Alcohol-Derived Acetaldehyde Exposure in the Oral Cavity


Abstract
1. Introduction
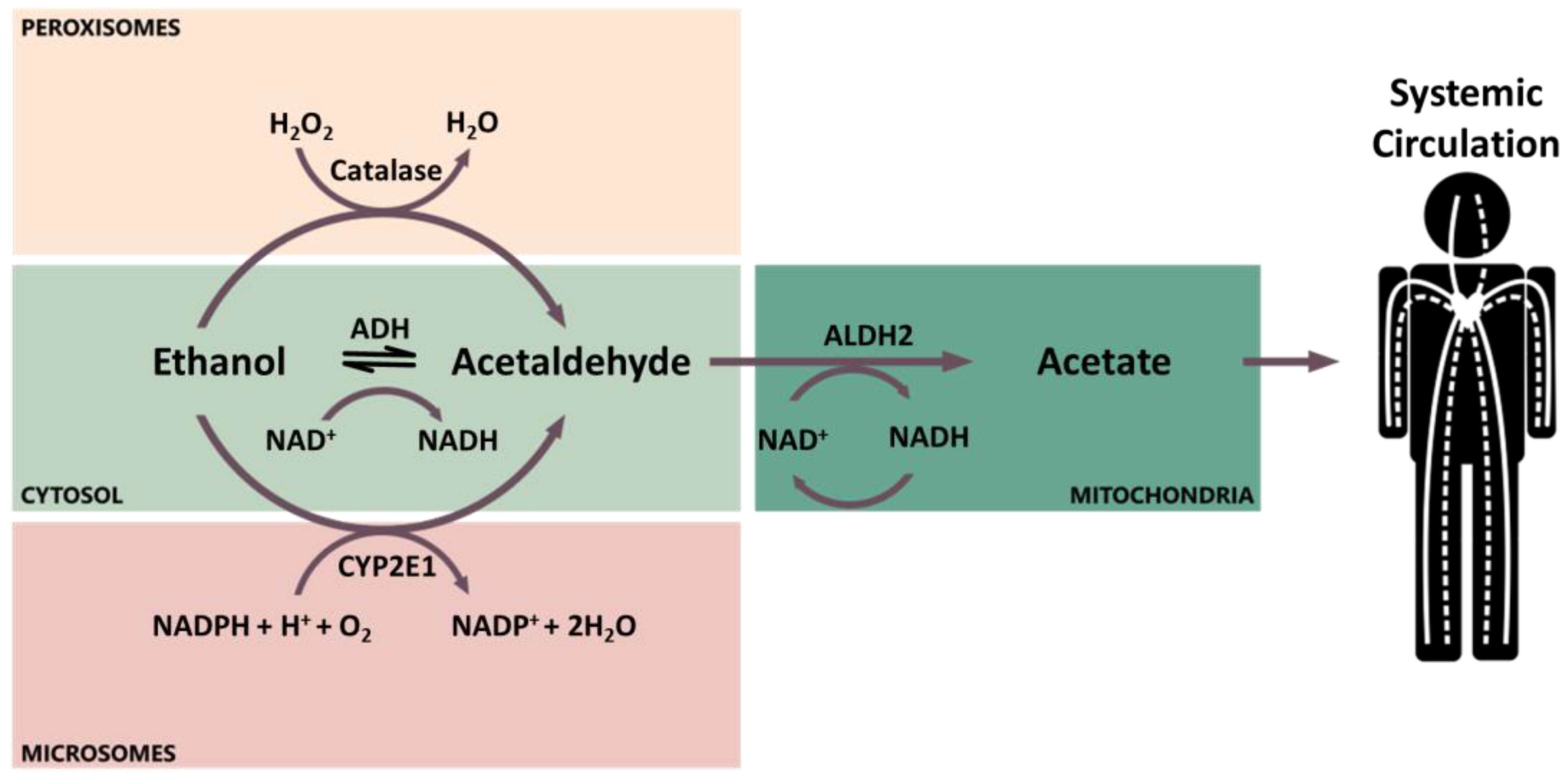
2. Ethanol Pharmacokinetics and Metabolism
2.1. Ethanol Pharmacokinetics
2.2. Genetic Variants and Enzyme Induction Influencing Ethanol Metabolism
3. Factors Influencing Acetaldehyde Exposure in the Oral Cavity
3.1. Genetic Polymorphisms
3.2. Poor Oral Hygiene
3.3. Nutritional and Environmental Factors
3.4. Other Factors
4. Ethanol Metabolism by Oral Mucosa Cells, Salivary Glands, and Oral Microbiome
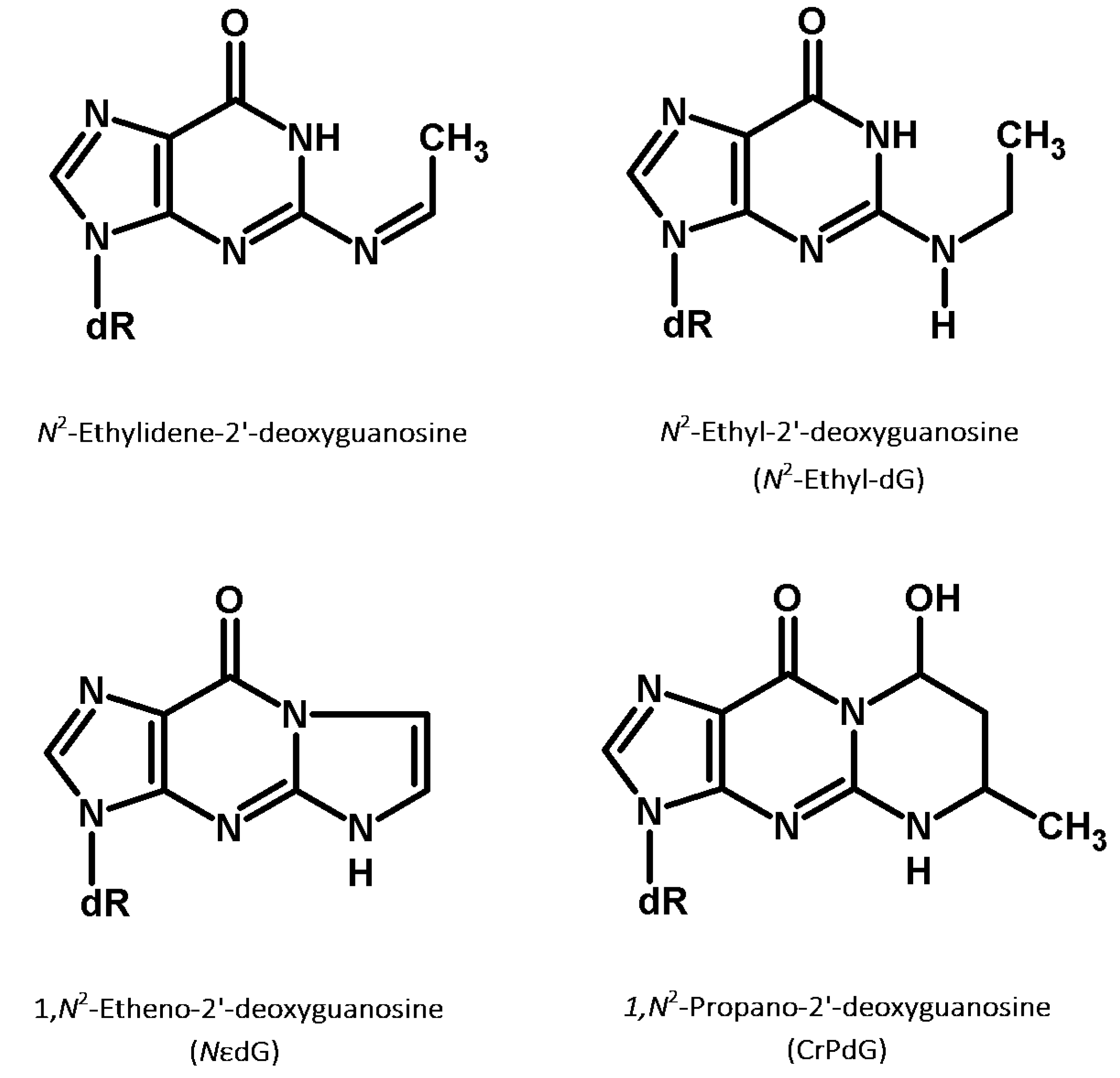
5. Acetaldehyde-Induced DNA Damage
5.1. N2-Ethyl-dG: A Biomarker of Acetaldehyde-Induced DNA Damage
5.2. Other Acetaldehyde-Induced DNA Adducts
6. Future Developments and Challenges
Acknowledgments
Conflicts of Interest
References
- Llewellyn, C.D.; Johnson, N.W.; Warnakulasuriya, K.A. Risk factors for squamous cell carcinoma of the oral cavity in young people-a comprehensive literature review. Oral Oncol. 2001, 37, 401–418. [Google Scholar] [CrossRef]
- Bartsch, H.; Ohshima, H.; Shuker, D.E.; Pignatelli, B.; Calmels, S. Exposure of humans to endogenous n-nitroso compounds: Implications in cancer etiology. Mutat. Res. 1990, 238, 255–267. [Google Scholar] [CrossRef]
- Homann, N.; Tillonen, J.; Meurman, J.H.; Rintamaki, H.; Lindqvist, C.; Rautio, M.; Jousimies-Somer, H.; Salaspuro, M. Increased salivary acetaldehyde levels in heavy drinkers and smokers: A microbiological approach to oral cavity cancer. Carcinogenesis 2000, 21, 663–668. [Google Scholar] [CrossRef] [PubMed]
- Seitz, H.K.; Stickel, F. Molecular mechanisms of alcohol-mediated carcinogenesis. Natl. Rev. Cancer 2007, 7, 599–612. [Google Scholar] [CrossRef] [PubMed]
- Anantharaman, D.; Marron, M.; Lagiou, P.; Samoli, E.; Ahrens, W.; Pohlabeln, H.; Slamova, A.; Schejbalova, M.; Merletti, F.; Richiardi, L.; et al. Population attributable risk of tobacco and alcohol for upper aerodigestive tract cancer. Oral Oncol. 2011, 47, 725–731. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Nanavati, R.; Modi, T.G.; Dobariya, C. Oral cancer: Etiology and risk factors: A review. J. Cancer Res. Ther. 2016, 12, 458–463. [Google Scholar] [CrossRef] [PubMed]
- Gustavsson, P.; Jakobsson, R.; Johansson, H.; Lewin, F.; Norell, S.; Rutkvist, L.E. Occupational exposures and squamous cell carcinoma of the oral cavity, pharynx, larynx, and oesophagus: A case-control study in Sweden. Occup. Environ. Med. 1998, 55, 393–400. [Google Scholar] [CrossRef] [PubMed]
- Barul, C.; Fayosse, A.; Carton, M.; Pilorget, C.; Woronoff, A.S.; Stucker, I.; Luce, D.; Icare Study Group. Occupational exposure to chlorinated solvents and risk of head and neck cancer in men: A population-based case-control study in France. Environ. Health 2017, 16, 77. [Google Scholar] [CrossRef] [PubMed]
- Negri, E.; La Vecchia, C.; Franceschi, S.; Tavani, A. Attributable risk for oral cancer in Northern Italy. Cancer Epidemiol. Biomark. Prev. 1993, 2, 189–193. [Google Scholar]
- International Agency for Research on Cancer. Tobacco smoke and involuntary smoking. In IARC Monographs on the Evaluation of Carcinogenic Risks to Humans; IARC: Lyon, France, 1986; Volume 38. [Google Scholar]
- International Agency for Research on Cancer. Tobacco smoke and involuntary smoking. In IARC Monographs on the Evaluation of Carcinogenic Risks to Humans; IARC: Lyon, France, 2004; Volume 83. [Google Scholar]
- International Agency for Research on Cancer. A review of human carcinogens. In IARC Monographs on the Evaluation of Carcinogenic Risks to Humans; IARC: Lyon, France, 2012; Volume 100 E. [Google Scholar]
- Xue, J.; Yang, S.; Seng, S. Mechanisms of cancer induction by tobacco-specific nnk and nnn. Cancers 2014, 6, 1138–1156. [Google Scholar] [CrossRef] [PubMed]
- Abro, B.; Pervez, S. Smoking and Oral Cancer in: Development of Oral Cancer. Risk Factors and Prevention Strategies; Springer International Publishing AG: Cham, Switzerland, 2017. [Google Scholar]
- Secretan, B.; Straif, K.; Baan, R.; Grosse, Y.; El Ghissassi, F.; Bouvard, V.; Benbrahim-Tallaa, L.; Guha, N.; Freeman, C.; Galichet, L.; et al. A review of human carcinogens-part E: Tobacco, areca nut, alcohol, coal smoke, and salted fish. Lancet Oncol. 2009, 10, 1033–1034. [Google Scholar] [CrossRef]
- International Agency for Research on Cancer. Alcohol consumption and ethyl carbamate. In IARC Monographs on the Evaluation of Carcinogenic Risks to Humans; IARC: Lyon, France, 2010; Volume 96. [Google Scholar]
- Baan, R.; Straif, K.; Grosse, Y.; Secretan, B.; El Ghissassi, F.; Bouvard, V.; Altieri, A.; Cogliano, V. World health organization. International agency for research on cancer monograph working group. Carcinogenicity of alcoholic beverages. Lancet Oncol. 2007, 8, 292–293. [Google Scholar] [CrossRef]
- Balbo, S.; Juanes, R.C.; Khariwala, S.; Baker, E.J.; Daunais, J.B.; Grant, K.A. Increased levels of the acetaldehyde-derived DNA adduct n 2-ethyldeoxyguanosine in oral mucosa DNA from Rhesus monkeys exposed to alcohol. Mutagenesis 2016, 31, 553–558. [Google Scholar] [CrossRef] [PubMed]
- International Agency for Research on Cancer. Re-evaluation of some organic chemicals, hydrazine and hydrogen peroxide. In IARC Monographs on the Evaluation of Carcinogenic Risks to Humans; IARC: Lyon, France, 1999; Volume 71. [Google Scholar]
- Seitz, H.K.; Becker, P. Alcohol metabolism and cancer risk. Alcohol Res. Health 2007, 30, 38–47. [Google Scholar] [PubMed]
- Cederbaum, A.I. Alcohol metabolism. Clin. Liver Dis. 2012, 16, 667–685. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, A.; Tsutsumi, E.; Imazeki, H.; Suwa, Y.; Nakamura, C.; Mizukami, T.; Yokoyama, T. Salivary acetaldehyde concentration according to alcoholic beverage consumed and aldehyde dehydrogenase-2 genotype. Alcohol. Clin. Exp. Res. 2008, 32, 1607–1614. [Google Scholar] [CrossRef] [PubMed]
