
Single-Molecular Förster Resonance Energy Transfer Measurement on Structures and Interactions of Biomolecules


Abstract
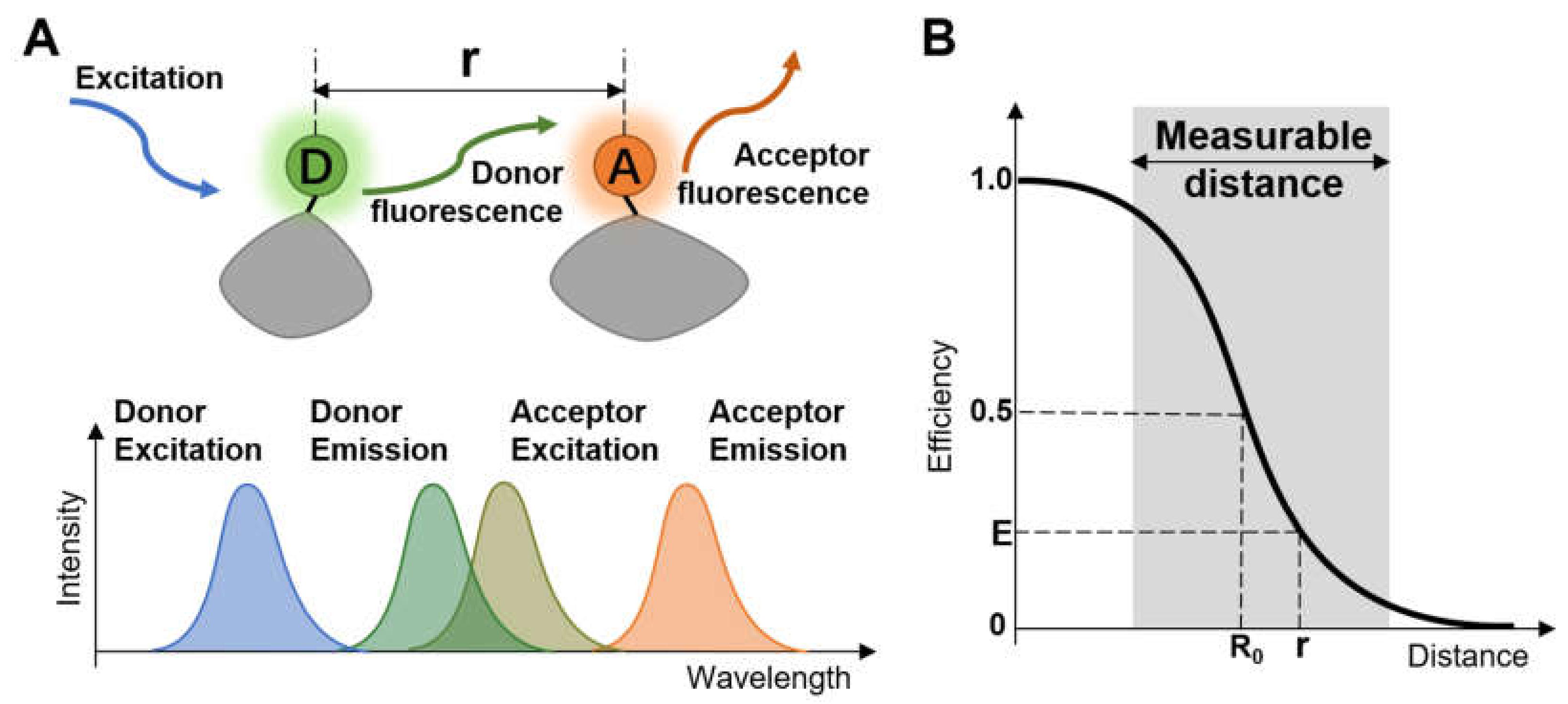
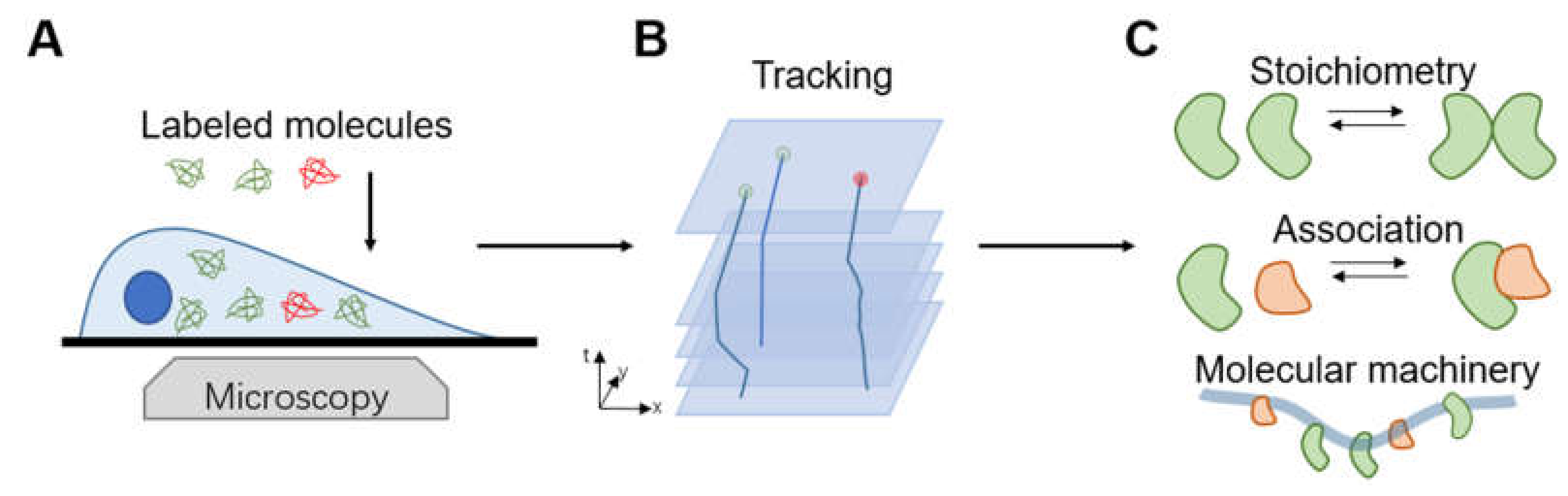
1. Introduction
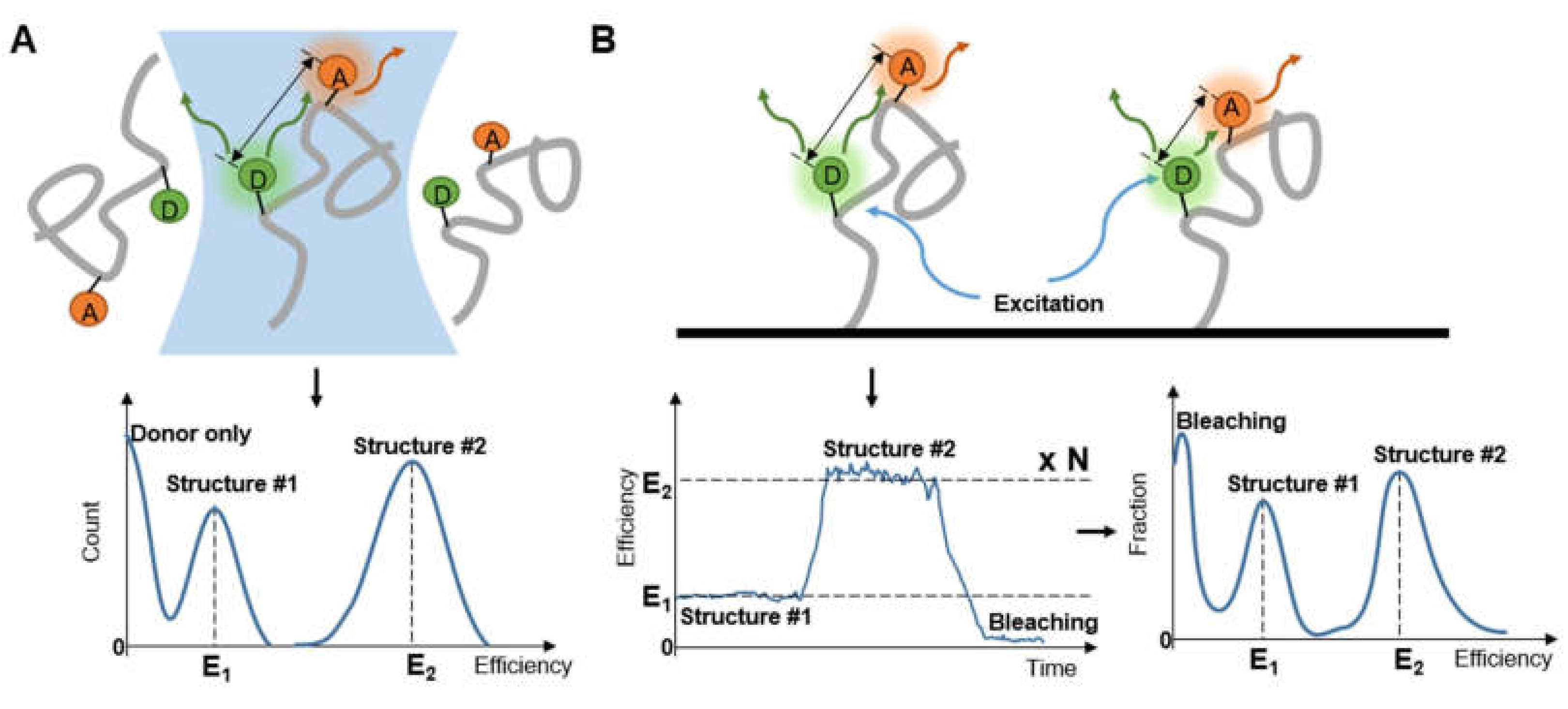
2. Setting a Single-Molecule FRET Measurement
2.1. Imaging Strategies for smFRET
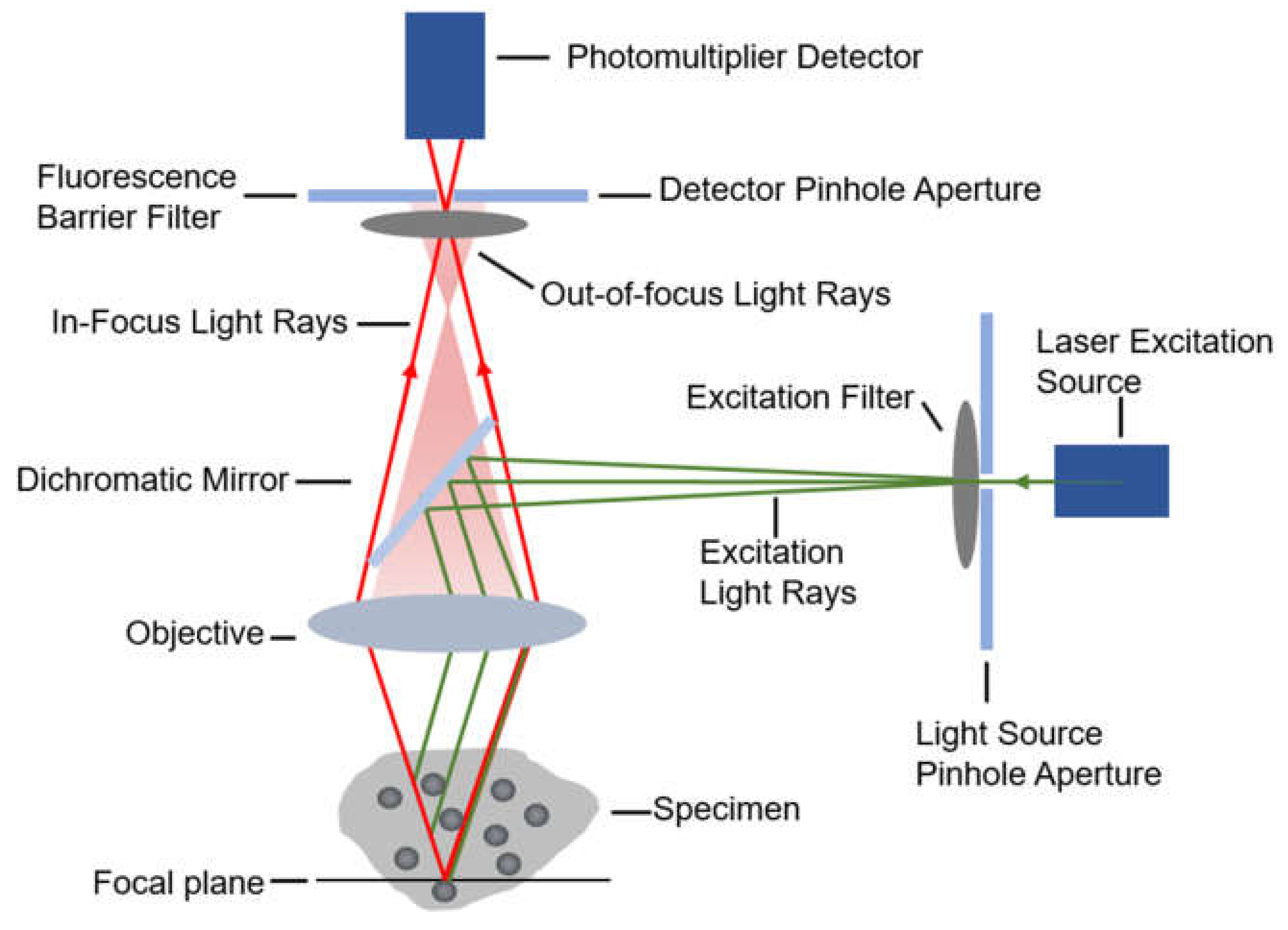
2.1.1. Confocal Microscopy
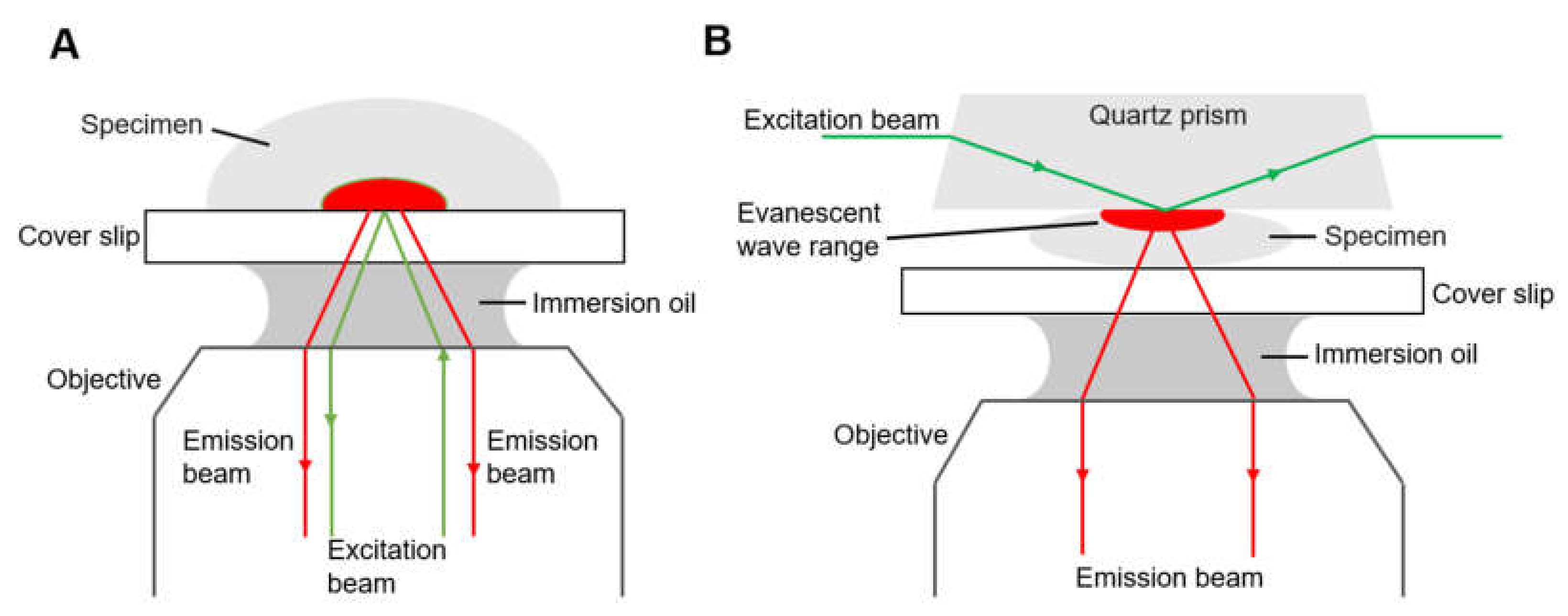
2.1.2. Total Internal Reflection Fluorescence Microscopy
2.2. Fluorescent Labeling
2.2.1. Choices of Fluorophore Pairs
2.2.2. Labeling with Low Impact
2.3. Refined Structure Determination via smFRET
3. Investigating the Structural Changes under Various Circumstances
3.1. Screening Conformational Changes of Proteins
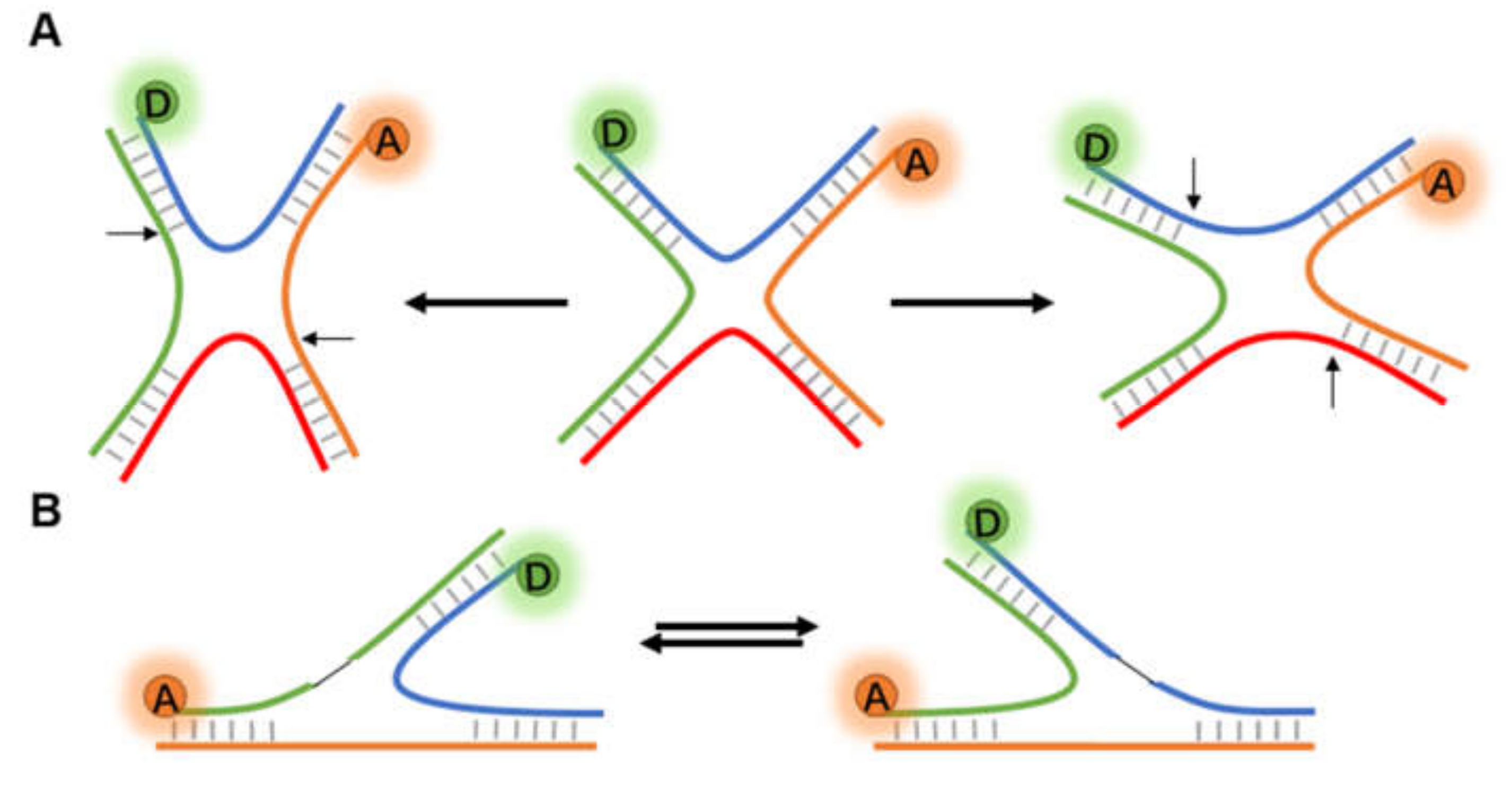
3.2. Dynamic Changes in Nucleic Acid Structure
3.2.1. Non-Helix Secondary Structures of DNA
3.2.2. Secondary Structures of RNA
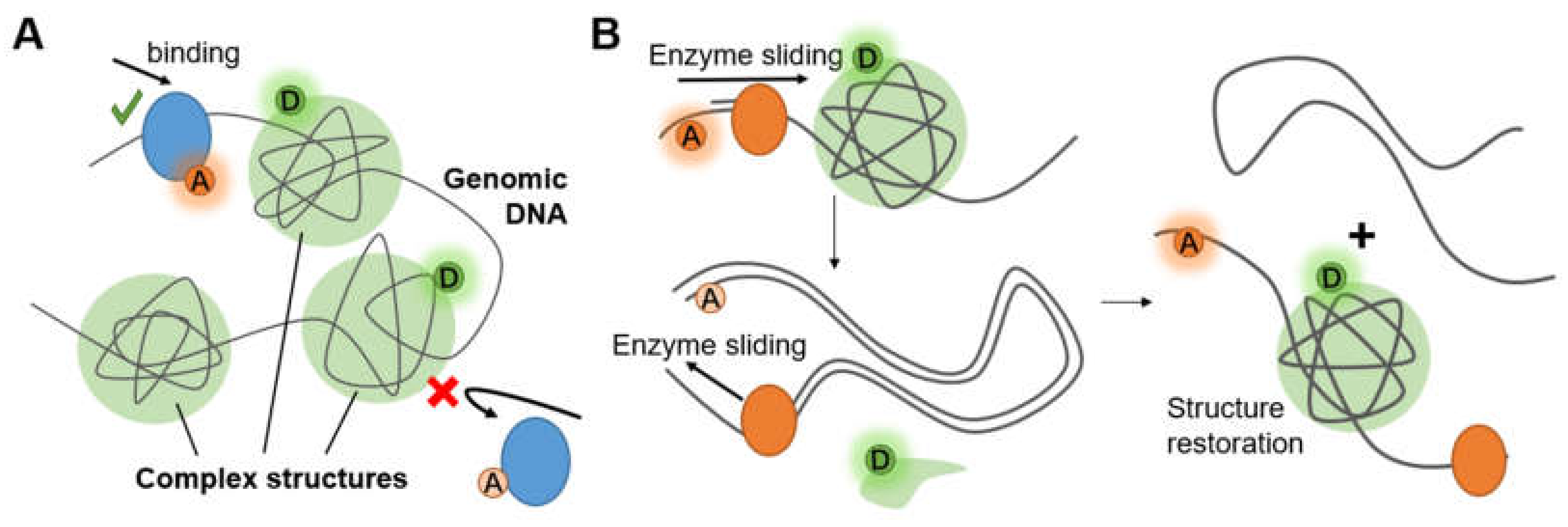
3.3. Complex Chromosome Structures
3.3.1. G-Quadruplex of Telomeres
3.3.2. Histone–DNA Complex of Nucleosomes
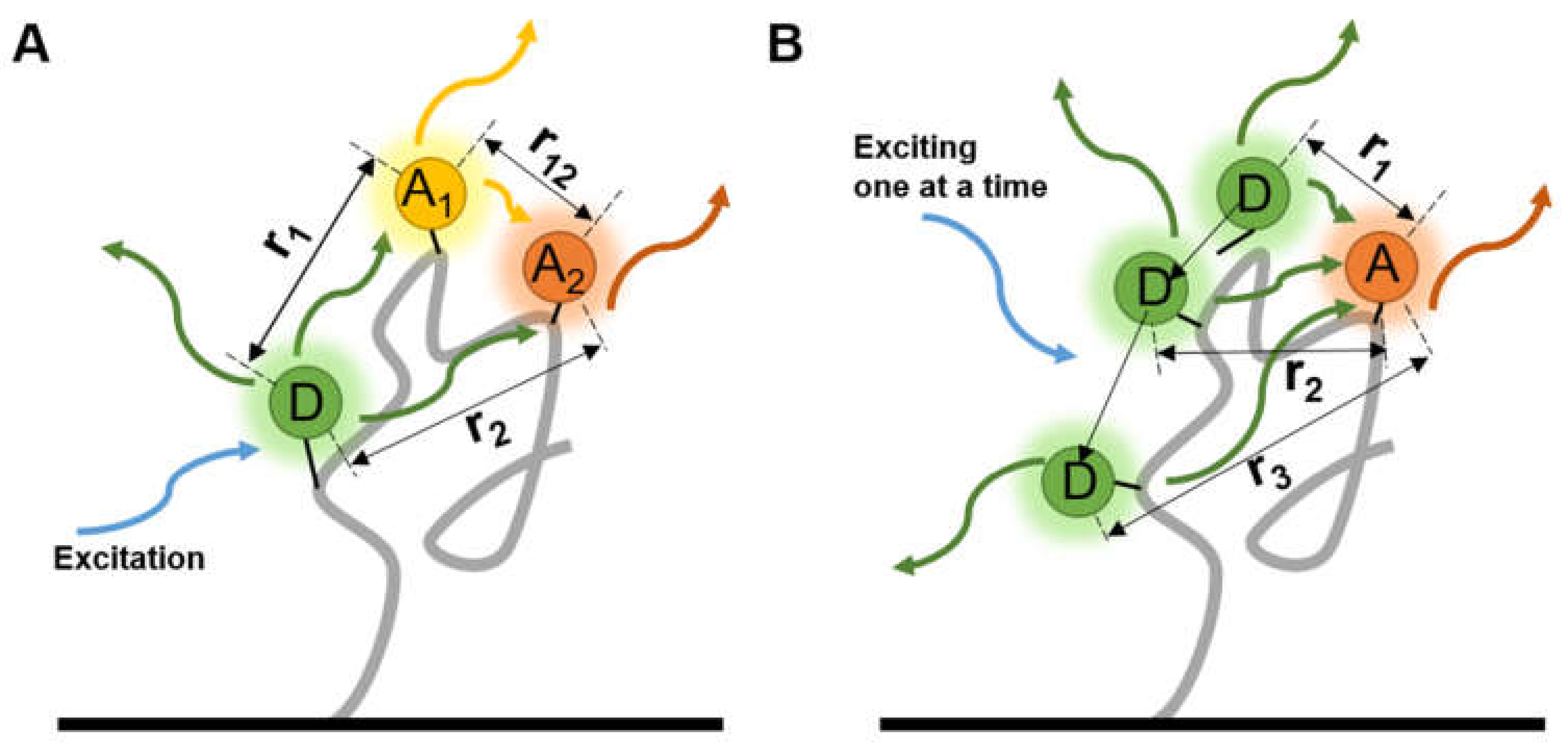
4. Tracking the Interactions between Biomolecules
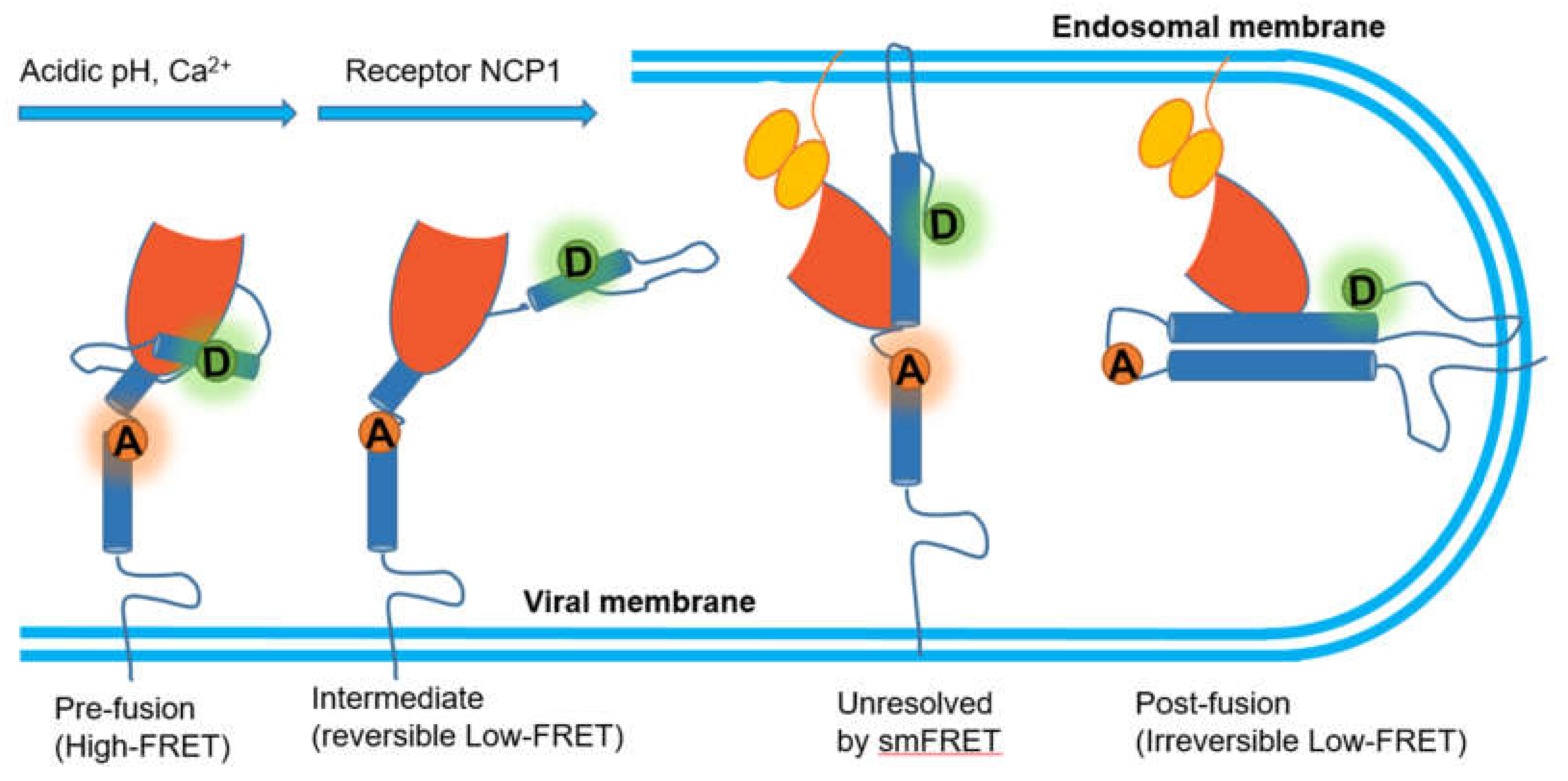
4.1. Virus Spike–Host Interaction
4.2. Dynamics of Membrane Proteins Folding
4.3. Protein Aggregation
4.4. Synthesis of DNA Strands
4.5. Imaging Molecular Behavior in Living Cells
5. Conclusions and Outlook
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Zhang, C.Y.; Liu, H.; Sun, F.; Lin, Y.; Ma, W.Q.; Chen, T. Automated E-FRET microscope for dynamical live-cell FRET imaging. J. Microsc. 2019, 274, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Lu, S.; Lei, L.; Lamberto, I.; Wang, Y.; Pasquale, E.B.; Wang, Y. Genetically encoded FRET biosensor for visualizing Epha4 activity in different compartments of the plasma membrane. ACS Sens. 2019, 4, 294–300. [Google Scholar] [CrossRef] [PubMed]
- Pelicci, S.; Diaspro, A.; Lanzano, L. Chromatin nanoscale compaction in live cells visualized by acceptor-to-donor ratio corrected forster resonance energy transfer between DNA dyes. J. Biophotonics 2019, 12, e201900164. [Google Scholar] [CrossRef] [PubMed]
- Yue, X.; Qiao, Y.; Gu, D.; Wu, Z.; Zhao, W.; Li, X.; Yin, Y.; Zhao, W.; Kong, D.; Xi, R.; et al. Reliable FRET-on imaging of telomerase in living cells by a tetrahedral DNA nanoprobe integrated with structure-switchable molecular beacon. Sens. Actuators B Chem. 2020, 312. [Google Scholar] [CrossRef]
- Forster, T. Zwischenmolekulare energiewanderung und fluoreszenz. Ann. Phys. 1948, 2, 55–75. [Google Scholar] [CrossRef]
- Edelhoch, H.; Brand, L.; Wilchek, M. Fluorescence studies with tryptophyl peptides. Biochemistry 1967, 6, 547. [Google Scholar] [CrossRef]
- Zeug, A.; Woehler, A.; Neher, E.; Ponimaskin, E.G. Intensity-based FRET approaches—A comparative snapshot. Biophys. J. 2012, 103, 1821–1827. [Google Scholar] [CrossRef]
- Mastop, M.; Bindels, D.S.; Shaner, N.C.; Postma, M.; Gadella, T.W.J., Jr.; Goedhart, J. Characterization of a spectrally diverse set of fluorescent proteins as FRET acceptors for mTurquoise2. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef]
- Kubitscheck, U.; Kircheis, M.; Schweitzerstenner, R.; Dreybrodt, W.; Jovin, T.M.; Pecht, I. Fluorescence resonance energy-transfer on single living cells–Application to binding of monovalent haptens to cell-bound immunoglobulin-E. Biophys. J. 1991, 60, 307–318. [Google Scholar] [CrossRef]
- Szollosi, J.; Alexander, D.R. The application of fluorescence resonance energy transfer to the investigation of phosphatases. Protein Phosphatases 2003, 366, 203–224. [Google Scholar]
- Truong, K.; Ikura, M. The use of FRET imaging microscopy to detect protein-protein interactions and protein conformational changes in vivo. Curr. Opin. Struct. Biol. 2001, 11, 573–578. [Google Scholar] [CrossRef]
- Shih, W.M.; Gryczynski, Z.; Lakowicz, J.R.; Spudich, J.A. A FRET-based sensor reveals large Atp hydrolysis-induced conformational changes and three distinct states of the molecular motor myosin. Cell 2000, 102, 683–694. [Google Scholar] [CrossRef]
- Mohamed, Z.H.; Rhein, C.; Saied, E.M.; Kornhuber, J.; Arenz, C. Probes for measuring sphingolipid metabolizing enzyme activity. Chem. Phys. Lipids 2018, 216, 152–161. [Google Scholar] [CrossRef] [PubMed]
- Heitkamp, T.; Deckers-Hebestreit, G.; Börsch, M. Observing single F0f1-Atp synthase at work using an improved fluorescent protein mneongreen as FRET donor. In Single Molecule Spectroscopy and Superresolution Imaging Ix; Enderlein, J., Gregor, I., Gryczynski, Z.K., Erdmann, R., Koberling, F., Eds.; SPIE: Bellingham, WA, USA, 2016. [Google Scholar]
- King, C.; Raicu, V.; Hristova, K. Understanding the FRET signatures of interacting membrane proteins. J. Biol. Chem. 2017, 292, 5291–5310. [Google Scholar] [CrossRef]
- King, C.; Sarabipour, S.; Byrne, P.; Leahy, D.J.; Hristova, K. The FRET signatures of noninteracting proteins in membranes: Simulations and experiments. Biophys. J. 2014, 106, 1309–1317. [Google Scholar] [CrossRef]
- Aoki, K.; Kiyokawa, E.; Nakamura, T.; Matsuda, M. Visualization of growth signal transduction cascades in living cells with genetically encoded probes based on forster resonance energy transfer. Philos. Trans. R. Soc. B Biol. Sci. 2008, 363, 2143–2151. [Google Scholar] [CrossRef]
