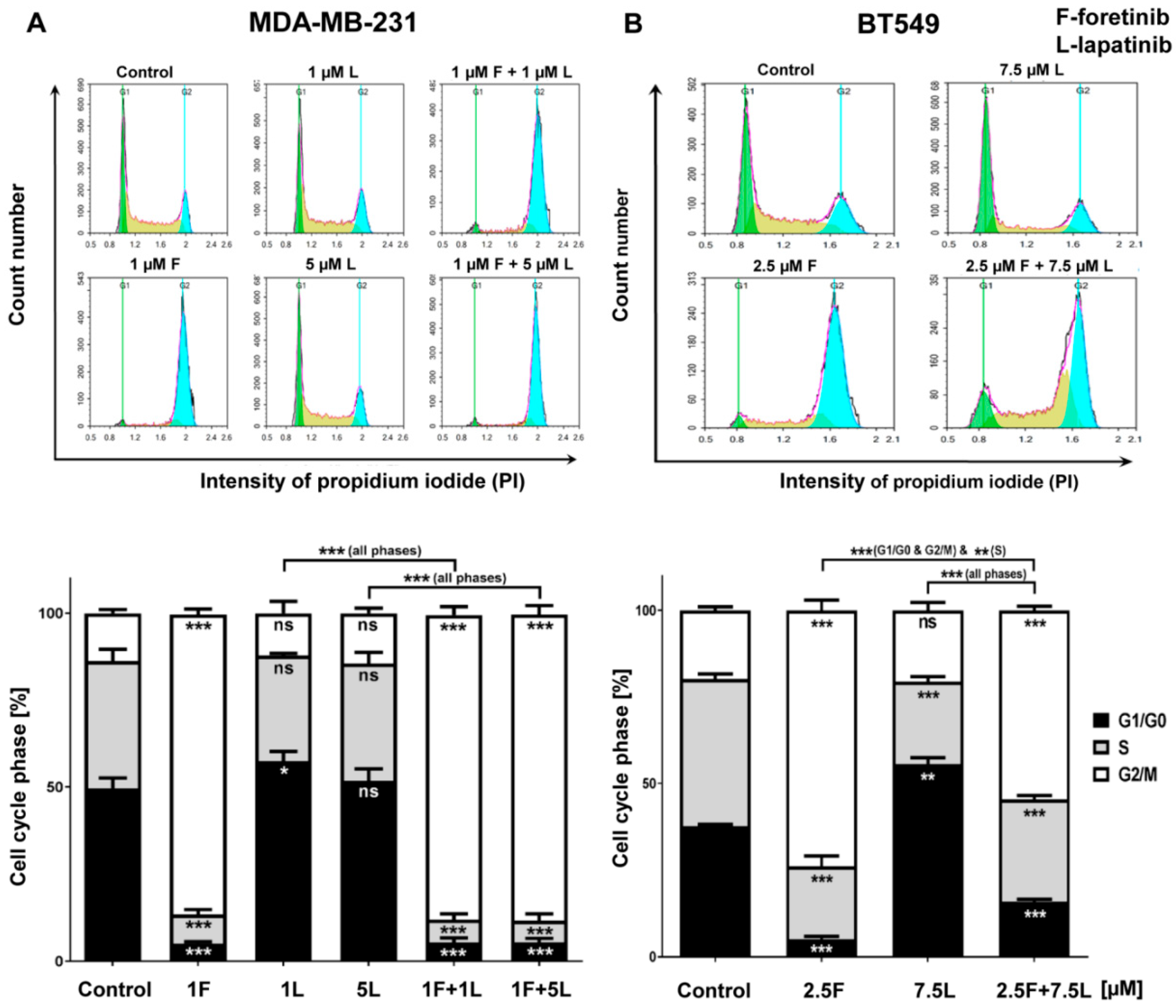
We also noticed that foretinib and its combination with lapatinib caused an accumulation of cells in the G2/M phase (G2/M arrest) (
Figure 3). Additionally, we observed the appearance of many tetraploid cells after inhibitor treatment, which suggests that foretinib is able to impair the regulation of cell division. A similar effect was observed by Dufies et al. who showed that foretinib-treated chronic myelogenous leukemia cells exhibited increased