- Lachenmeier, D.W.; Monakhova, Y.B. Short-term salivary acetaldehyde increase due to direct exposure to alcoholic beverages as an additional cancer risk factor beyond ethanol metabolism. J. Exp. Clin. Cancer Res. 2011, 30, 3. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, A.; Tsutsumi, E.; Imazeki, H.; Suwa, Y.; Nakamura, C.; Yokoyama, T. Polymorphisms of alcohol dehydrogenase-1b and aldehyde dehydrogenase-2 and the blood and salivary ethanol and acetaldehyde concentrations of Japanese alcoholic men. Alcohol. Clin. Exp. Res. 2010, 34, 1246–1256. [Google Scholar] [CrossRef] [PubMed]
- Homann, N.; Jousimies-Somer, H.; Jokelainen, K.; Heine, R.; Salaspuro, M. High acetaldehyde levels in saliva after ethanol consumption: Methodological aspects and pathogenetic implications. Carcinogenesis 1997, 18, 1739–1743. [Google Scholar] [CrossRef] [PubMed]
- Sarkola, T.; Iles, M.R.; Kohlenberg-Mueller, K.; Eriksson, C.J. Ethanol, acetaldehyde, acetate, and lactate levels after alcohol intake in white men and women: Effect of 4-methylpyrazole. Alcohol. Clin. Exp. Res. 2002, 26, 239–245. [Google Scholar] [CrossRef] [PubMed]
- Balbo, S.; Meng, L.; Bliss, R.L.; Jensen, J.A.; Hatsukami, D.K.; Hecht, S.S. Time course of DNA adduct formation in peripheral blood granulocytes and lymphocytes after drinking alcohol. Mutagenesis 2012, 27, 485–490. [Google Scholar] [CrossRef] [PubMed]
- Balbo, S.; Meng, L.; Bliss, R.L.; Jensen, J.A.; Hatsukami, D.K.; Hecht, S.S. Kinetics of DNA adduct formation in the oral cavity after drinking alcohol. Cancer Epidemiol. Biomark. Prev. 2012, 21, 601–608. [Google Scholar] [CrossRef] [PubMed]
- Vakevainen, S.; Tillonen, J.; Agarwal, D.P.; Srivastava, N.; Salaspuro, M. High salivary acetaldehyde after a moderate dose of alcohol in ALDH2-deficient subjects: Strong evidence for the local carcinogenic action of acetaldehyde. Alcohol. Clin. Exp. Res. 2000, 24, 873–877. [Google Scholar] [CrossRef] [PubMed]
- Vakevainen, S.; Tillonen, J.; Salaspuro, M. 4-methylpyrazole decreases salivary acetaldehyde levels in aldh2-deficient subjects but not in subjects with normal ALDH2. Alcohol. Clin. Exp. Res. 2001, 25, 829–834. [Google Scholar] [CrossRef] [PubMed]
- Visapaa, J.P.; Gotte, K.; Benesova, M.; Li, J.; Homann, N.; Conradt, C.; Inoue, H.; Tisch, M.; Horrmann, K.; Vakevainen, S.; et al. Increased cancer risk in heavy drinkers with the alcohol dehydrogenase 1c*1 allele, possibly due to salivary acetaldehyde. Gut 2004, 53, 871–876. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, A.; Tsutsumi, E.; Imazeki, H.; Suwa, Y.; Nakamura, C.; Yokoyama, T. Contribution of the alcohol dehydrogenase-1b genotype and oral microorganisms to high salivary acetaldehyde concentrations in Japanese alcoholic men. Int. J. Cancer 2007, 121, 1047–1054. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, A.; Muramatsu, T.; Ohmori, T.; Yokoyama, T.; Okuyama, K.; Takahashi, H.; Hasegawa, Y.; Higuchi, S.; Maruyama, K.; Shirakura, K.; et al. Alcohol-related cancers and aldehyde dehydrogenase-2 in Japanese alcoholics. Carcinogenesis 1998, 19, 1383–1387. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, A.; Muramatsu, T.; Omori, T.; Yokoyama, T.; Matsushita, S.; Higuchi, S.; Maruyama, K.; Ishii, H. Alcohol and aldehyde dehydrogenase gene polymorphisms and oropharyngolaryngeal, esophageal and stomach cancers in Japanese alcoholics. Carcinogenesis 2001, 22, 433–439. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.H.; Lee, J.M.; Wu, D.C.; Goan, Y.G.; Chou, S.H.; Wu, I.C.; Kao, E.L.; Chan, T.F.; Huang, M.C.; Chen, P.S.; et al. Carcinogenetic impact of ADH1B and ALDH2 genes on squamous cell carcinoma risk of the esophagus with regard to the consumption of alcohol, tobacco and betel quid. Int. J. Cancer 2008, 122, 1347–1356. [Google Scholar] [CrossRef] [PubMed]
- Homann, N.; Tillonen, J.; Rintamaki, H.; Salaspuro, M.; Lindqvist, C.; Meurman, J.H. Poor dental status increases acetaldehyde production from ethanol in saliva: A possible link to increased oral cancer risk among heavy drinkers. Oral Oncol. 2001, 37, 153–158. [Google Scholar] [CrossRef]
- Salaspuro, V.; Salaspuro, M. Synergistic effect of alcohol drinking and smoking on in vivo acetaldehyde concentration in saliva. Int. J. Cancer 2004, 111, 480–483. [Google Scholar] [CrossRef] [PubMed]
- Lachenmeier, D.W.; Salaspuro, M. ALDH2-deficiency as genetic epidemiologic and biochemical model for the carcinogenicity of acetaldehyde. Regul. Toxicol. Pharmacol. 2017, 86, 128–136. [Google Scholar] [CrossRef] [PubMed]
- Seitz, H.K.; Stickel, F. Acetaldehyde as an underestimated risk factor for cancer development: Role of genetics in ethanol metabolism. Genes Nutr. 2010, 5, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Paiano, V.; Bianchi, G.; Davoli, E.; Negri, E.; Fanelli, R.; Fattore, E. Risk assessment for the Italian population of acetaldehyde in alcoholic and non-alcoholic beverages. Food Chem. 2014, 154, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Lachenmeier, D.W.; Kanteres, F.; Rehm, J. Carcinogenicity of acetaldehyde in alcoholic beverages: Risk assessment outside ethanol metabolism. Addiction 2009, 104, 533–550. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.Q.; Pilone, G.J. An overview of formation and roles of acetaldehyde in winemaking with emphasis on microbiological implications. Int. J. Food Sci. Technol. 2000, 35, 49–61. [Google Scholar] [CrossRef]
- Hooper, S.J.; Wilson, M.J.; Crean, S.J. Exploring the link between microorganisms and oral cancer: A systematic review of the literature. Head Neck 2009, 31, 1228–1239. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, M.P.; Willoughby, D. Alcohol consumption: The good, the bad, and the indifferent. Appl. Physiol. Nutr. Metab. 2008, 33, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Lieber, C.S. Ethanol metabolism, cirrhosis and alcoholism. Clin. Chim. Acta 1997, 257, 59–84. [Google Scholar] [CrossRef]