- Herbst, K.; Qiang Ni, J.; Zhang, J. Dynamic visualization of signal transduction in living cells: From second messengers to kinases. IUBMB Life 2009, 61, 902–908. [Google Scholar] [CrossRef]
- Lomas, O.; Brescia, M.; Carnicer, R.; Monterisi, S.; Surdo, N.C.; Zaccolo, M. Adenoviral transduction of FRET-based biosensors for cAMP in primary adult mouse cardiomyocytes. Methods Mol. Biol. 2015, 1294, 103–115. [Google Scholar]
- Wan-Lung, C.L.; Ermolenko, D.N. Ensemble and single-molecule FRET studies of protein synthesis. Methods 2018, 137, 37–48. [Google Scholar]
- Ha, T.; Enderle, T.; Ogletree, D.F.; Chemla, D.S.; Selvin, P.R.; Weiss, S. the interaction between two single molecules: Fluorescence resonance energy transfer between a single donor and a single acceptor. Proc. Natl. Acad. Sci. USA 1996, 93, 6264–6268. [Google Scholar] [CrossRef]
- Li, C.C.; Li, Y.; Zhang, Y.; Zhang, C.Y. Single-molecule fluorescence resonance energy transfer and its biomedical applications. Trac Trends Anal. Chem. 2020, 122. [Google Scholar] [CrossRef]
- Lerner, E.; Cordes, T.; Ingargiola, A.; Alhadid, Y.; Chung, S.; Michalet, X.; Weiss, S. Toward dynamic structural biology: Two decades of single-molecule Forster resonance energy transfer. Science 2018, 359, 288. [Google Scholar] [CrossRef] [PubMed]
- Dustin, M.L.; Depoil, D. New insights into the T cell synapse from single molecule techniques. Nat. Rev. Immunol. 2011, 11, 672–684. [Google Scholar] [CrossRef]
- Axmann, M.; Huppa, J.B.; Davis, M.M.; Schutz, G.J. Determination of interaction kinetics between the T cell receptor and peptide-loaded MHC Class II via single-molecule diffusion measurements. Biophys. J. 2012, 103, L17–L19. [Google Scholar] [CrossRef]
- Diao, J.; Su, Z.; Ishitsuka, Y.; Lu, B.; Lee, K.S.; Lai, Y.; Shin, Y.Y.; Ha, T. A single-vesicle content mixing assay for SNARE-mediated membrane fusion. Nat. Commun. 2010, 1. [Google Scholar] [CrossRef]
- Sasmal, D.K.; Pulido, L.E.; Kasal, S.; Huang, J. Single-molecule fluorescence resonance energy transfer in molecular biology. Nanoscale 2016, 8, 19928–19944. [Google Scholar] [CrossRef]
- Borner, R.; Howerko, D.; Miserachs, H.G.; Schaffer, M.F.; Sigel, R.K.O. Metal ion induced heterogeneity in RNA folding studied by smFRET. Coord. Chem. Rev. 2016, 327, 123–142. [Google Scholar] [CrossRef]
- Zhao, R.; Rueda, D. RNA folding dynamics by single-molecule fluorescence resonance energy transfer. Methods 2009, 49, 112–117. [Google Scholar] [CrossRef]
- Chen, J.; Poddar, N.K.; Tauzin, L.J.; Cooper, D.; Kolomeisky, A.B.; Landes, C.F. Single-molecule FRET studies of HIV TAR-DNA hairpin unfolding dynamics. J. Phys. Chem. B 2014, 118, 12130–12139. [Google Scholar] [CrossRef]
- Bokinsky, G.; Rueda, D.; Misra, V.K.; Rhodes, M.M.; Gordus, A.; Babcock, H.P.; Walter, N.G.; Zhuang, X.W. Single-molecule transition-state analysis of RNA folding. Proc. Natl. Acad. Sci. USA 2003, 100, 9302–9307. [Google Scholar] [CrossRef]
- Nahas, M.K.; Wilson, T.J.; Hohng, S.; Jarvie, K.; Lilley, D.M.J.; Ha, T. Observation of internal cleavage and ligation reactions of a ribozyme. Nat. Struct. Mol. Biol. 2004, 11, 1107–1113. [Google Scholar] [CrossRef]
- Hyeon, C.; Lee, J.; Yoon, J.; Hohng, S.; Thirumalai, D. Hidden complexity in the isomerization dynamics of Holliday junctions. Nat. Chem. 2012, 4, 907–914. [Google Scholar] [CrossRef]
- Daher, M.; Rueda, D. Fluorescence characterization of the transfer RNA-like domain of transfer messenger RNA in complex with small binding protein, B. Biochemistry 2012, 51, 3531–3538. [Google Scholar] [CrossRef][Green Version]
- Ying, L.M.; Green, J.J.; Li, H.T.; Klenerman, D.; Balasubramanian, S. Studies on the structure and dynamics of the human telomeric G quadruplex by single-molecule fluorescence resonance energy transfer. Proc. Natl. Acad. Sci. USA 2003, 100, 14629–14634. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Yoon, J.; Kihm, H.W.; Kim, D.S. Structural diversity and extreme stability of unimolecular Oxytricha nova telomeric G-quadruplex. Biochemistry 2008, 47, 3389–3396. [Google Scholar] [CrossRef] [PubMed]
- Shirude, P.S.; Okumus, B.; Ying, L.; Ha, T.; Balasubramanian, S. Single-molecule conformational analysis of G-quadruplex formation in the promoter DNA duplex of the proto-oncogene C-Kit. J. Am. Chem. Soc. 2007, 129, 7484. [Google Scholar] [CrossRef] [PubMed]
- Deniz, A.A.; Dahan, M.; Grunwell, J.R.; Ha, T.J.; Faulhaber, A.E.; Chemla, D.S.; Weiss, S.; Schultz, P.G. Single-pair fluorescence resonance energy transfer on freely diffusing molecules: Observation of forster distance dependence and subpopulations. Proc. Natl. Acad. Sci. USA 1999, 96, 3670–3675. [Google Scholar] [CrossRef]
- Deniz, A.A.; Laurence, T.A.; Beligere, G.S.; Dahan, M.; Martin, A.B.; Chemla, D.S.; Dawson, P.E.; Schultz, P.G.; Weiss, S. Single-molecule protein folding: Diffusion fluorescence resonance energy transfer studies of the denaturation of chymotrypsin inhibitor 2. Proc. Natl. Acad. Sci. USA 2000, 97, 5179–5184. [Google Scholar] [CrossRef]