size, spindle assembly checkpoint anomalies, and enhanced ploidy that collectively resulted in a mitotic catastrophe [
32]. After foretinib and foretinib/lapatinib treatment, we also observed an increased cell size (
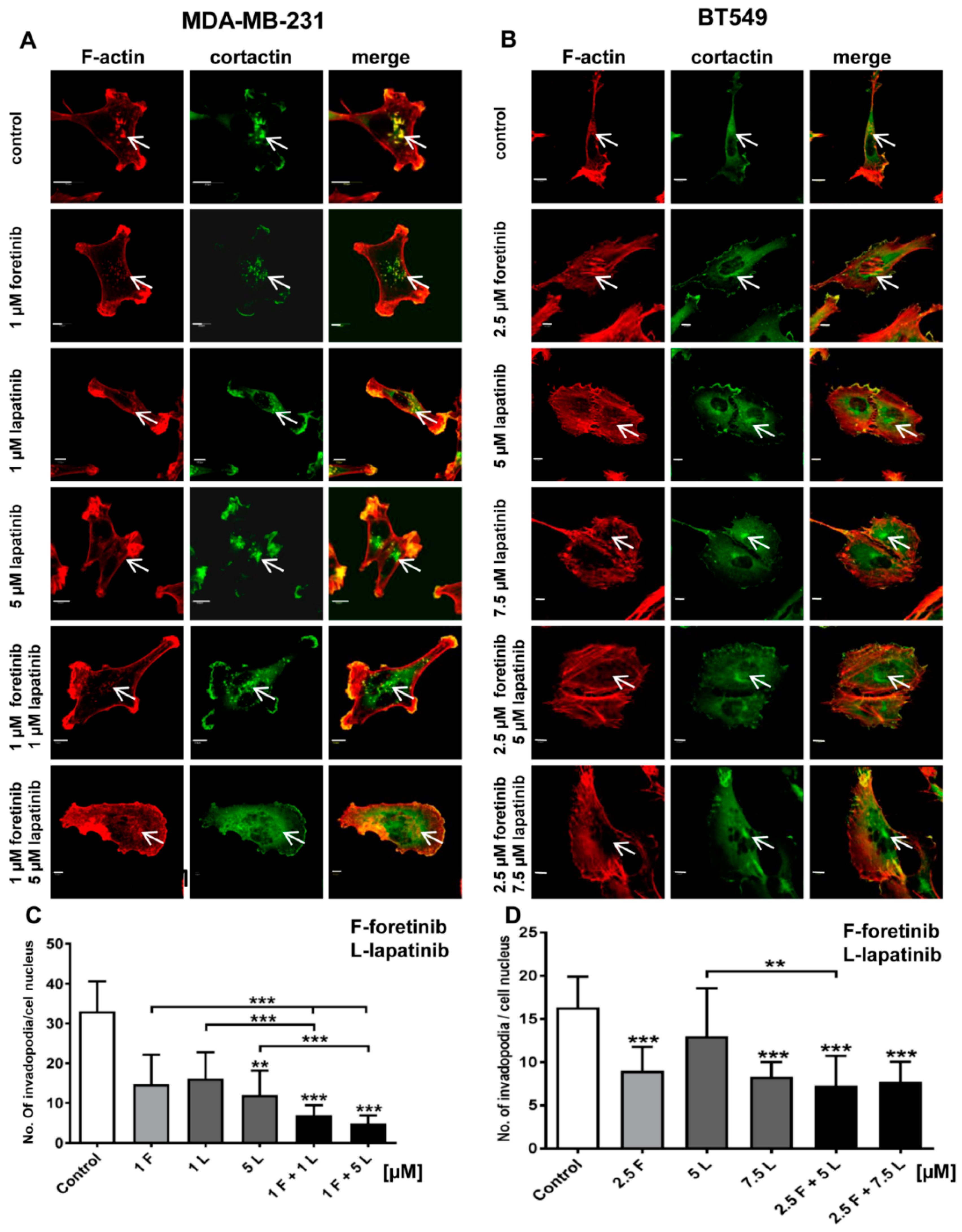
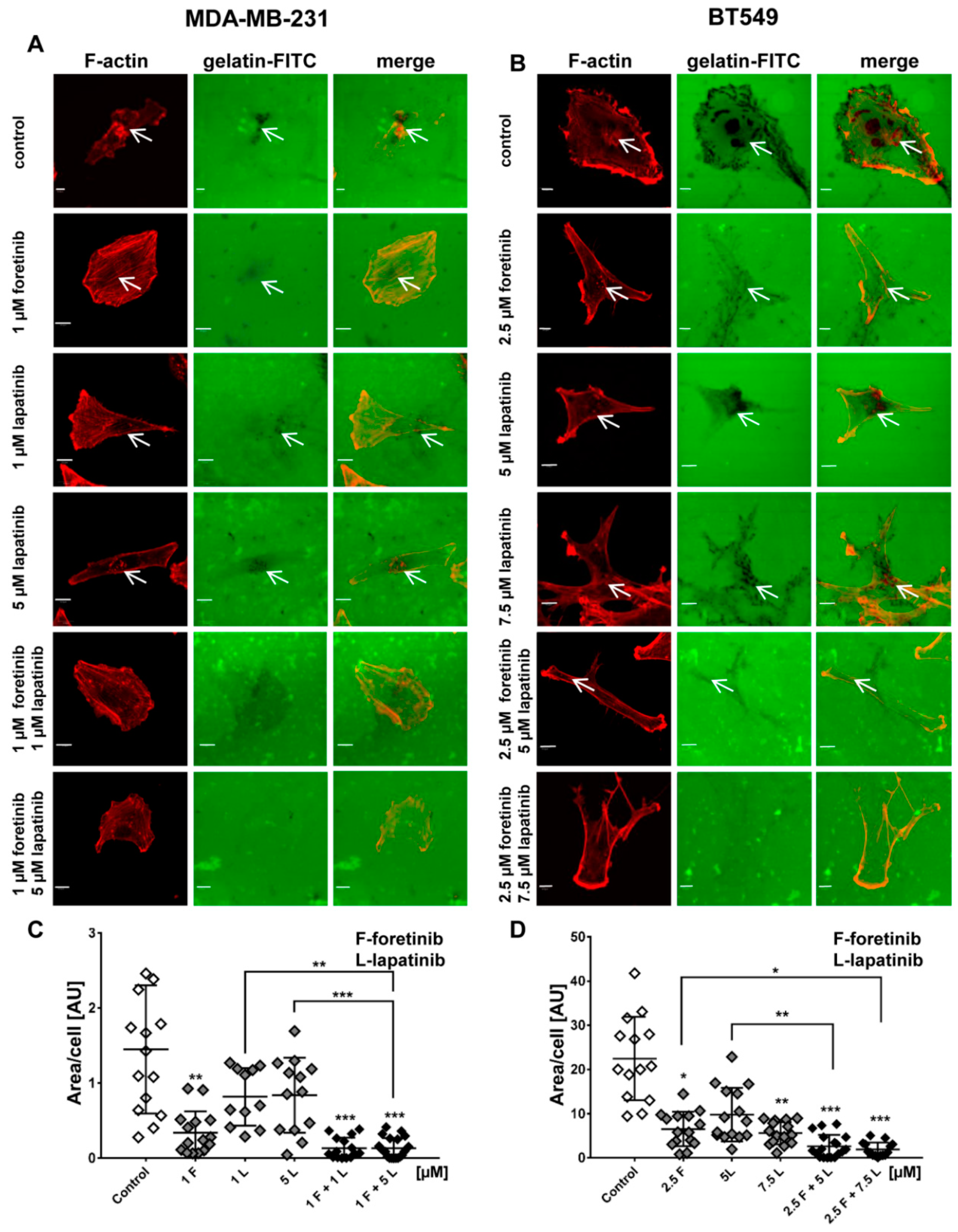
Figure 4) and appearance of cells with an elevated number of nuclei, which confirms data obtained by flow cytometry. Both cell lines also formed a lower number of invadopodia after incubation with a combination of inhibitors (