- Paton, A. Alcohol in the body. BMJ 2005, 330, 85–87. [Google Scholar] [CrossRef] [PubMed]
- Julkunen, R.J.; Tannenbaum, L.; Baraona, E.; Lieber, C.S. First pass metabolism of ethanol: An important determinant of blood levels after alcohol consumption. Alcohol 1985, 2, 437–441. [Google Scholar] [CrossRef]
- Ramchandani, V.A.; Bosron, W.F.; Li, T.K. Research advances in ethanol metabolism. Pathol. Biol. 2001, 49, 676–682. [Google Scholar] [CrossRef]
- Jones, A.W.; Jonsson, K.A.; Neri, A. Peak blood-ethanol concentration and the time of its occurrence after rapid drinking on an empty stomach. J. Forensic Sci. 1991, 36, 376–385. [Google Scholar] [CrossRef] [PubMed]
- Quertemont, E. Genetic polymorphism in ethanol metabolism: Acetaldehyde contribution to alcohol abuse and alcoholism. Mol. Psychiatry 2004, 9, 570–581. [Google Scholar] [CrossRef] [PubMed]
- Salaspuro, M.P.; Lieber, C.S. Non-uniformity of blood ethanol elimination: Its exaggeration after chronic consumption. Ann. Clin. Res. 1978, 10, 294–297. [Google Scholar] [PubMed]
- Matsumoto, H.; Fukui, Y. Pharmacokinetics of ethanol: A review of the methodology. Addict. Biol. 2002, 7, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Holford, N.H. Clinical pharmacokinetics of ethanol. Clin. Pharmacokinet. 1987, 13, 273–292. [Google Scholar] [CrossRef] [PubMed]
- Enomoto, N.; Takase, S.; Yasuhara, M.; Takada, A. Acetaldehyde metabolism in different aldehyde dehydrogenase-2 genotypes. Alcohol. Clin. Exp. Res. 1991, 15, 141–144. [Google Scholar] [CrossRef] [PubMed]
- Harty, L.C.; Caporaso, N.E.; Hayes, R.B.; Winn, D.M.; Bravo-Otero, E.; Blot, W.J.; Kleinman, D.V.; Brown, L.M.; Armenian, H.K.; Fraumeni, J.F., Jr.; et al. Alcohol dehydrogenase 3 genotype and risk of oral cavity and pharyngeal cancers. J. Natl. Cancer Inst. 1997, 89, 1698–1705. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, A.; Kamada, Y.; Imazeki, H.; Hayashi, E.; Murata, S.; Kinoshita, K.; Yokoyama, T.; Kitagawa, Y. Effects of ADH1B and ALDH2 genetic polymorphisms on alcohol elimination rates and salivary acetaldehyde levels in intoxicated Japanese alcoholic men. Alcohol. Clin. Exp. Res. 2016, 40, 1241–1250. [Google Scholar] [CrossRef] [PubMed]
- Coutelle, C.; Ward, P.J.; Fleury, B.; Quattrocchi, P.; Chambrin, H.; Iron, A.; Couzigou, P.; Cassaigne, A. Laryngeal and oropharyngeal cancer, and alcohol dehydrogenase 3 and glutathione s-transferase m1 polymorphisms. Hum. Genet. 1997, 99, 319–325. [Google Scholar] [CrossRef] [PubMed]
- Peters, E.S.; McClean, M.D.; Liu, M.; Eisen, E.A.; Mueller, N.; Kelsey, K.T. The ADH1C polymorphism modifies the risk of squamous cell carcinoma of the head and neck associated with alcohol and tobacco use. Cancer Epidemiol. Biomark. Prev. 2005, 14, 476–482. [Google Scholar] [CrossRef] [PubMed]
- Bouchardy, C.; Hirvonen, A.; Coutelle, C.; Ward, P.J.; Dayer, P.; Benhamou, S. Role of alcohol dehydrogenase 3 and cytochrome p-4502e1 genotypes in susceptibility to cancers of the upper aerodigestive tract. Int. J. Cancer 2000, 87, 734–740. [Google Scholar] [CrossRef]
- Olshan, A.F.; Weissler, M.C.; Watson, M.A.; Bell, D.A. Risk of head and neck cancer and the alcohol dehydrogenase 3 genotype. Carcinogenesis 2001, 22, 57–61. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Q.; Weiner, H.; Crabb, D.W. The mutation in the mitochondrial aldehyde dehydrogenase (ALDH2) gene responsible for alcohol-induced flushing increases turnover of the enzyme tetramers in a dominant fashion. J. Clin. Investig. 1996, 98, 2027–2032. [Google Scholar] [CrossRef] [PubMed]
- Goedde, H.W.; Agarwal, D.P.; Fritze, G.; Meier-Tackmann, D.; Singh, S.; Beckmann, G.; Bhatia, K.; Chen, L.Z.; Fang, B.; Lisker, R.; et al. Distribution of ADH2 and ALDH2 genotypes in different populations. Hum. Genet. 1992, 88, 344–346. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, A.; Muramatsu, T.; Ohmori, T.; Makuuchi, H.; Higuchi, S.; Matsushita, S.; Yoshino, K.; Maruyama, K.; Nakano, M.; Ishii, H. Multiple primary esophageal and concurrent upper aerodigestive tract cancer and the aldehyde dehydrogenase-2 genotype of Japanese alcoholics. Cancer 1996, 77, 1986–1990. [Google Scholar] [CrossRef]
- Yokoyama, A.; Muramatsu, T.; Ohmori, T.; Higuchi, S.; Hayashida, M.; Ishii, H. Esophageal cancer and aldehyde dehydrogenase-2 genotypes in Japanese males. Cancer Epidemiol. Biomark. Prev. 1996, 5, 99–102. [Google Scholar]
- Maejima, R.; Iijima, K.; Kaihovaara, P.; Hatta, W.; Koike, T.; Imatani, A.; Shimosegawa, T.; Salaspuro, M. Effects of ALDH2 genotype, ppi treatment and l-cysteine on carcinogenic acetaldehyde in gastric juice and saliva after intragastric alcohol administration. PLoS ONE 2015, 10, e0120397. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, T.; Yabushita, H.; Kanaly, R.A.; Shibutani, S.; Yokoyama, A. Increased DNA damage in ALDH2-deficient alcoholics. Chem. Res. Toxicol. 2006, 19, 1374–1378. [Google Scholar] [CrossRef] [PubMed]
- Seitz, H.K.; Wang, X.D. The role of cytochrome p450 2e1 in ethanol-mediated carcinogenesis. Subcell. Biochem. 2013, 67, 131–143. [Google Scholar]
- Hayashi, S.; Watanabe, J.; Kawajiri, K. Genetic polymorphisms in the 5’-flanking region change transcriptional regulation of the human cytochrome p450IIe1 gene. J. Biochem. 1991, 110, 559–565. [Google Scholar] [CrossRef] [PubMed]
- Fairbrother, K.S.; Grove, J.; de Waziers, I.; Steimel, D.T.; Day, C.P.; Crespi, C.L.; Daly, A.K. Detection and characterization of novel polymorphisms in the CYP2e1 gene. Pharmacogenetics 1998, 8, 543–552. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, J.; Hayashi, S.; Kawajiri, K. Different regulation and expression of the human CYP2e1 gene due to the Rsai polymorphism in the 5’-flanking region. J. Biochem. 1994, 116, 321–326. [Google Scholar] [CrossRef] [PubMed]