- Sauer, M.; Arden-Jacob, J.; Drexhage, K.H.; Gobel, F.; Lieberwirth, U.; Muhlegger, K.; Muller, R.; Wolfrum, J.; Zander, C. Time-resolved identification of individual mononucleotide molecules in aqueous solution with pulsed semiconductor lasers. Bioimaging 1998, 6, 14–24. [Google Scholar] [CrossRef]
- Eggeling, C.; Fries, J.R.; Brand, L.; Gunther, R.; Seidel, C.A.M. Monitoring conformational dynamics of a single molecule by selective fluorescence spectroscopy. Proc. Natl. Acad. Sci. USA 1998, 95, 1556–1561. [Google Scholar] [CrossRef]
- Tychinskii, V.P.; Kufal, G.E.; Vyshenskaya, T.V.; Perevedentseva, E.V.; Nikandrov, S.L. Measurements of submicron structures with the Airyscan laser phase microscope. Quantum Electron. 1997, 27, 735–739. [Google Scholar] [CrossRef]
- Sivaguru, M.; Urban, M.A.; Fried, G.; Wesseln, C.J.; Mander, L.; Punyasena, S.W. Comparative performance of Airyscan and structured illumination superresolution microscopy in the study of the surface texture and 3D shape of pollen. Microsc. Res. Tech. 2018, 81, 101–114. [Google Scholar] [CrossRef]
- Hu, J.; Wu, M.; Jiang, L.; Zhong, Z.; Zhou, Z.; Rujiralai, T.; Ma, J. Combining gold nanoparticle antennas with single-molecule fluorescence resonance energy transfer (smFRET) to study DNA hairpin dynamics. Nanoscale 2018, 10, 6611–6619. [Google Scholar] [CrossRef]
- Ramachandran, S.; Cohen, D.A.; Quist, A.P.; Lal, R. High performance, led powered, waveguide based total internal reflection microscopy. Sci. Rep. 2013, 3, 2133. [Google Scholar] [CrossRef]
- Asanov, A.; Zepeda, A.; Vaca, L. A novel form of total internal reflection fluorescence microscopy (Lg-Tirfm) reveals different and independent lipid raft domains in living cells. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2010, 1801, 147–155. [Google Scholar] [CrossRef]
- Ha, T.; Zhuang, X.W.; Kim, H.D.; Orr, J.W.; Williamson, J.R.; Chu, S. Ligand-induced conformational changes observed in single RNA molecules. Proc. Natl. Acad. Sci. USA 1999, 96, 9077–9082. [Google Scholar] [CrossRef]
- Graneli, A.; Yeykal, C.C.; Prasad, T.K.; Greene, E.C. Organized arrays of individual DNA molecules tethered to supported lipid bilayers. Langmuir 2006, 22, 292–299. [Google Scholar] [CrossRef]
- Roy, R.; Hohng, S.; Ha, T. A practical guide to single-molecule FRET. Nat. Methods 2008, 5, 507–516. [Google Scholar] [CrossRef]
- Ha, T.; Rasnik, I.; Cheng, W.; Babcock, H.P.; Gauss, G.H.; Lohman, T.M.; Chu, S. Initiation and Re-Initiation of DNA unwinding by the Escherichia Coli rep helicase. Nature 2002, 419, 638–641. [Google Scholar] [CrossRef]
- Aleman, E.A.; Pedini, H.S.; Rueda, D. Covalent-bond-based immobilization approaches for single-molecule fluorescence. Chembiochem 2009, 10, 2862–2866. [Google Scholar] [CrossRef]
- Boukobza, E.; Sonnenfeld, A.; Haran, G. Immobilization in surface-tethered lipid vesicles as a new tool for single biomolecule spectroscopy. J. Phys. Chem. B 2001, 105, 12165–12170. [Google Scholar] [CrossRef]
- Dempsey, G.T.; Vaughan, J.C.; Chen, K.H.; Bates, M.; Zhuang, X. Evaluation of fluorophores for optimal performance in localization-based super-resolution imaging. Nat. Methods 2011, 8, 1027–1036. [Google Scholar] [CrossRef] [PubMed]
- Bajar, B.T.; Wang, E.S.; Zhang, S.; Lin, M.Z.; Chu, J. A guide to fluorescent protein FRET pairs. Sensors 2016, 16, 9. [Google Scholar] [CrossRef] [PubMed]
- Quast, R.B.; Margeat, E. Single-molecule FRET on its way to structural biology in live cells. Nat. Methods 2021, 18, 344–345. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Zhao, R.; Sun, Y.; Ye, Z.; He, K.; Fang, X. Single-Molecule Imaging and Tracking of Molecular Dynamics in Living Cells. Natl. Sci. Rev. 2017, 4, 739–760. [Google Scholar] [CrossRef]
- Cardoso Dos Santos, M.; Algar, W.R.; Medintz, I.L.; Hildebrandt, N. Quantum dots for Förster Resonance Energy Transfer (FRET). TrAC Trends Anal. Chem. 2020, 125. [Google Scholar] [CrossRef]
- Yoo, J.; Louis, J.M.; Gopich, I.V.; Chung, H.S. Three-Color Single-Molecule FRET and fluorescence lifetime analysis of fast protein folding. J. Phys. Chem. B 2018, 122, 11702–11720. [Google Scholar] [CrossRef]
- Filius, M.; Kim, S.H.; Severins, I.; Joo, C. High-resolution single-molecule FRET via DNA exchange (FRET X). Nano Lett. 2021. [Google Scholar] [CrossRef]
- Kim, S.H.; Kim, H.; Jeong, H.; Yoon, T.Y. Encoding multiple virtual signals in DNA barcodes with single-molecule FRET. Nano Lett. 2021, 21, 1694–1701. [Google Scholar] [CrossRef]
- Hartmann, A.; Krainer, G.; Schlierf, M. Different fluorophore labeling strategies and designs affect millisecond kinetics of DNA hairpins. Molecules 2014, 19, 13735–13754. [Google Scholar] [CrossRef]
- Blanchard, D.J.; Cservenyi, T.Z.; Manderville, R.A. Dual fluorescent deoxyguanosine mimics for FRET detection of G-quadruplex folding. Chem. Commun. 2015, 51, 16829–16831. [Google Scholar] [CrossRef]
- Hagen, T.; Malinowska, A.L.; Lightfoot, H.L.; Bigatti, M.; Hall, J. Site-specific fluorophore labeling of guanosines in RNA G-quadruplexes. ACS Omega 2019, 4, 8472–8479. [Google Scholar] [CrossRef]
- Voelz, V.A.; Singh, V.R.; Wedemeyer, W.J.; Lapidus, L.J.; Pande, V.S. Unfolded-state dynamics and structure of protein L characterized by simulation and experiment. J. Am. Chem. Soc. 2010, 132, 4702–4709. [Google Scholar] [CrossRef]