Figure 4). Additionally, we noticed that treatment with foretinib and lapatinib simultaneously reduced the ability of both types of cells to digest fluorescently-labeled gelatin (
Figure 5). These processes were accompanied by a reduced level of pAKT. Our results are in line with data obtained by Yamaguchi et al. who indicated that active AKT is necessary for efficient invadopodia assembly. Overexpression of this kinase led to increased invadopodia formation and gelatin degradation [
7]. Other researchers have also shown that elements of the PIK3/AKT signaling pathway, e.g., PDK1, are involved in active invadopodia assembly [
8,
9]. Under the treatment of the pair of inhibitors, migration and invasion abilities of examined cells were also reduced (
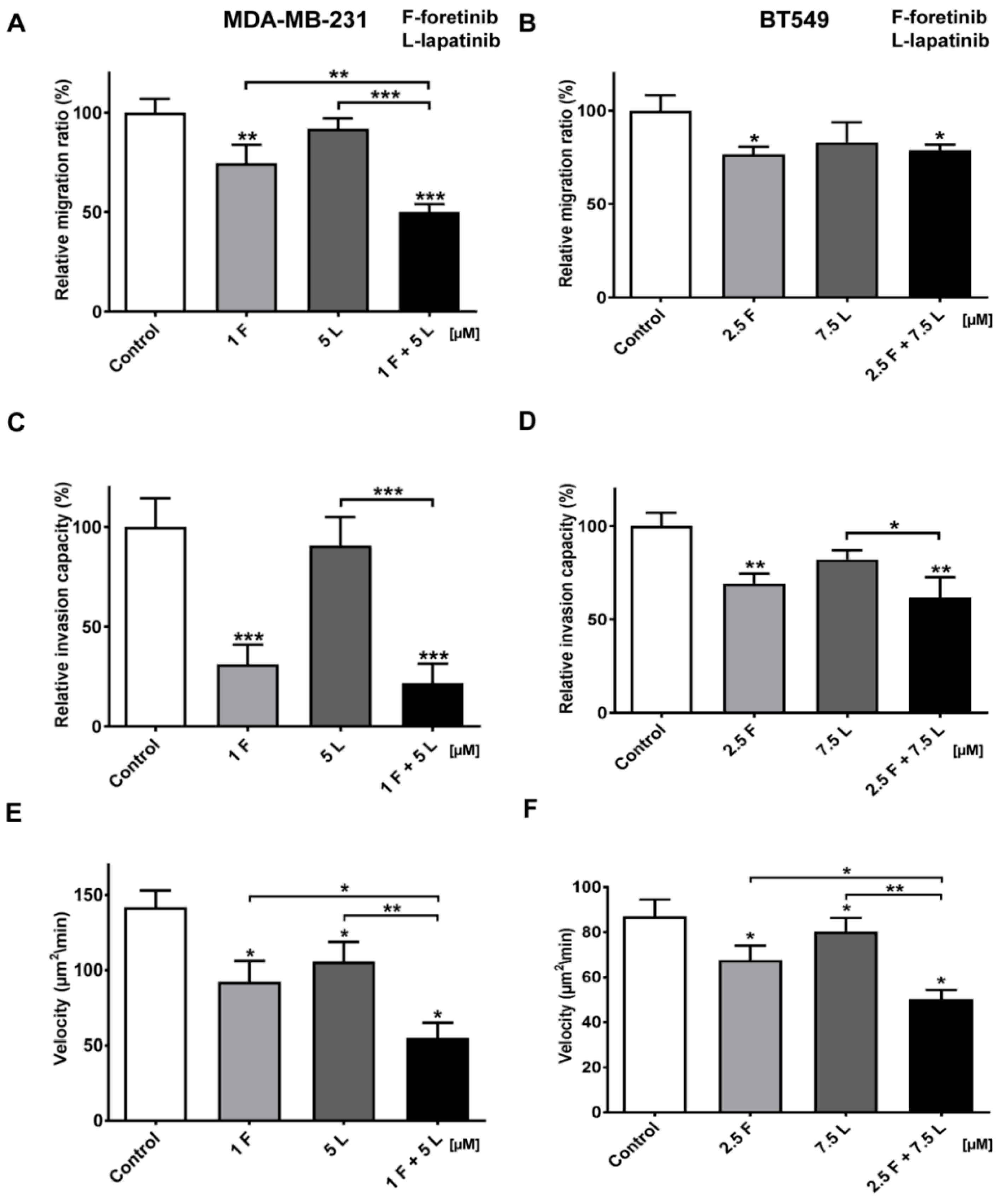
Figure 6). The obtained data indicates that the combination of EGFR and MET inhibitors is able to reduce TNBC invasion more effectively than monotherapy.