- Cadoni, G.; Boccia, S.; Petrelli, L.; Di Giannantonio, P.; Arzani, D.; Giorgio, A.; De Feo, E.; Pandolfini, M.; Galli, P.; Paludetti, G.; et al. A review of genetic epidemiology of head and neck cancer related to polymorphisms in metabolic genes, cell cycle control and alcohol metabolism. Acta Otorhinol. Ital. 2012, 32, 1–11. [Google Scholar]
- Yoshimura, K.; Hanaoka, T.; Ohnami, S.; Ohnami, S.; Kohno, T.; Liu, Y.; Yoshida, T.; Sakamoto, H.; Tsugane, S. Allele frequencies of single nucleotide polymorphisms (snps) in 40 candidate genes for gene-environment studies on cancer: Data from population-based Japanese random samples. J. Hum. Genet. 2003, 48, 654–658. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Yin, L.H.; Pu, Y.P. Association of combined CYP2e1 gene polymorphism with the risk for esophageal squamous cell carcinoma in Huai’an population, China. Chin. Med. J. 2007, 120, 1797–1802. [Google Scholar] [PubMed]
- Kato, S.; Shields, P.G.; Caporaso, N.E.; Hoover, R.N.; Trump, B.F.; Sugimura, H.; Weston, A.; Harris, C.C. Cytochrome p450IIe1 genetic polymorphisms, racial variation, and lung cancer risk. Cancer Res. 1992, 52, 6712–6715. [Google Scholar] [PubMed]
- Uematsu, F.; Kikuchi, H.; Motomiya, M.; Abe, T.; Sagami, I.; Ohmachi, T.; Wakui, A.; Kanamaru, R.; Watanabe, M. Association between restriction fragment length polymorphism of the human cytochrome p450IIe1 gene and susceptibility to lung cancer. Jpn. J. Cancer Res. 1991, 82, 254–256. [Google Scholar] [CrossRef] [PubMed]
- Uematsu, F.; Ikawa, S.; Kikuchi, H.; Sagami, I.; Kanamaru, R.; Abe, T.; Satoh, K.; Motomiya, M.; Watanabe, M. Restriction fragment length polymorphism of the human CYP2e1 (cytochrome p450IIe1) gene and susceptibility to lung cancer: Possible relevance to low smoking exposure. Pharmacogenetics 1994, 4, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Parsian, A.; Cloninger, C.R.; Zhang, Z.H. Association studies of polymorphisms of CYP2e1 gene in alcoholics with cirrhosis, antisocial personality, and normal controls. Alcohol. Clin. Exp. Res. 1998, 22, 888–891. [Google Scholar] [CrossRef] [PubMed]
- Hildesheim, A.; Anderson, L.M.; Chen, C.J.; Cheng, Y.J.; Brinton, L.A.; Daly, A.K.; Reed, C.D.; Chen, I.H.; Caporaso, N.E.; Hsu, M.M.; et al. CYP2e1 genetic polymorphisms and risk of nasopharyngeal carcinoma in Taiwan. J. Natl. Cancer Inst. 1997, 89, 1207–1212. [Google Scholar] [CrossRef] [PubMed]
- Wong, N.A.; Rae, F.; Simpson, K.J.; Murray, G.D.; Harrison, D.J. Genetic polymorphisms of cytochrome p4502e1 and susceptibility to alcoholic liver disease and hepatocellular carcinoma in a white population: A study and literature review, including meta-analysis. Mol. Pathol. 2000, 53, 88–93. [Google Scholar] [CrossRef] [PubMed]
- Garte, S.; Gaspari, L.; Alexandrie, A.K.; Ambrosone, C.; Autrup, H.; Autrup, J.L.; Baranova, H.; Bathum, L.; Benhamou, S.; Boffetta, P.; et al. Metabolic gene polymorphism frequencies in control populations. Cancer Epidemiol. Biomark. Prev. 2001, 10, 1239–1248. [Google Scholar]
- Ronis, M.J.J.; Huang, J.; Crouch, J.; Mercado, C.; Irby, D.; Valentine, C.R.; Lumpkin, C.K.; Ingelman-Sundberg, M.; Badger, T.M. Cytochrome p450 CYP 2e1 induction during chronic alcohol exposure occurs by a two-step mechanism associated with blood alcohol concentrations in rats1. J. Pharmacol. Exp. Ther. 1993, 264, 944–950. [Google Scholar] [PubMed]
- Roberts, B.J.; Song, B.J.; Soh, Y.; Park, S.S.; Shoaf, S.E. Ethanol induces CYP2e1 by protein stabilization. Role of ubiquitin conjugation in the rapid degradation of CYP2e1. J. Biol. Chem. 1995, 270, 29632–29635. [Google Scholar] [PubMed]
- Chien, J.Y.; Thummel, K.E.; Slattery, J.T. Pharmacokinetic consequences of induction of CYP2e1 by ligand stabilization. Drug Metab. Dispos. 1997, 25, 1165–1175. [Google Scholar] [PubMed]
- Lieber, C.S.; DeCarli, L.M. Hepatic microsomal ethanol-oxidizing system. In vitro characteristics and adaptive properties in vivo. J. Biol. Chem. 1970, 245, 2505–2512. [Google Scholar] [PubMed]
- Oneta, C.M.; Lieber, C.S.; Li, J.; Ruttimann, S.; Schmid, B.; Lattmann, J.; Rosman, A.S.; Seitz, H.K. Dynamics of cytochrome p4502e1 activity in man: Induction by ethanol and disappearance during withdrawal phase. J. Hepatol. 2002, 36, 47–52. [Google Scholar] [CrossRef]
- Danko, I.M.; Chaschin, N.A. Association of CYP2e1 gene polymorphism with predisposition to cancer development. Exp. Oncol. 2005, 27, 248–256. [Google Scholar] [PubMed]
- Ruwali, M.; Khan, A.J.; Shah, P.P.; Singh, A.P.; Pant, M.C.; Parmar, D. Cytochrome p450 2e1 and head and neck cancer: Interaction with genetic and environmental risk factors. Environ. Mol. Mutagen. 2009, 50, 473–482. [Google Scholar] [CrossRef] [PubMed]
- Gattas, G.J.; de Carvalho, M.B.; Siraque, M.S.; Curioni, O.A.; Kohler, P.; Eluf-Neto, J.; Wunsch-Filho, V. Genetic polymorphisms of CYP1a1, CYP2e1, GSTM1, and GSTT1 associated with head and neck cancer. Head Neck 2006, 28, 819–826. [Google Scholar] [CrossRef] [PubMed]
- Hung, H.C.; Chuang, J.; Chien, Y.C.; Chern, H.D.; Chiang, C.P.; Kuo, Y.S.; Hildesheim, A.; Chen, C.J. Genetic polymorphisms of CYP2e1, GSTM1, and GSTT1; environmental factors and risk of oral cancer. Cancer Epidemiol. Biomark. Prev. 1997, 6, 901–905. [Google Scholar]
- Katoh, T.; Kaneko, S.; Kohshi, K.; Munaka, M.; Kitagawa, K.; Kunugita, N.; Ikemura, K.; Kawamoto, T. Genetic polymorphisms of tobacco- and alcohol-related metabolizing enzymes and oral cavity cancer. Int. J. Cancer 1999, 83, 606–609. [Google Scholar] [CrossRef]
- Yang, C.X.; Matsuo, K.; Ito, H.; Hirose, K.; Wakai, K.; Saito, T.; Shinoda, M.; Hatooka, S.; Mizutani, K.; Tajima, K. Esophageal cancer risk by ALDH2 and ADH2 polymorphisms and alcohol consumption: Exploration of gene-environment and gene-gene interactions. Asian Pac. J. Cancer Prev. 2005, 6, 256–262. [Google Scholar] [PubMed]