- Jacob, J.; Krantz, B.; Dothager, R.S.; Thiyagarajan, P.; Sosnick, T.R. Early collapse is not an obligate step in protein folding. J. Mol. Biol. 2004, 338, 369–382. [Google Scholar] [CrossRef]
- Fuertes, G.; Banterle, N.; Ruff, K.M.; Chowdhury, A.; Mercadante, D.; Koehler, C.; Kachala, M.; Estrada Girona, G.; Milles, S.; Mishra, A.; et al. Decoupling of size and shape fluctuations in heteropolymeric sequences reconciles discrepancies in Saxs Vs. FRET measurements. Proc. Natl. Acad. Sci. USA 2017, 114, E6342–E6351. [Google Scholar] [CrossRef]
- Riback, J.A.; Bowman, M.A.; Zmyslowski, A.M.; Plaxco, K.W.; Clark, P.L.; Sosnick, T.R. Commonly used FRET fluorophores promote collapse of an otherwise disordered protein. Proc. Natl. Acad. Sci. USA 2019, 116, 8889–8894. [Google Scholar] [CrossRef]
- Sikkema, H.R.; Poolman, B. In silico method for selecting residue pairs for single-molecule microscopy and spectroscopy. Sci. Rep. 2021, 11, 5756. [Google Scholar] [CrossRef]
- Henzler-Wildman, K.A.; Thai, V.; Lei, M.; Ott, M.; Wolf-Watz, M.; Fenn, T.; Pozharski, E.; Wilson, M.A.; Petsko, G.A.; Karplus, M.; et al. Intrinsic motions along an enzymatic reaction trajectory. Nature 2007, 450, 838–844. [Google Scholar] [CrossRef]
- Szalai, A.M.; Siarry, B.; Lukin, J.; Giusti, S.; Unsain, N.; Caceres, A.; Steiner, F.; Tinnefeld, P.; Refojo, D.; Jovin, T.M.; et al. Super-resolution imaging of energy transfer by intensity-based sted-FRET. Nano Lett. 2021, 21, 2296–2303. [Google Scholar] [CrossRef]
- Nettels, D.; Gopich, I.V.; Hoffmann, A.; Schuler, B. Ultrafast dynamics of protein collapse from single-molecule photon statistics. Proc. Natl. Acad. Sci. USA 2007, 104, 2655–2660. [Google Scholar] [CrossRef]
- Borgia, A.; Wensley, B.G.; Soranno, A.; Nettels, D.; Borgia, M.B.; Hoffmann, A.; Pfeil, S.H.; Lipman, E.A.; Clarke, J.; Schuler, B. Localizing internal friction along the reaction coordinate of protein folding by combining ensemble and single-molecule fluorescence spectroscopy. Nat. Commun. 2012, 3, 1195. [Google Scholar] [CrossRef]
- Weninger, K.; Qiu, R.; Ou, E.; Milikisiyants, S.; Sanabria, H.; Smirnova, T.I. SmFRET and deer distance measurements as applied to disordered and structured proteins. Biophys. J. 2016, 110, 559A. [Google Scholar] [CrossRef]
- Krainer, G.; Hartmann, A.; Bogatyr, V.; Nielsen, J.; Schlierf, M.; Otzen, D.E. SDS-induced multi-stage unfolding of a small globular protein through different denatured states revealed by single-molecule fluorescence. Chem. Sci. 2020, 11, 9141–9153. [Google Scholar] [CrossRef]
- Wu, S.; Hong, L.; Wang, Y.; Yu, J.; Yang, J.; Yang, J.; Zhang, H.; Perrett, S. Kinetics of the conformational cycle of Hsp70 reveals the importance of the dynamic and heterogeneous nature of Hsp70 for its function. Proc. Natl. Acad. Sci. USA 2020, 117, 7814–7823. [Google Scholar] [CrossRef]
- Dunker, A.K.; Brown, C.J.; Lawson, J.D.; Iakoucheva, L.M.; Obradovic, Z. Intrinsic disorder and protein function. Biochemistry 2002, 41, 6573–6582. [Google Scholar] [CrossRef]
- Darling, A.L.; Uversky, V.N. Intrinsic disorder and posttranslational modifications: The darker side of the biological dark matter. Front. Genet. 2018, 9, 158. [Google Scholar] [CrossRef]
- Choi, U.B.; Xiao, S.; Wollmuth, L.P.; Bowen, M.E. Effect of Src Kinase Phosphorylation on Disordered C-terminal domain of N-Methyl-D-Aspartic Acid (Nmda) receptor subunit Glun2b protein. J. Biol. Chem. 2011, 286, 29904–29912. [Google Scholar] [CrossRef]
- Goldschen-Ohm, M.P.; White, D.S.; Klenchin, V.A.; Chanda, B.; Goldsmith, R.H. Observing single-molecule dynamics at millimolar concentrations. Angew. Chem. Int. Ed. Engl. 2017, 56, 2399–2402. [Google Scholar] [CrossRef]
- Gomes, G.W.; Krzeminski, M.; Namini, A.; Martin, E.W.; Mittag, T.; Head-Gordon, T.; Forman-Kay, J.D.; Gradinaru, C.C. Conformational ensembles of an intrinsically disordered protein consistent with Nmr, Saxs, and single-molecule FRET. J. Am. Chem. Soc. 2020, 142, 15697–15710. [Google Scholar] [CrossRef]
- Holliday, R. A mechanism for gene conversion in fungi (Reprinted). Genet. Res. 2007, 89, 285–307. [Google Scholar] [CrossRef]
- Bennett, R.J.; West, S.C. Resolution of Holliday junctions in genetic recombination: Ruvc Protein Nicks DNA at the point of strand exchange. Proc. Natl. Acad. Sci. USA 1996, 93, 12217–12222. [Google Scholar] [CrossRef] [PubMed]
- McKinney, S.A.; Declais, A.C.; Lilley, D.M.J.; Ha, T. Structural dynamics of individual Holliday junctions. Nat. Struct. Biol. 2003, 10, 93–97. [Google Scholar] [CrossRef] [PubMed]
- Joo, C.; McKinney, S.A.; Lilley, D.M.J.; Ha, T. Exploring rare conformational species and ionic effects in DNA Holliday junctions using single-molecule spectroscopy. J. Mol. Biol. 2004, 341, 739–751. [Google Scholar] [CrossRef] [PubMed]
- Benson, F.E.; Illing, G.T.; Sharples, G.J.; Lloyd, R.G. Nucleotide Sequencing of the Ruv Region of Escherichia-Coli K-12 reveals a lexa regulated operon encoding 2 genes. Nucleic Acids Res. 1988, 16, 1541–1549. [Google Scholar] [CrossRef] [PubMed]