In summary, the tested drug combination could be applied to overcome intrinsic EGFR resistance in TNBC. Furthermore, our studies support the findings of targeting multiple RTKs to prevent signaling crosstalk and resistance to kinase inhibitors. These results help to understand the RTK signaling in TNBC and indicate potential therapeutic strategies for patients with this type of cancer. These findings provide the rationale for testing foretinib in combination with lapatinib in clinical trials in TNBC. Potential biomarkers to identify patients that might benefit from this approach with high levels of EGFR and/or MET expression are still needed.
4. Materials and Methods
4.1. Chemicals
Rabbit polyclonal anti-cortactin antibodies were obtained from Santa Cruz Biotechnology (Dallas, TX, USA). Alexa Fluor 568-conjugated phalloidin, secondary anti-rabbit antibodies conjugated with Alexa Fluor 488, gelatin conjugated with FITC, fetal bovine serum (FBS), trypsin, glutamine, penicillin/streptomycin, and DMEM were purchased from Invitrogen (Carlsbad, CA, USA). Dako fluorescent mounting medium was obtained from Dako (Agilent, Santa Clara, CA, USA). The epidermal growth factor (EGF) and rat-tail collagen (acid-extracted, non-pepsin treated) type I were obtained from BD Biosciences (Agilent, Santa Clara, CA, USA). Cell Proliferation Kit II was purchased from Roche (Basel, Switzerland). Antibodies directed against EGFR (SC-03), MET (SC-10), AKT1/2/3 (SC-8312), and phospho-AKT1/2/3 (S473; SC-135651) were purchased from Santa Cruz Biotechnologies. Anti-phospho-EGFR (Y1069; catalogue no. 3777), anti-phospho-MET (Y1234/Y1235; no. 3077), anti-ERK1/2 (no. 9102), and anti-phospho-ERK1/2 (T202/Y204; no. 9101) antibodies were from Cell Signaling Technologies (Danvers, MA, USA). HGF was obtained from Sigma Aldrich (Saint Louis, MO, USA), foretinib from Santa Cruz Biotechnologies, and lapatinib from Selleckchem (Houston, TX, USA). All other chemicals were classified as analytical grade reagents.
4.2. Cell Culture
The human triple negative breast cancer cell lines, MDA-MB-231, and BT549 (both confirmed by LGC Cell Line Authentication Service) were grown in DMEM medium containing 10% FBS, 2 mM glutamine, and antibiotics (100 U/mL penicillin, 100 μg/mL streptomycin). The cells were cultured in 25 cm2 tissue culture flasks (Sarstedt, Nümbrecht, Germany)) at 37 °C in 5% CO2/95% humidified air and passaged twice a week using a 0.25% trypsin/0.05% EDTA solution.
4.3. Treatment of Cells with Inhibitors
The cells were incubated with the EGFR inhibitor lapatinib and the MET inhibitor foretinib separately or with a combination of both for 12, 24 or 48 h. The inhibitor concentrations used in the experiments were selected based on XTT experiments and matched to the sensitivity of a given cell line. For each assay, cells were stimulated with 5 nM EGF and 30 ng/mL HGF to imitate conditions present in the breast cancer microenvironment. Cells incubated only with growth factors and 0.1% DMSO (inhibitors solvent) were used as a control.
4.4. Collagen Matrix Preparation
The solution for collagen matrix preparation consisted of type I collagen (2 mg/mL, final concentration), MEM, and 8.3 mM NaHCO3 in Hank’s Balanced Salt Solution and its pH was adjusted to a pH of 7.4–8 with 1 M NaOH.