- Guo, Y.; Zhou, S.; Liu, F.; Zhang, B. CYP2e1 Rsai/Psti polymorphisms contributed to oral cancer susceptibility: A meta-analysis. Int. J. Clin. Exp. Pathol. 2015, 8, 14685–14692. [Google Scholar] [PubMed]
- Lieber, C.S. Alcohol and the liver: 1994 update. Gastroenterology 1994, 106, 1085–1105. [Google Scholar] [CrossRef]
- Helminen, A.; Vakevainen, S.; Salaspuro, M. ALDH2 genotype has no effect on salivary acetaldehyde without the presence of ethanol in the systemic circulation. PLoS ONE 2013, 8, e74418. [Google Scholar] [CrossRef] [PubMed]
- Salaspuro, M. Inhibitors of alcohol metabolism. Acta Med. Scand. Suppl. 1985, 703, 219–224. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, F.; Yamamoto, K.; Suzuki, S.; Inoue, H.; Tsurumaru, M.; Kajiyama, Y.; Kato, H.; Igaki, H.; Furuta, K.; Fujita, H.; et al. Strong interaction between the effects of alcohol consumption and smoking on oesophageal squamous cell carcinoma among individuals with ADH1B and/or ALDH2 risk alleles. Gut 2010, 59, 1457–1464. [Google Scholar] [CrossRef] [PubMed]
- Kocaelli, H.; Apaydin, A.; Aydil, B.; Ayhan, M.; Karadeniz, A.; Ozel, S.; Yilmaz, E.; Akgun, B.; Eren, B. Evaluation of potential salivary acetaldehyde production from ethanol in oral cancer patients and healthy subjects. Hippokratia 2014, 18, 269–274. [Google Scholar] [PubMed]
- Linderborg, K.; Salaspuro, M.; Vakevainen, S. A single sip of a strong alcoholic beverage causes exposure to carcinogenic concentrations of acetaldehyde in the oral cavity. Food Chem. Toxicol. 2011, 49, 2103–2106. [Google Scholar] [CrossRef] [PubMed]
- Boffetta, P.; Kaihovaara, P.; Rudnai, P.; Znaor, A.; Lissowska, J.; Swiatkowska, B.; Mates, D.; Pandics, T.; Salaspuro, M. Acetaldehyde level in spirits from central European countries. Eur. J. Cancer Prev. 2011, 20, 526–529. [Google Scholar] [CrossRef] [PubMed]
- Lachenmeier, D.W.; Sohnius, E.M. The role of acetaldehyde outside ethanol metabolism in the carcinogenicity of alcoholic beverages: Evidence from a large chemical survey. Food Chem. Toxicol. 2008, 46, 2903–2911. [Google Scholar] [CrossRef] [PubMed]
- Linderborg, K.; Joly, J.P.; Visapaa, J.P.; Salaspuro, M. Potential mechanism for Calvados-related oesophageal cancer. Food Chem. Toxicol. 2008, 46, 476–479. [Google Scholar] [CrossRef] [PubMed]
- Miyake, T.; Shibamoto, T. Quantitative-analysis of acetaldehyde in foods and beverages. J. Agric. Food Chem. 1993, 41, 1968–1970. [Google Scholar] [CrossRef]
- Lachenmeier, D.W.; Gumbel-Mako, S.; Sohnius, E.M.; Keck-Wilhelm, A.; Kratz, E.; Mildau, G. Salivary acetaldehyde increase due to alcohol-containing mouthwash use: A risk factor for oral cancer. Int. J. Cancer 2009, 125, 730–735. [Google Scholar] [CrossRef] [PubMed]
- Salaspuro, V.; Hietala, J.; Kaihovaara, P.; Pihlajarinne, L.; Marvola, M.; Salaspuro, M. Removal of acetaldehyde from saliva by a slow-release buccal tablet of L-cysteine. Int. J. Cancer 2002, 97, 361–364. [Google Scholar] [CrossRef] [PubMed]
- Uebelacker, M.; Lachenmeier, D.W. Quantitative determination of acetaldehyde in foods using automated digestion with simulated gastric fluid followed by headspace gas chromatography. J. Autom. Methods Manag. Chem. 2011, 907317. [Google Scholar] [CrossRef] [PubMed]
- LaVecchia, C.; Tavani, A.; Franceschi, S.; Levi, F.; Corrao, G.; Negri, E. Epidemiology and prevention of oral cancer. Oral Oncol. 1997, 33, 302–312. [Google Scholar] [CrossRef]
- Blot, W.J.; McLaughlin, J.K.; Winn, D.M.; Austin, D.F.; Greenberg, R.S.; Preston-Martin, S.; Bernstein, L.; Schoenberg, J.B.; Stemhagen, A.; Fraumeni, J.F., Jr. Smoking and drinking in relation to oral and pharyngeal cancer. Cancer Res. 1988, 48, 3282–3287. [Google Scholar] [PubMed]
- Brugere, J.; Guenel, P.; Leclerc, A.; Rodriguez, J. Differential effects of tobacco and alcohol in cancer of the larynx, pharynx, and mouth. Cancer 1986, 57, 391–395. [Google Scholar] [CrossRef]
- Mashberg, A.; Boffetta, P.; Winkelman, R.; Garfinkel, L. Tobacco smoking, alcohol drinking, and cancer of the oral cavity and oropharynx among U.S. veterans. Cancer 1993, 72, 1369–1375. [Google Scholar] [CrossRef]
- Boffetta, P.; Mashberg, A.; Winkelmann, R.; Garfinkel, L. Carcinogenic effect of tobacco smoking and alcohol drinking on anatomic sites of the oral cavity and oropharynx. Int. J. Cancer 1992, 52, 530–533. [Google Scholar] [CrossRef] [PubMed]
- Dator, R.; Carra, A.; Maertens, L.; Guidolin, V.; Villalta, P.W.; Balbo, S. A high resolution/accurate mass (HRAM) data-dependent MS(3) neutral loss screening, classification, and relative quantitation methodology for carbonyl compounds in saliva. J. Am. Soc. Mass Spectrom. 2017, 28, 608–618. [Google Scholar] [CrossRef] [PubMed]
- Bhatti, S.A.; Walsh, T.F.; Douglas, C.W. Ethanol and pH levels of proprietary mouthrinses. Community Dent. Health 1994, 11, 71–74. [Google Scholar] [PubMed]
- O’Reilly, P.; McCartan, B.E.; Clancy, J. Alcohol content of proprietary mouthwashes. Ir. J. Med. Sci. 1994, 163, 178–181. [Google Scholar] [CrossRef] [PubMed]
- Gagari, E.; Kabani, S. Adverse effects of mouthwash use: A review. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. Endod. 1995, 80, 432–439. [Google Scholar] [CrossRef]
- La Vecchia, C. Mouthwash and oral cancer risk: An update. Oral Oncol. 2009, 45, 198–200. [Google Scholar] [CrossRef] [PubMed]
- Sprince, H.; Parker, C.M.; Smith, G.G.; Gonzales, L.J. Protection against acetaldehyde toxicity in the rat by l-cysteine, thiamin and l-2-methylthiazolidine-4-carboxylic acid. Agents Actions 1974, 4, 125–130. [Google Scholar] [CrossRef] [PubMed]
- FDA. Code for Federal Regulations Title 21. Available online: https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/CFRSearch.cfm?fr=184.1271 (accessed on 21 September 2017).