- Yamada, K.; Miyata, T.; Tsuchiya, D.; Oyama, T.; Fujiwara, Y.; Ohnishi, T.; Iwasaki, H.; Shinagawa, H.; Ariyoshi, M.; Mayanagi, K.; et al. Crystal structure of the Ruva-Ruvb complex: A structural basis for the Holliday junction migrating motor machinery. Mol. Cell 2002, 10, 671–681. [Google Scholar] [CrossRef]
- Gibbs, D.R.; Dhakal, S. Single-molecule imaging reveals conformational manipulation of Holliday junction DNA by the junction processing protein RuvA. Biochemistry 2018, 57, 3616–3624. [Google Scholar] [CrossRef] [PubMed]
- Ray, S.; Pal, N.; Walter, N.G. Single bacterial resolvases first exploit, then constrain intrinsic dynamics of the Holliday junction to direct recombination. Nucleic Acids Res. 2021, 49, 2803–2815. [Google Scholar] [CrossRef]
- Bellendir, S.P.; Rognstad, D.J.; Morris, L.P.; Zapotoczny, G.; Walton, W.G.; Redinbo, M.R.; Ramsden, D.A.; Sekelsky, J.; Erie, D.A. Substrate preference of gen endonucleases highlights the importance of branched structures as DNA damage repair intermediates. Nucleic Acids Res. 2017, 45, 5333–5348. [Google Scholar] [CrossRef]
- Gibbs, D.R.; Dhakal, S. Homologous recombination under the single-molecule fluorescence microscope. Int. J. Mol. Sci. 2019, 20, 6102. [Google Scholar] [CrossRef]
- Sobhy, M.A.; Bralic, A.; Raducanu, V.S.; Takahashi, M.; Tehseen, M.; Rashid, F.; Zaher, M.S.; Hamdan, S.M. Resolution of the Holliday Junction recombination intermediate by human Gen1 at the single-molecule level. Nucleic Acids Res. 2019, 47, 1935–1949. [Google Scholar] [CrossRef]
- Leveille, M.P.; Tran, T.; Dingillo, G.; Cannon, B. Detection of Mg2+-Dependent, Coaxial Stacking Rearrangements in a Bulged Three-Way DNA junction by single-molecule FRET. Biophys. Chem. 2019, 245, 25–33. [Google Scholar] [CrossRef]
- Hu, T.; Morten, M.J.; Magennis, S.W. Conformational and migrational dynamics of slipped-strand DNA three-way junctions containing trinucleotide repeats. Nat. Commun. 2021, 12, 204. [Google Scholar] [CrossRef]
- Shaw, E.; St-Pierre, P.; McCluskey, K.; Lafontaine, D.A.; Penedo, J.C. Using Sm-FRET and denaturants to reveal folding landscapes. In Riboswitch Discovery, Structure and Function; BurkeAguero, D.H., Ed.; Academic Press: Cambridge, MA, USA, 2014; Volume 549, pp. 313–341. [Google Scholar]
- Elenko, M.P.; Szostak, J.W.; van Oijen, A.M. Single-molecule imaging of an in vitro-evolved Rna aptamer reveals homogeneous ligand binding kinetics. J. Am. Chem. Soc. 2009, 131, 9866–9867. [Google Scholar] [CrossRef]
- Mundigala, H.; Michaux, J.B.; Feig, A.L.; Ennifar, E.; Rueda, D. HIV-1 dis stem loop forms an obligatory bent kissing intermediate in the dimerization pathway. Nucleic Acids Res. 2014, 42, 7281–7289. [Google Scholar] [CrossRef]
- Hodak, J.H.; Fiore, J.L.; Nesbitt, D.J.; Downey, C.D.; Pardi, A. Docking kinetics and equilibrium of a Gaaa tetra loop-receptor motif probed by single-molecule FRET. Proc. Natl. Acad. Sci. USA 2005, 102, 10505–10510. [Google Scholar] [CrossRef]
- Walter, F.; Murchie, A.I.H.; Lilley, D.M.J. Folding of the four-way Rna junction of the hairpin ribozyme. Biochemistry 1998, 37, 17629–17636. [Google Scholar] [CrossRef]
- Wang, Y.; Xiao, M.; Li, Y. Heterogeneity of Single Molecule FRET signals reveals multiple active ribosome subpopulations. Proteins Struct. Funct. Bioinform. 2014, 82, 1–9. [Google Scholar] [CrossRef]
- Cisse, I.I.; Kim, H.; Ha, T. A rule of seven in Watson-Crick base-pairing of mismatched sequences. Nature Struct. Mol. Biol. 2012, 19, 623–627. [Google Scholar] [CrossRef]
- Bisaria, N.; Herschlag, D. Probing the kinetic and thermodynamic consequences of the tetraloop/tetraloop receptor monovalent ion-binding site in P4-P6 Rna by SmFRET. Biochem. Soc. Trans. 2015, 43, 172–178. [Google Scholar] [CrossRef]
- Sengupta, A.; Sung, H.L.; Nesbitt, D.J. Amino acid specific effects on RNA tertiary interactions: Single-molecule kinetic and thermodynamic studies. J. Phys. Chem. B 2016, 120, 10615–10627. [Google Scholar] [CrossRef]
- Nicholson, D.A.; Sengupta, A.; Nesbitt, D.J. Chirality-dependent amino acid modulation of RNA folding. J. Phys. Chem. B 2020, 124, 11561–11572. [Google Scholar] [CrossRef]
- Holmstrom, E.D.; Dupuis, N.F.; Nesbitt, D.J. Kinetic and thermodynamic origins of osmolyte-influenced nucleic acid folding. J. Phys. Chem. B 2015, 119, 3687–3696. [Google Scholar] [CrossRef]
- Ueyama, H.; Takagi, M.; Takenaka, S. A novel potassium sensing in aqueous media with a synthetic oligonucleotide derivative. Fluorescence resonance energy transfer associated with guanine quartet-potassium ion complex formation. J. Am. Chem. Soc. 2002, 124, 14286–14287. [Google Scholar] [CrossRef]
- Luo, Y.; Granzhan, A.; Verga, D.; Mergny, J.L. FRET-Mc: A fluorescence melting competition assay for studying G4 Structures in Vitro. Biopolymers 2020. [Google Scholar] [CrossRef]
- Zhao, H.; Hu, W.; Jing, J.; Zhang, X. One-step G-quadruplex-based fluorescence resonance energy transfer sensing method for ratiometric detection of uracil-DNA glycosylase activity. Talanta 2021, 21. [Google Scholar] [CrossRef]
- Long, X.; Stone, M.D. Kinetic partitioning modulates human telomere DNA G-quadruplex structural polymorphism. PLoS ONE 2013, 8, e83420. [Google Scholar] [CrossRef]