4.5. Western Blot Analysis
Cells were lysed in urea-containing buffer and were supplemented with protease and phosphatase inhibitor cocktails (Sigma Aldrich). The protein concentration of the cell lysates was determined by a standard Bradford procedure [
33]. Additionally, 24 h after seeding in 60 mm culture dishes, cells (1 × 10
6) were treated for 4 h with 1 to 2.5 μM of foretinib, 1 to 7.5 μM of lapatinib, or a combination of the two in medium containing 30 ng/mL HGF and 5 nM EGF. Samples were analyzed by SDS-PAGE, according to the procedure described by Laemmli [
34], and transferred to nitrocellulose sheets, according to Towbin et al [
35]. Anti-MET, anti-p-MET, anti-EGFR, anti-pEGFR, anti-AKT1/2/3, anti-pAKT1/2/3, anti-ERK 1/2, and anti-pERK 1/2 were used as primary antibodies. Goat anti-mouse or goat anti-rabbit antibody conjugated to horseradish peroxidase (HRP) was applied according to the manufacturer’s protocols. Immunoblots were developed using the Luminol Reagent (Bio-rad, Hercules, CA, USA) and scanned by using the ChemiDoc gel scanner (Bio-Rad). Quantitative analysis was performed by using Image Lab software (Bio-Rad). Bands for phosphorylated proteins were normalized to total protein content (Ponceau S staining) and compared to control cells. All experiments were done in triplicate.
4.6. Cell Cytotoxicity and Proliferation Assay in 2D and 3D
The Cell Proliferation Kit II (XTT) (Roche, Hercules, CA, USA) was used according to the manufacturer’s protocol. The XTT labeling mixture was added in parallel samples at time 0 (t0) and after 24 or 48 h of cell growth in the absence or presence of inhibitors (foretinib, lapatinib, or their combination) at the indicated concentrations. For both 2D and 3D matrix-embedded cultures, absorbance was measured three hours after XTT addition. All conditions were prepared in four replicates. The exact protocol of seeding cells in 2D and 3D conditions, the execution of the test, and the calculation of cytotoxicity and the proliferation rate was described earlier by Huyck et al. (2012).
4.7. Calculation of the Combination Index
Combination analysis of treatment with foretinib and lapatinib was performed using the Calcusyn software program (Biosoft, Cambridge, UK), which calculates a combination index, according to Chou and Talalay-derived equations [
36]. CI <1, =1, and >1 represent synergistic, additive, and antagonist effects, respectively.
4.8. Cell Cycle Analysis
BT549 (3.5 × 105) and MDA-MB-231 (2.5 × 105) cells were plated on six-well plates. After 24 h, the medium was replaced with a medium containing growth factors (5 nM EGF and 30 ng/mL HGF) in combination with either DMSO (0.1% final concentration, mock treatment), foretinib (1–2.5 μM), lapatinib (1–7.5 μM), or a combination of the two inhibitors. Cells were incubated in the presence of these compounds for 24 h. Afterwards, the medium was discarded, cells were washed with PBS without Ca2+/Mg2+, trypsinized, centrifuged (100× g, 5 min), and fixed with ice-cold 70% ethanol for at least 24 h in −20 °C. Then, cells were washed three times with PBS (800× g, 5 min), incubated with RNase A (final concentration 10 μg/mL, 45 min, room temperature), stained with propidium iodide (final concentration 50 μg/mL, 30 min, 4 °C), and subsequently analyzed with a NovoCyte flow cytometer and ACEA NovoExpress software (ver. 1.2.4; ACEA Biosciences, San Diego, CA, USA).
4.9. Immunofluorescence
The subcellular distribution of actin filaments and cortactin was examined by immunofluorescence. Cells (3 × 104 per coverslip) were seeded on collagen type I-coated coverslips in 24-well plates and grown for 24 h. To obtain a coating of collagen, prepared as described in the collagen matrix preparation section, the coverslips were incubated for 30 min at 37 °C and 5% CO2. Next, the cells were fixed with 4% formaldehyde for 20 min at room temperature and permeabilized with 0.1% Triton X-100 in PBS for 5 min. Coverslips were blocked for 30 min with 1% bovine serum albumin in PBS. Anti-cortactin antibodies, followed by Alexa Fluor 488-conjugated anti-rabbit secondary antibodies, were applied to visualize this protein. Actin filaments were stained with Alexa Fluor 568-labeled phalloidin. After incubation and washing steps, coverslips were mounted with Dako fluorescent mounting medium. For each condition, about 25 cells were imaged (Olympus FV500 confocal laser scanning microscope, Tokio, Japan) in three independent experiments and representative cells are shown. Quantitative analysis of the number of invadopodia per nuclei was performed using ImageJ software. Only invadopodia positive for F-actin and cortactin were scored and 30 cells were analyzed per condition.