- EFSA. Opinion of the scientific panel on food additives, flavourings, processing aids and materials in contact with food on a request from the commission related to the use of l-cysteine in foods intended for infants and young children. EFSA J. 2006, 390, 1–7. [Google Scholar]
- Kera, Y.; Kiriyama, T.; Komura, S. Conjugation of acetaldehyde with cysteinylglycine, the first metabolite in glutathione breakdown by gamma-glutamyltranspeptidase. Agents Actions 1985, 17, 48–52. [Google Scholar] [CrossRef] [PubMed]
- Irie, K.; Tomofuji, T.; Tamaki, N.; Sanbe, T.; Ekuni, D.; Azuma, T.; Maruyama, T.; Yamamoto, T. Effects of ethanol consumption on periodontal inflammation in rats. J. Dent. Res. 2008, 87, 456–460. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.M.; Schell, T.D.; Richie, J.P., Jr.; Sun, Y.W.; Zhang, S.M.; Calcagnotto, A.; Aliaga, C.; Gowda, K.; Amin, S.; El-Bayoumy, K. Effects of chronic alcohol consumption on DNA damage and immune regulation induced by the environmental pollutant dibenzo[a,l]pyrene in oral tissues of mice. J. Environ. Sci. Health Part C 2017, 35, 213–222. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.J.; Peng, T.K.; Yin, S.J. Expression and activities of class iv alcohol dehydrogenase and class iii aldehyde dehydrogenase in human mouth. Alcohol 1996, 13, 257–262. [Google Scholar] [CrossRef]
- Salaspuro, M.P. Acetaldehyde, microbes, and cancer of the digestive tract. Crit. Rev. Clin. Lab. Sci. 2003, 40, 183–208. [Google Scholar] [CrossRef] [PubMed]
- Salaspuro, M. Microbial metabolism of ethanol and acetaldehyde and clinical consequences. Addict. Biol. 1997, 2, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Nosova, T.; Jokelainen, K.; Kaihovaara, P.; Jousimies-Somer, H.; Siitonen, A.; Heine, R.; Salaspuro, M. Aldehyde dehydrogenase activity and acetate production by aerobic bacteria representing the normal flora of human large intestine. Alcohol Alcohol. 1996, 31, 555–564. [Google Scholar] [CrossRef] [PubMed]
- Nosova, T.; Jokelainen, K.; Kaihovaara, P.; Heine, R.; Jousimies-Somer, H.; Salaspuro, M. Characteristics of aldehyde dehydrogenases of certain aerobic bacteria representing human colonic flora. Alcohol Alcohol. 1998, 33, 273–280. [Google Scholar] [CrossRef] [PubMed]
- Muto, M.; Hitomi, Y.; Ohtsu, A.; Shimada, H.; Kashiwase, Y.; Sasaki, H.; Yoshida, S.; Esumi, H. Acetaldehyde production by non-pathogenic neisseria in human oral microflora: Implications for carcinogenesis in upper aerodigestive tract. Int. J. Cancer 2000, 88, 342–350. [Google Scholar] [CrossRef]
- Kurkivuori, J.; Salaspuro, V.; Kaihovaara, P.; Kari, K.; Rautemaa, R.; Gronroos, L.; Meurman, J.H.; Salaspuro, M. Acetaldehyde production from ethanol by oral streptococci. Oral Oncol. 2007, 43, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Uittamo, J.; Siikala, E.; Kaihovaara, P.; Salaspuro, M.; Rautemaa, R. Chronic candidosis and oral cancer in apeced-patients: Production of carcinogenic acetaldehyde from glucose and ethanol by Candida albicans. Int. J. Cancer 2009, 124, 754–756. [Google Scholar] [CrossRef] [PubMed]
- Marttila, E.; Bowyer, P.; Sanglard, D.; Uittamo, J.; Kaihovaara, P.; Salaspuro, M.; Richardson, M.; Rautemaa, R. Fermentative 2-carbon metabolism produces carcinogenic levels of acetaldehyde in Candida albicans. Mol. Oral Microbiol. 2013, 28, 281–291. [Google Scholar] [CrossRef] [PubMed]
- Nieminen, M.T.; Uittamo, J.; Salaspuro, M.; Rautemaa, R. Acetaldehyde production from ethanol and glucose by non-Candida albicans yeasts in vitro. Oral Oncol. 2009, 45, e245–e248. [Google Scholar] [CrossRef] [PubMed]
- Tillonen, J.; Homann, N.; Rautio, M.; Jousimies-Somer, H.; Salaspuro, M. Role of yeasts in the salivary acetaldehyde production from ethanol among risk groups for ethanol-associated oral cavity cancer. Alcohol. Clin. Exp. Res. 1999, 23, 1409–1415. [Google Scholar] [CrossRef] [PubMed]
- Moritani, K.; Takeshita, T.; Shibata, Y.; Ninomiya, T.; Kiyohara, Y.; Yamashita, Y. Acetaldehyde production by major oral microbes. Oral Dis. 2015, 21, 748–754. [Google Scholar] [CrossRef] [PubMed]
- Pavlova, S.I.; Jin, L.; Gasparovich, S.R.; Tao, L. Multiple alcohol dehydrogenases but no functional acetaldehyde dehydrogenase causing excessive acetaldehyde production from ethanol by oral streptococci. Microbiology 2013, 159, 1437–1446. [Google Scholar] [CrossRef] [PubMed]
- Keijser, B.J.; Zaura, E.; Huse, S.M.; van der Vossen, J.M.; Schuren, F.H.; Montijn, R.C.; ten Cate, J.M.; Crielaard, W. Pyrosequencing analysis of the oral microflora of healthy adults. J. Dent. Res. 2008, 87, 1016–1020. [Google Scholar] [CrossRef] [PubMed]
- Stoltenberg, J.L.; Osborn, J.B.; Pihlstrom, B.L.; Herzberg, M.C.; Aeppli, D.M.; Wolff, L.F.; Fischer, G.E. Association between cigarette smoking, bacterial pathogens, and periodontal status. J. Periodontol. 1993, 64, 1225–1230. [Google Scholar] [CrossRef] [PubMed]
- Arendorf, T.M.; Walker, D.M. The prevalence and intra-oral distribution of Candida albicans in man. Arch. Oral Biol. 1980, 25, 1–10. [Google Scholar] [CrossRef]
- Holmstrup, P.; Bessermann, M. Clinical, therapeutic, and pathogenic aspects of chronic oral multifocal candidiasis. Oral Surg. Oral Med. Oral Pathol. 1983, 56, 388–395. [Google Scholar] [CrossRef]
- Colman, G.; Beighton, D.; Chalk, A.J.; Wake, S. Cigarette smoking and the microbial flora of the mouth. Aust. Dent. J. 1976, 21, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Marttila, E.; Uittamo, J.; Rusanen, P.; Lindqvist, C.; Salaspuro, M.; Rautemaa, R. Acetaldehyde production and microbial colonization in oral squamous cell carcinoma and oral lichenoid disease. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. 2013, 116, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Alnuaimi, A.D.; Ramdzan, A.N.; Wiesenfeld, D.; O’Brien-Simpson, N.M.; Kolev, S.D.; Reynolds, E.C.; McCullough, M.J. Candida virulence and ethanol-derived acetaldehyde production in oral cancer and non-cancer subjects. Oral Dis. 2016, 22, 805–814. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; McIntee, E.J.; Cheng, G.; Shi, Y.; Villalta, P.W.; Hecht, S.S. Identification of DNA adducts of acetaldehyde. Chem. Res. Toxicol. 2000, 13, 1149–1157. [Google Scholar] [CrossRef] [PubMed]