- Mustafa, G.; Shiekh, S.; Gc, K.; Abeysirigunawardena, S.; Balci, H. Interrogating accessibility of telomeric sequences with FRET-PAINT: Evidence for length-dependent telomere compaction. Nucleic Acids Res. 2021, 49, 3371–3380. [Google Scholar] [CrossRef] [PubMed]
- Paudel, B.P.; Moye, A.L.; Abou Assi, H.; El-Khoury, R.; Cohen, S.B.; Holien, J.K.; Birrento, M.L.; Samosorn, S.; Intharapichai, K.; Tomlinson, C.G.; et al. A Mechanism for the Extension and Unfolding of Parallel Telomeric G-quadruplexes by human telomerase at single-molecule resolution. eLife 2020, 9, e56428. [Google Scholar] [CrossRef] [PubMed]
- Parks, J.W.; Kappel, K.; Das, R.; Stone, M.D. Single-molecule FRET-Rosetta Reveals Rna structural rearrangements during human telomerase catalysis. RNA 2017, 23, 175–188. [Google Scholar] [CrossRef] [PubMed]
- Fyodorov, D.V.; Zhou, B.R.; Skoultchi, A.I.; Bai, Y. Emerging roles of linker histones in regulating chromatin structure and function. Nat. Rev. Mol. Cell Biol. 2018, 19, 192–206. [Google Scholar] [CrossRef]
- Rudnizky, S.; Malik, O.; Bavly, A.; Pnueli, L.; Melamed, P.; Kaplan, A. Nucleosome mobility and the regulation of gene expression: Insights from single-molecule studies. Protein Sci. 2017, 26, 1266–1277. [Google Scholar] [CrossRef]
- Tomschik, M.; Zheng, H.C.; van Holde, K.; Zlatanova, J.; Leuba, S.H. Fast, long-range, reversible conformational fluctuations in nucleosomes revealed by single-pair fluorescence resonance energy transfer. Proc. Natl. Acad. Sci. USA 2005, 102, 3278–3283. [Google Scholar] [CrossRef]
- Gansen, A.; Valeri, A.; Hauger, F.; Felekyan, S.; Kalinin, S.; Toth, K.; Langowski, J.; Seidel, C.A.M. Nucleosome disassembly intermediates characterized by single-molecule FRET. Proc. Natl. Acad. Sci. USA 2009, 106, 15308–15313. [Google Scholar] [CrossRef]
- Toth, K.; Bohm, V.; Sellmann, C.; Danner, M.; Hanne, J.; Berg, M.; Barz, I.; Gansen, A.; Langowski, J. Histone- and DNA sequence-dependent stability of nucleosomes studied by single-pair FRET. Cytom. Part A 2013, 83, 839–846. [Google Scholar] [CrossRef]
- Sultanov, D.C.; Gerasimova, N.S.; Kudryashova, K.S.; Maluchenko, N.V.; Kotova, E.Y.; Langelier, M.F.; Pascal, J.M.; Kirpichnikov, M.P.; Feofanov, A.V.; Studitsky, V.M. Unfolding of core nucleosomes by Parp-1 revealed by SpFRET microscopy. AIMS Genet. 2017, 4, 21–31. [Google Scholar] [CrossRef]
- Gansen, A.; Felekyan, S.; Kuhnemuth, R.; Lehmann, K.; Toth, K.; Seidel, C.A.M.; Langowski, J. High precision FRET studies reveal reversible transitions in nucleosomes between microseconds and minutes. Nat. Commun. 2018, 9, 4628. [Google Scholar] [CrossRef]
- Lehmann, K.; Felekyan, S.; Kuhnemuth, R.; Dimura, M.; Toth, K.; Seidel, C.A.M.; Langowski, J. Dynamics of the nucleosomal histone H3 N-terminal tail revealed by high precision single-molecule FRET. Nucleic Acids Res. 2020, 48, 1551–1571. [Google Scholar] [CrossRef]
- Lou, J.; Scipioni, L.; Wright, B.K.; Bartolec, T.K.; Zhang, J.; Masamsetti, V.P.; Gaus, K.; Gratton, E.; Cesare, A.J.; Hinde, E. Phasor histone flim-FRET Microscopy Quantifies Spatiotemporal Rearrangement of Chromatin Architecture During the DNA damage response. Proc. Natl. Acad. Sci. USA 2019, 116, 7323–7332. [Google Scholar] [CrossRef]
- Harrison, S.C. Viral membrane fusion. Nat. Struct. Mol. Biol. 2008, 15, 690–698. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krueger, N.; Herrler, T.; Erichsen, S.; Tobias, S.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-COV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 2020, 181, 271–280. [Google Scholar] [CrossRef]
- Walls, A.C.; Park, Y.J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, function, and antigenicity of the SARS-COV-2 spike glycoprotein. Cell 2020, 181, 281–292. [Google Scholar] [CrossRef]
- Lu, M.; Uchil, P.D.; Li, W.; Zheng, D.; Terry, D.S.; Gorman, J.; Shi, W.; Zhang, B.; Zhou, T.; Ding, S.; et al. Real-time conformational dynamics of SARS-COV-2 spikes on virus particles. Cell Host Microbe 2020, 28, 880–891. [Google Scholar] [CrossRef]
- Salata, C.; Calistri, A.; Alvisi, G.; Celestino, M.; Parolin, C.; Palu, G. Ebola virus entry: From molecular characterization to drug discovery. Viruses 2019, 11, 274. [Google Scholar] [CrossRef]
- Moller-Tank, S.; Maury, W. Ebola virus entry: A curious and complex series of events. PLoS Pathog. 2015, 11, 4. [Google Scholar] [CrossRef]
- Carette, J.E.; Raaben, M.; Wong, A.C.; Herbert, A.S.; Obernosterer, G.; Mulherkar, N.; Kuehne, A.I.; Kranzusch, P.J.; Griffin, A.M.; Ruthel, G.; et al. Ebola virus entry requires the cholesterol transporter Niemann-Pick C1. Nature 2011, 477, 340–343. [Google Scholar] [CrossRef]
- Cote, M.; Misasi, J.; Ren, T.; Bruchez, A.; Lee, K.; Filone, C.M.; Hensley, L.; Li, Q.; Ory, D.; Chandran, K.; et al. Small molecule inhibitors reveal Niemann-Pick C1 is essential for Ebola virus infection. Nature 2011, 477, 344–348. [Google Scholar] [CrossRef]
- Das, D.K.; Bulow, U.; Diehl, W.E.; Durham, N.D.; Senjobe, F.; Chandran, K.; Luban, J.; Munro, J.B. Conformational changes in the Ebola virus membrane fusion machine induced by pH, Ca2+, and receptor binding. PLoS Biol. 2020, 18. [Google Scholar] [CrossRef]
- Borgia, A.; Williams, P.M.; Clarke, J. Single-molecule studies of protein folding. Annu. Rev. Biochem. 2008, 77, 101–125. [Google Scholar] [CrossRef]