4.10. Fluorescent-Gelatin Degradation Assay
The test was conducted according to the procedure described by Artym [
37]. Poly-L-lysine-coated coverslips were washed with PBS and fixed with 0.5% glutaraldehyde for 15 min, which was followed by extensive washing. Afterward, the coverslips were inverted on an 80 μL drop of gelatin conjugated with FITC and incubated for 10 min at room temperature. After washing with PBS, the residual reactive groups were quenched with 5 mg/mL sodium borohydride for 1 min followed by extensive washing with PBS. Then, 4 × 10
4 cells were plated in 24-well plates containing a coverslip coated with fluorescent gelatin matrix and incubated in the presence of foretinib, lapatinib, or a combination at 37 °C. After 12 h, cells were fixed with 4% formaldehyde in PBS for 20 min and permeabilized for 5 min with 0.5% Triton X-100. Cells were stained for filamentous actin. Confocal images were collected using an Olympus FV500 confocal laser scanning microscope. The sites of degraded matrix were visible as dark areas (spots) in the bright fluorescent gelatin matrix. The area of gelatin digestion was calculated for 15 cells per condition using ImageJ software [
38].
4.11. Migration Assay
Cell migration tests were performed using Transwell™ filters (BD Biosciences, Franklin Lakes, NJ, USA) in a 24-well plate. Prior to the assay, cells were starved for 6 h in serum-free DMEM medium. The lower compartment of the Transwell™ contained 500 μL of medium consisting of 80% DMEM, 20% FBS, 5 nM EGF, and 30 ng/mL of HGF. Cells (5 × 104) were seeded onto a rehydrated Transwell™ insert in medium without FBS in the absence (control) or presence of foretinib, lapatinib, or foretinib/lapatinib. After 24 h, non-migrating cells on the upper side of the filters were removed. Cells that migrated through the membrane were fixed with 4% formaldehyde, stained with Hoechst 33342 (Invitrogen), and counted under a fluorescent microscope Olympus FV500 (Tokio, Japan). The results are showed as a relative migration factor (%) where the number of control cells that migrated through the Transwell™ filters are presented as 100%. The experiments were performed three times. Each independent experiment consisted of three measurements.
4.12. Invasion Assay
MDA-MB-231 and BT549 cells (5 × 104) were seeded after 6 h of starvation in serum-free DMEM onto Transwell™ filters coated with a collagen gel (1 mg/mL) and prepared as described in the collagen matrix preparation section. Medium containing 20% fetal bovine serum with EGF and HGF was present in the lower compartment as a chemoattractant. After 24 h, non-invading cells and collagen on the upper side of the filters were removed. Cells that had migrated through the membrane were fixed with 4% formaldehyde, stained with Hoechst 33342, and counted under a fluorescent microscope Olympus FV500 (Tokio, Japan). The results are presented as a relative invasion factor (%) versus the control. The number of control cells that invaded through the Transwell™ filters is set as 100%.
4.13. Timelapse Invasion Assay
The 3D collagen matrix was prepared as described in the collagen coating preparation section. 4.5 × 10
3 cells were seeded in 96-well plates on top of a thin layer of collagen gel and around a shielded central zone in the middle of the well, according to the ORIS cell invasion protocol [
27]. The cells and the central cell-free zone were covered with a second collagen gel layer (40 µL volume). After polymerization, 100 μL of growth medium was added on top of the 3D collagen I gel containing the cell layer. Phase-contrast time-lapse movies were recorded with a time interval of 20 min using a 10 × UPlanFL objective (N.A. 0.30) on a CellM system with an IX81 microscope (Olympus). Control cells and cells treated with inhibitors were allowed to invade the central zone for 48 h. Image and data analyses were done using CELLMIA image processing software [
27] and the data management and analysis software CellMissy [
28], respectively. The cell invasion velocity is based on the increase in areas covered by the cells in a given time.
4.14. Statistical Analysis
All data are given as means ± SD. Their significance was determined with GraphPad Prism 7 Software using one-way ANOVA followed by the Tukey test (i.e., cytotoxicity and proliferation assay, cell cycle analysis, and migration and invasion assays) or the Kruskal–Wallis method, which was followed by the Dunn’s post-hoc test (i.e., number of invadopodia and FITC-gelatin digestion).

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}