- Vaca, C.E.; Fang, J.L.; Schweda, E.K. Studies of the reaction of acetaldehyde with deoxynucleosides. Chem. Biol. Int. 1995, 98, 51–67. [Google Scholar] [CrossRef]
- Wang, M.; Yu, N.; Chen, L.; Villalta, P.W.; Hochalter, J.B.; Hecht, S.S. Identification of an acetaldehyde adduct in human liver DNA and quantitation as n2-ethyldeoxyguanosine. Chem. Res. Toxicol. 2006, 19, 319–324. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.C.; Greenberg, D.M. Enzymatic breakdown of threonine by threonine aldolase. J. Gen. Physiol. 1954, 38, 181–196. [Google Scholar] [CrossRef] [PubMed]
- Jinsmaa, Y.; Florang, V.R.; Rees, J.N.; Anderson, D.G.; Strack, S.; Doorn, J.A. Products of oxidative stress inhibit aldehyde oxidation and reduction pathways in dopamine catabolism yielding elevated levels of a reactive intermediate. Chem. Res. Toxicol. 2009, 22, 835–841. [Google Scholar] [CrossRef] [PubMed]
- Fowler, A.K.; Hewetson, A.; Agrawal, R.G.; Dagda, M.; Dagda, R.; Moaddel, R.; Balbo, S.; Sanghvi, M.; Chen, Y.; Hogue, R.J.; et al. Alcohol-induced one-carbon metabolism impairment promotes dysfunction of DNA base excision repair in adult brain. J. Biol. Chem. 2012, 287, 43533–43542. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, T.; Matsumoto, A.; Uchida, M.; Kanaly, R.A.; Misaki, K.; Shibutani, S.; Kawamoto, T.; Kitagawa, K.; Nakayama, K.I.; Tomokuni, K.; et al. Increased formation of hepatic n-2-ethylidene-2’-deoxyguanosine DNA adducts in aldehyde dehydrogenase 2-knockout mice treated with ethanol. Carcinogenesis 2007, 28, 2363–2366. [Google Scholar] [CrossRef] [PubMed]
- Nagayoshi, H.; Matsumoto, A.; Nishi, R.; Kawamoto, T.; Ichiba, M.; Matsuda, T. Increased formation of gastric n-2-ethylidene-2’-deoxyguanosine DNA adducts in aldehyde dehydrogenase-2 knockout mice treated with ethanol. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2009, 673, 74–77. [Google Scholar] [CrossRef] [PubMed]
- Yukawa, Y.; Ohashi, S.; Amanuma, Y.; Nakai, Y.; Tsurumaki, M.; Kikuchi, O.; Miyamoto, S.; Oyama, T.; Kawamoto, T.; Chiba, T.; et al. Impairment of aldehyde dehydrogenase 2 increases accumulation of acetaldehyde-derived DNA damage in the esophagus after ethanol ingestion. Am. J. Cancer Res. 2014, 4, 279–284. [Google Scholar] [PubMed]
- Brooks, P.J.; Zakhari, S. Acetaldehyde and the genome: Beyond nuclear DNA adducts and carcinogenesis. Environ. Mol. Mutagen. 2014, 55, 77–91. [Google Scholar] [CrossRef] [PubMed]
- Terashima, I.; Matsuda, T.; Fang, T.W.; Suzuki, N.; Kobayashi, J.; Kohda, K.; Shibutani, S. Miscoding potential of the N2-ethyl-2′-deoxyguanosine DNA adduct by the exonuclease-free Klenow fragment of Escherichia coli DNA polymerase i. Biochemistry 2001, 40, 4106–4114. [Google Scholar] [CrossRef] [PubMed]
- Perrino, F.W.; Blans, P.; Harvey, S.; Gelhaus, S.L.; McGrath, C.; Akman, S.A.; Jenkins, G.S.; LaCourse, W.R.; Fishbein, J.C. The n2-ethylguanine and the o6-ethyl- and o6-methylguanine lesions in DNA: Contrasting responses from the “bypass” DNA polymerase eta and the replicative DNA polymerase alpha. Chem. Res. Toxicol. 2003, 16, 1616–1623. [Google Scholar] [CrossRef] [PubMed]
- Pence, M.G.; Blans, P.; Zink, C.N.; Hollis, T.; Fishbein, J.C.; Perrino, F.W. Lesion bypass of n2-ethylguanine by human DNA polymerase iota. J. Biol. Chem. 2009, 284, 1732–1740. [Google Scholar] [CrossRef] [PubMed]
- Upton, D.C.; Wang, X.Y.; Blans, P.; Perrino, F.W.; Fishbein, J.C.; Akman, S.A. Replication of n-2-ethyldeoxyguanosine DNA adducts in the human embryonic kidney cell line 293. Chem. Res. Toxicol. 2006, 19, 960–967. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, T.; Terashima, I.; Matsumoto, Y.; Yabushita, H.; Matsui, S.; Shibutani, S. Effective utilization of n2-ethyl-2’-deoxyguanosine triphosphate during DNA synthesis catalyzed by mammalian replicative DNA polymerases. Biochemistry 1999, 38, 929–935. [Google Scholar] [CrossRef] [PubMed]
- Garcia, C.C.; Angeli, J.P.; Freitas, F.P.; Gomes, O.F.; de Oliveira, T.F.; Loureiro, A.P.; Di Mascio, P.; Medeiros, M.H. [13C2]-acetaldehyde promotes unequivocal formation of 1,n2-propano-2’-deoxyguanosine in human cells. J. Am. Chem. Soc. 2011, 133, 9140–9143. [Google Scholar] [CrossRef] [PubMed]
- Theruvathu, J.A.; Jaruga, P.; Nath, R.G.; Dizdaroglu, M.; Brooks, P.J. Polyamines stimulate the formation of mutagenic 1,n2-propanodeoxyguanosine adducts from acetaldehyde. Nucleic Acids Res. 2005, 33, 3513–3520. [Google Scholar] [CrossRef] [PubMed]
- Stein, S.; Lao, Y.; Yang, I.Y.; Hecht, S.S.; Moriya, M. Genotoxicity of acetaldehyde- and crotonaldehyde-induced 1,n2-propanodeoxyguanosine DNA adducts in human cells. Mutat. Res. 2006, 608, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.Y.; Zang, H.; Angel, K.C.; Kozekov, I.D.; Goodenough, A.K.; Rizzo, C.J.; Guengerich, F.P. Translesion synthesis across 1,n2-ethenoguanine by human DNA polymerases. Chem. Res. Toxicol. 2006, 19, 879–886. [Google Scholar] [CrossRef] [PubMed]
- Balbo, S.; Hecht, S.S.; Upadhyaya, P.; Villalta, P.W. Application of a high-resolution mass-spectrometry-based DNA adductomics approach for identification of DNA adducts in complex mixtures. Anal. Chem. 2014, 86, 1744–1752. [Google Scholar] [CrossRef] [PubMed]
- Stornetta, A.; Villalta, P.W.; Hecht, S.S.; Sturla, S.J.; Balbo, S. Screening for DNA alkylation mono and cross-linked adducts with a comprehensive LC-MS3 adductomic approach. Anal. Chem. 2015, 87, 11706–11713. [Google Scholar] [CrossRef] [PubMed]