- Otzen, D. Protein-surfactant interactions: A tale of many states. Biochim. Biophys. Acta Proteins Proteom. 2011, 1814, 562–591. [Google Scholar] [CrossRef] [PubMed]
- Yano, Y.; Kondo, K.; Kitani, R.; Yamamoto, A.; Matsuzaki, K. Cholesterol-induced lipophobic interaction between transmembrane helices using ensemble and single-molecule fluorescence resonance energy transfer. Biochemistry 2015, 54, 1371–1379. [Google Scholar] [CrossRef] [PubMed]
- Yano, Y.; Kondo, K.; Watanabe, Y.; Zhang, T.O.; Ho, J.-J.; Oishi, S.; Fujii, N.; Zanni, M.T.; Matsuzaki, K. GXXXG-mediated parallel and antiparallel dimerization of transmembrane helices and its inhibition by cholesterol: Single-pair FRET and 2D IR studies. Angew. Chem. 2017, 56, 1756–1759. [Google Scholar] [CrossRef] [PubMed]
- Krainer, G.; Treff, A.; Hartmann, A.; Stone, T.A.; Schenkel, M.; Keller, S.; Deber, C.M.; Schlierf, M. A minimal helical-hairpin motif provides molecular-level insights into misfolding and pharmacological rescue of CFTR. Commun. Biol. 2018, 1. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Brettmann, J.B.; Nichols, C.G. Studying structural dynamics of potassium channels by single-molecule FRET. Methods Mol. Biol. 2018, 1684, 163–180. [Google Scholar] [CrossRef]
- Kedrov, A.; Sustarsic, M.; de Keyzer, J.; Caumanns, J.J.; Wu, Z.C.; Driessen, A.J.M. Elucidating the native architecture of the YidC: Ribosome complex. J. Mol. Biol. 2013, 425, 4112–4124. [Google Scholar] [CrossRef]
- Taufik, I.; Kedrov, A.; Exterkate, M.; Driessen, A.J.M. Monitoring the activity of single translocons. J. Mol. Biol. 2013, 425, 4145–4153. [Google Scholar] [CrossRef][Green Version]
- Winckler, P.; Lartigue, L.; Giannone, G.; De Giorgi, F.; Ichas, F.; Sibarita, J.-B.; Lounis, B.; Cognet, L. Identification and super-resolution imaging of ligand-activated receptor dimers in live cells. Sci. Rep. 2013, 3. [Google Scholar] [CrossRef]
- Zaman, M.; Khan, A.N.; Wahiduzzaman; Zakariya, S.M.; Khan, R.H. Protein misfolding, aggregation and mechanism of amyloid cytotoxicity: An overview and therapeutic strategies to inhibit aggregation. Int. J. Biol. Macromol. 2019, 134, 1022–1037. [Google Scholar] [CrossRef]
- Birol, M.; Melo, A.M. Untangling the conformational polymorphism of disordered proteins associated with neurodegeneration at the single-molecule level. Front. Mol. Neurosci. 2019, 12, 309. [Google Scholar] [CrossRef]
- Wickramasinghe, S.P.; Lempart, J.; Merens, H.E.; Murphy, J.; Huettemann, P.; Jakob, U.; Rhoades, E. Polyphosphate Initiates Tau Aggregation through Intra- and Intermolecular Scaffolding. Biophys. J. 2019, 117, 717–728. [Google Scholar] [CrossRef]
- Melo, A.M.; Coraor, J.; Alpha-Cobb, G.; Elbaum-Garfinkle, S.; Nath, A.; Rhoades, E. A functional role for intrinsic disorder in the tau-tubulin complex. Proc. Natl. Acad. Sci. USA 2016, 113, 14336–14341. [Google Scholar] [CrossRef]
- Elbaum-Garfinkle, S.; Rhoades, E. Identification of an aggregation-prone structure of tau. J. Am. Chem. Soc. 2012, 134, 16607–16613. [Google Scholar] [CrossRef]
- Cremades, N.; Cohen, S.I.A.; Deas, E.; Abramov, A.Y.; Chen, A.Y.; Orte, A.; Sandal, M.; Clarke, R.W.; Dunne, P.; Aprile, F.A.; et al. Direct observation of the interconversion of normal and toxic forms of alpha-synuclein. Cell 2012, 149, 1048–1059. [Google Scholar] [CrossRef]
- Horrocks, M.H.; Tosatto, L.; Dear, A.J.; Garcia, G.A.; Iljina, M.; Cremades, N.; Dalla Serra, M.; Knowles, T.P.; Dobson, C.M.; Klenerman, D. Fast flow microfluidics and single-molecule fluorescence for the rapid characterization of alpha-synuclein oligomers. Anal. Chem. 2015, 87, 8818–8826. [Google Scholar] [CrossRef]
- Christian, T.D.; Romano, L.J.; Rueda, D. Single-molecule measurements of synthesis by DNA polymerase with base-pair resolution. Proc. Natl. Acad. Sci. USA 2009, 106, 21109–21114. [Google Scholar] [CrossRef]
- Fijen, C.; Monton Silva, A.; Hochkoeppler, A.; Hohlbein, J. A single-molecule FRET sensor for monitoring DNA synthesis in real time. Phys. Chem. Chem. Phys. 2017, 19, 4222–4230. [Google Scholar] [CrossRef]
- Pauszek, R.F., 3rd; Lamichhane, R.; Rajkarnikar Singh, A.; Millar, D.P. Single-molecule view of coordination in a multi-functional DNA polymerase. eLife 2021, 10. [Google Scholar] [CrossRef]
- Cognet, L.; Leduc, C.; Lounis, B. Advances in live-cell single-particle tracking and dynamic super-resolution imaging. Curr. Opin. Chem. Biol. 2014, 20, 78–85. [Google Scholar] [CrossRef]
- Avruch, J.; Zhang, X.F.; Kyriakis, J.M. Raf meets Ras–Completing the framework of a signal-transduction pathway. Trends Biochem. Sci. 1994, 19, 279–283. [Google Scholar] [CrossRef]
- Daum, G.; Eisenmanntappe, I.; Fries, H.W.; Troppmair, J.; Rapp, U.R. The ins and outs of Raf kinases. Trends Biochem. Sci. 1994, 19, 474–480. [Google Scholar] [CrossRef]
- Murakoshi, H.; Iino, R.; Kobayashi, T.; Fujiwara, T.; Ohshima, C.; Yoshimura, A.; Kusumi, A. Single-molecule imaging analysis of Ras activation in living cells. Proc. Natl. Acad. Sci. USA 2004, 101, 7317–7322. [Google Scholar] [CrossRef]
- Hibino, K.; Shibata, T.; Yanagida, T.; Sako, Y. A RasGTP-induced conformational change in C-Raf is essential for accurate molecular recognition. Biophys. J. 2009, 97, 1277–1287. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, K.; Hibino, K.; Sako, Y. In-cell single-molecule FRET measurements reveal three conformational state changes in RAF protein. Biochim. Biophys. Acta Gen. Subj. 2020, 1864, 129358. [Google Scholar] [CrossRef] [PubMed]
- Sako, Y.; Minoguchi, S.; Yanagida, T. Single-molecule imaging of EGFR signalling on the surface of living cells. Nat. Cell Biol. 2000, 2, 168–172. [Google Scholar] [CrossRef]
- Asher, W.B.; Geggier, P.; Holsey, M.D.; Gilmore, G.T.; Pati, A.K.; Meszaros, J.; Terry, D.S.; Mathiasen, S.; Kaliszewski, M.J.; McCauley, M.D.; et al. Single-molecule FRET imaging of GPCR dimers in living cells. Nat. Methods 2021. [Google Scholar] [CrossRef]
- Di Antonio, M.; Ponjavic, A.; Radzevicius, A.; Ranasinghe, R.T.; Catalano, M.; Zhang, X.; Shen, J.; Needham, L.M.; Lee, S.F.; Klenerman, D.; et al. Single-molecule visualization of DNA G-quadruplex formation in live cells. Nat. Chem. 2020, 12, 832–837. [Google Scholar] [CrossRef]
- Zou, Y.; Long, S.; Xiong, T.; Zhao, X.; Sun, W.; Du, J.; Fan, J.; Peng, X. Single-molecule forster resonance energy transfer-based photosensitizer for synergistic photodynamic/photothermal therapy. ACS Cent. Sci. 2021, 7, 327–334. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Variables | Key Findings and Significances | Ref. |
|---|---|---|---|
| Proteins with various states | SDS concentration | Transition among several sites was described under different solutions within a large concentration range | [74] |
| Posttranslational modification | Domains were immobilized to record the conformational change | [75,78] | |
| Protein regulation | Transition states of Hsp70 chaperone cycle was shown to associate with various activity levels | [75,78] | |
| The concentration of sample was increased largely with parallel small chambers of ZMW | [79] | ||
| DNA secondary structure | Solvent condition | Dynamics intrinsic to HJ were analyzed to find cognate sequence and achieve site-specific cleavage | [88] |
| Kinetic details of GEN1 dimers decomposed HJ were explained | [91] | ||
| Strand sequence | A model of reversible branch migration in mobile 3WJ with trinucleotide repeats was proposed, which may help the treatment of diseases | [93] | |
| RNA secondary structure | Metal cation | Na+ and K+ were proved to facilitate the formation of RNA tetraloop–tetraloop receptor tertiary motif | [101] |
| Small molecules in solution | Arginine and lysine interacted with nucleic acids in a manner similar to monovalent cations, and arginine had strong chirality dependence on the inhibition of TL-TLR folding | [102,103] | |
| TMAO and urea were demonstrated to alter the nucleic acid folding by osmotic pressure | [104] | ||
| Chromosome Structure | Strand Sequences | PNA–probe binding was detected repeatedly to evaluate the accessibility in the telomere area | [109] |
| Presence of telomerase | Enzymatic destruction of telomere was described and computer simulation was coupled | [110,111] | |
| Solvent condition | Primer-labeling method used to determine the dynamics of single nucleosomes | [114,115,116,117] | |
| DNA damage | Chromatin function in DNA damage response was observed dynamically in living cells | [89,120] |
| Interaction | Bioprocess | Key Findings and Significances | Ref. |
|---|---|---|---|
| Protein–Protein | Virus infection | smFRET imaging assay revealed structure arrangement of critical binding domains between the membrane receptor and virus spike | [124,129] |
| Protein aggregation | Intramolecular FRET showed the states of aggregation in various conditions quantitatively | [141,142,143,144,145] | |
| Intermolecular FRET exhibited the aggregating principle | [144,145] | ||
| Protein–Lipid | Cross-membrane transport | smFRET with long critical transfer distance detected both parallel and antiparallel dimers of transmembrane helix regulated by cholesterol | [132,133] |
| Protein–Nucleic acid | DNA synthesis | Drops in FRET efficiency indicated DNA synthesis with label on polymerase or on template | [146,147] |
| Various labeling strategies can present the states of different active domains on polymerase | [148] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qiao, Y.; Luo, Y.; Long, N.; Xing, Y.; Tu, J. Single-Molecular Förster Resonance Energy Transfer Measurement on Structures and Interactions of Biomolecules. Micromachines 2021, 12, 492. https://doi.org/10.3390/mi12050492
Qiao Y, Luo Y, Long N, Xing Y, Tu J. Single-Molecular Förster Resonance Energy Transfer Measurement on Structures and Interactions of Biomolecules. Micromachines. 2021; 12(5):492. https://doi.org/10.3390/mi12050492
Chicago/Turabian StyleQiao, Yi, Yuhan Luo, Naiyun Long, Yi Xing, and Jing Tu. 2021. "Single-Molecular Förster Resonance Energy Transfer Measurement on Structures and Interactions of Biomolecules" Micromachines 12, no. 5: 492. https://doi.org/10.3390/mi12050492
APA StyleQiao, Y., Luo, Y., Long, N., Xing, Y., & Tu, J. (2021). Single-Molecular Förster Resonance Energy Transfer Measurement on Structures and Interactions of Biomolecules. Micromachines, 12(5), 492. https://doi.org/10.3390/mi12050492