- Stornetta, A.; Villalta, P.W.; Gossner, F.; Wilson, W.R.; Balbo, S.; Sturla, S.J. DNA adduct profiles predict in vitro cell viability after treatment with the experimental anticancer prodrug pr104a. Chem. Res. Toxicol. 2017, 30, 830–839. [Google Scholar] [CrossRef] [PubMed]
- Carrà, A.; Villalta, P.W.; Dator, R.P.; Balbo, S. Screening for inflammation-induced DNA adducts with a comprehensive high resolution LC-MS3 adductomic approach. In Proceedings of the 64th ASMS Annual Meeting, San Antonio, TX, USA, 4–8 June 2016. [Google Scholar]



{kind=link}
{kind=link}
| Type of Alcoholic Beverage | Subcategory (If Present) | Acetaldehyde (µM) 1 | n 2 | Ref. |
|---|---|---|---|---|
| Apple wine/cider | 1123 ± 932 | 11 | [100] | |
| 2529 | 1 | [23] | ||
| Beer | 140 | 1 | [22] | |
| 120 | 1 | [23] | ||
| 205 ± 150 | 364 | [100] | ||
| 233 ± 281 | 6 | [101] | ||
| 172 ± 67 | 3 | [102] | ||
| 192 | 12 | [40] 8 | ||
| Fortified wines | 586 | 7 | [40] 8 | |
| 2686 ± 2728 | 133 | [100] | ||
| 2231 ± 2450 | 53 | [100] | ||
| Cherry spirit | 8522 | 1 | [23] | |
| Port | 1909 ± 3306 | 27 | [100] | |
| Sherry | 2583 | 1 | [23] | |
| 3537 ± 2482 | 53 | [100] | ||
| Liquors and spirits | 1251 ± 1155 | 12 | [101] | |
| 1541 ± 2344 | 834 | [100] | ||
| 972 | 61 | [40] 8 | ||
| Bacanora 3 | 7711 ± 5061 | 13 | [100] | |
| Brandy/Cognac | 1704 ± 1096 | 82 | [100] | |
| Cachaça 4 | 1149 ± 491 | 21 | [100] | |
| Calvados | 1781 ± 861 | 25 | [101] | |
| 600 | 1 | [22] | ||
| 753 ± 342 | 2 | [23] | ||
| 870 ± 334 | 27 | [100] | ||
| Chinese spirits | 7419 ± 3955 | 30 | [100] | |
| Fruit-based | 1953 ± 2704 | 315 | [100] | |
| 1414 | 17 | [40] 8 | ||
| Gin | 21 | 3 | [40] 8 | |
| Grape mark spirit | 12,903 ± 2697 | 4 | [23] | |
| Grappa 5 | 11,327 | 13 | [40] 8 | |
| Herb and spice-based | 638 | 11 | [40] 8 | |
| Mezcal | 2103 ± 2024 | 10 | [100] | |
| Rum | 3110 | 3 | [40] 8 | |
| 403 ± 321 | 38 | [100] | ||
| Sake | 717 ± 359 | 5 | [102] | |
| Shochu 6 | 600 | 1 | [22] | |
| Sotol 7 | 1876 ± 1346 | 16 | [100] | |
| Tequila | 530 | 1 | [23] | |
| 1371 ± 1960 | 70 | [100] | ||
| Vodka | 48 | 3 | [40] 8 | |
| 61 ± 70 | 72 | [100] | ||
| Whiskey | 1746 | 3 | [40] 8 | |
| 1410 ± 715 | 3 | [102] | ||
| 627 ± 448 | 37 | [100] | ||
| Wine | 275 ± 236 | 6 | [101] | |
| 474 | 1 | [23] | ||
| 773 ± 760 | 213 | [100] | ||
| 1140 ± 308 | 3 | [102] | ||
| 1544 | 60 | [40] 8 | ||
| Red wine | 1267 | 21 | [40] 8 | |
| 250 | 1 | [22] | ||
| Rose wine | 1855 | 3 | [40] 8 | |
| Sparkling wine | 2792 | 15 | [40] 8 | |
| White wine | 1521 | 21 | [40] 8 | |
| Pure alcohol | 56 | 1 | [40] 8 |
| Type | Species | Isolate | In Vitro Acetaldehyde Production from Ethanol | In Vitro Acetaldehyde Production from Glucose | Ref. |
|---|---|---|---|---|---|
| Yeasts | Candida albicans | Culture and subject strains | 157.43 ± 1.57 µM (ATCC 90029), 247.9 ± 4.2 µM (APECED), 280.2 ± 11.8 µM (cancer), 299.1 ± 12.7 (controls) µM | 38.02 ± 2.06 µM (ATCC 90029), 53.5 ± 2.3 µM (APECED), 33.7 ± 3.5 µM (cancer), 34.6 ± 1.9 µM (controls) | [129] |
| Subject strains | - | 619.4 µM (APECED), 716.6 µM (cancer), 654.0 µM (controls) | [130] | ||
| ATCC90029 | 235.1 ± 2.8 µM | 18.7 ± 2.9 µM | [131] | ||
| Subject strains | 73.7 ± 55.4 µM (high-producing saliva), 43.2 ± 22.3 µM (low-producing saliva) | - | [132] | ||
| C. dubliniensis | Isolates from culture collection strains | 139.0 ± 7.1 µM | 4.6 ± 1.1 µM | [131] | |
| C. glabrata | Isolates from culture collection strains | 185.6 ± 8.6 µM | 5.4 ± 1.1 µM | [131] | |
| TIMM 5512 | - | 83.1 ± 24.8 µM | [131] | ||
| C. guilliermondii | Isolates from culture collection strains | 190.2 ± 12.4 µM | 7.3 ± 1.7 µM | [131] | |
| C. kefyr | TIMM 0298 | 299.3 ± 80.9 µM | 80.1 ± 16.8 µM | [133] | |
| C. krusei | Isolates from culture collection strains | 54.6 ± 2.9 µM | 2.6 ± 0.9 µM | [131] | |
| C. parapsilosis | Isolates from culture collection strains | 243.3 ± 8.8 µM | 45.2 ± 2.1 µM | [131] | |
| C. tropicalis | isolates from culture collection strains | 252.3 ± 14.9 µM | 81.8 ± 2.9 µM | [131] | |
| TIMM 0313 | 248.9 ± 49.9 µM | - | [133] | ||
| Bacteria | Neisseria flava | ATCC 14221 | 94.4 ± 2.1 µM | - | [133] |
| N. flavescens | ATCC 13120 | 168.4 ± 8.6 µM | - | [133] | |
| N. mucosa | ATCC 19695 | 272.8 ± 65.5 µM | - | [133] | |
| N. sicca | ATCC29256 | 174.9 ± 39.4 nmol/min/1011 CFU | - | [127] | |
| N. subflava | 9903683 | 23,050.4 ± 1624.7 nmol/min/1011 CFU | - | [127] | |
| Prevotella histicola | JCM 15637 | 53.2 ± 10.1 µM | - | [133] | |
| Rothia mucilaginosa | JCM 10910 | 95.9 ± 5.9 µM | - | [133] | |
| Streptococcus australis | - | - | [133] | ||
| S. gordonii | - | - | [134] | ||
| S. intermedius | - | - | [128] | ||
| S. mitis | JCM 12971 | 90.2 ± 31.3 µM | - | [133] | |
| - | - | [128] | |||
| S. oralis | - | - | [134] | ||
| S. parasanguis | - | - | [133] | ||
| S. salivarius | T-42104 | 135.0-426.3 µM | - | [128] | |
| S. sanguinis | - | - | [134] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stornetta, A.; Guidolin, V.; Balbo, S. Alcohol-Derived Acetaldehyde Exposure in the Oral Cavity. Cancers 2018, 10, 20. https://doi.org/10.3390/cancers10010020
Stornetta A, Guidolin V, Balbo S. Alcohol-Derived Acetaldehyde Exposure in the Oral Cavity. Cancers. 2018; 10(1):20. https://doi.org/10.3390/cancers10010020
Chicago/Turabian StyleStornetta, Alessia, Valeria Guidolin, and Silvia Balbo. 2018. "Alcohol-Derived Acetaldehyde Exposure in the Oral Cavity" Cancers 10, no. 1: 20. https://doi.org/10.3390/cancers10010020
APA StyleStornetta, A., Guidolin, V., & Balbo, S. (2018). Alcohol-Derived Acetaldehyde Exposure in the Oral Cavity. Cancers, 10(1), 20. https://doi.org/10.3390/cancers10010020