Gelatin-Coated Microfluidic Channels for 3D Microtissue Formation: On-Chip Production and Characterization




Abstract
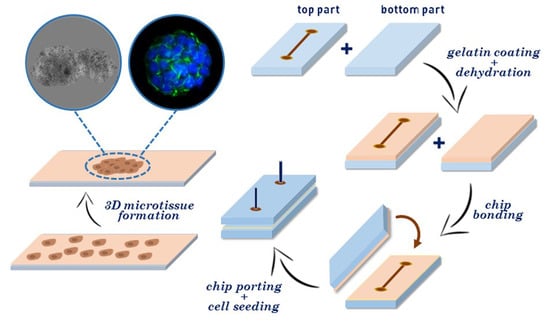
1. Introduction
2. Materials and Methods
2.1. Materials and Equipment
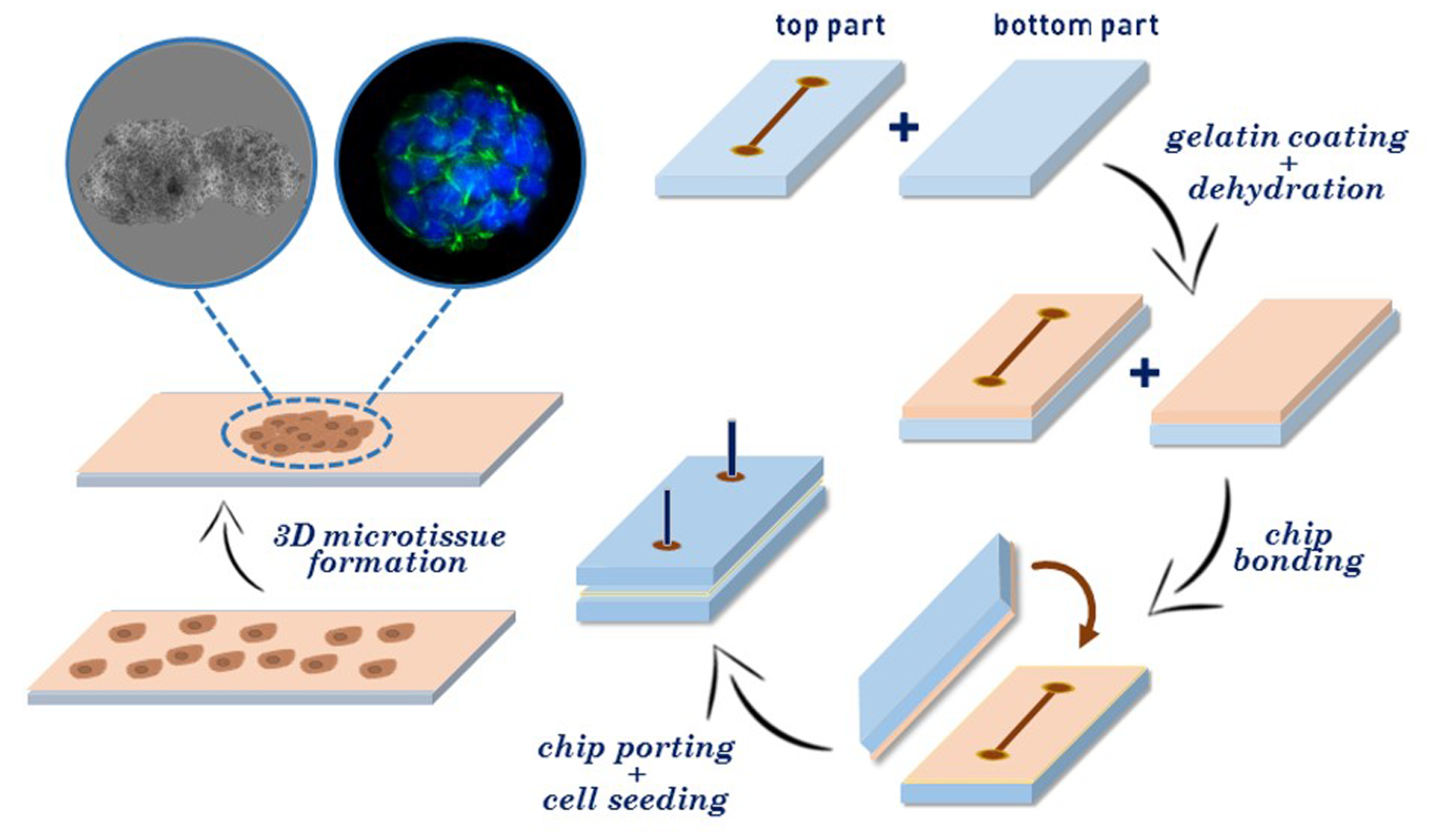
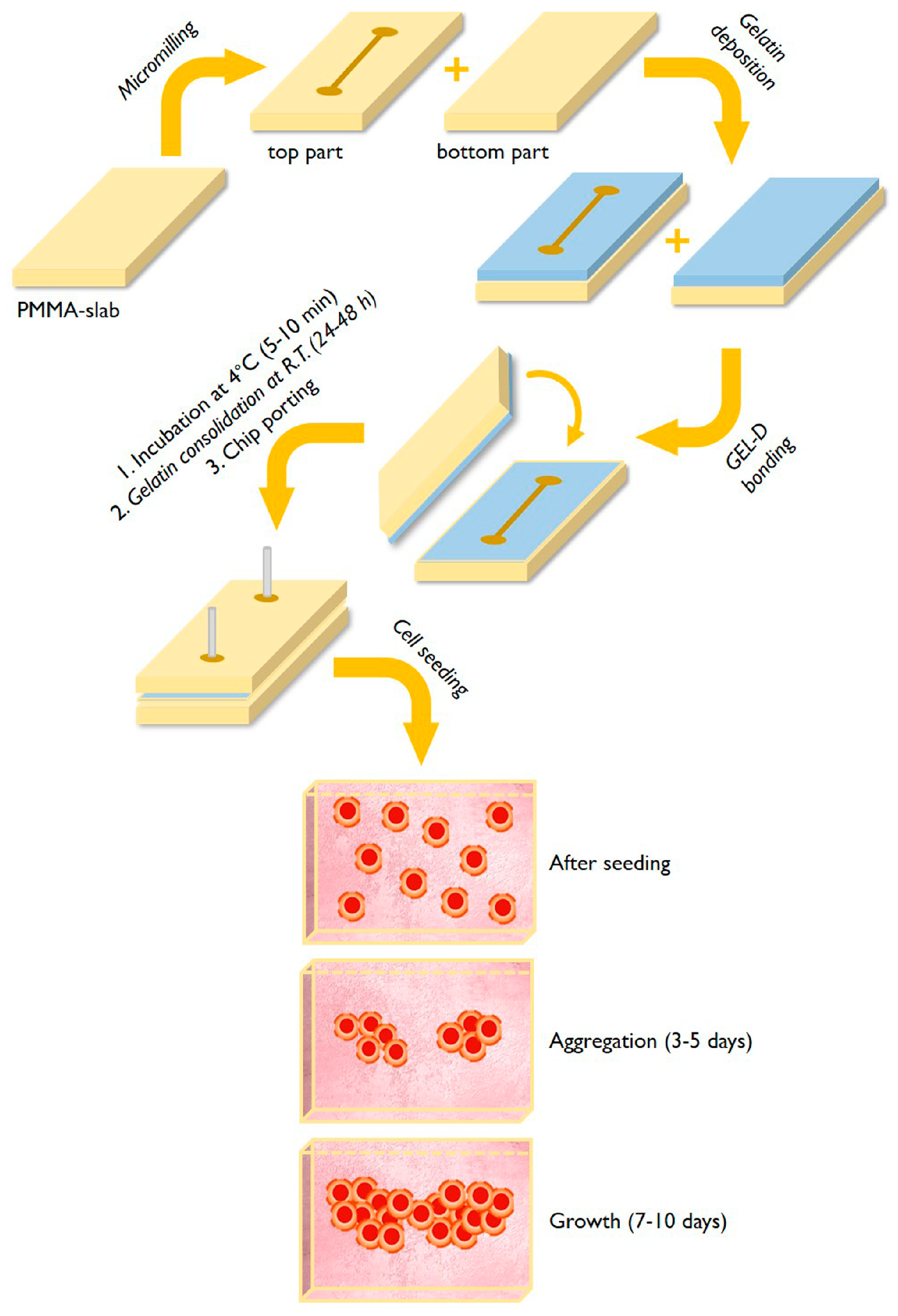
2.2. Fabrication of Microfluidic Chips and Gelatin Coating
2.3. Cell Seeding and Characterization
3. Results and Discussions
3.1. Fabrication of Gelatin-Coated Microfluidic Channels
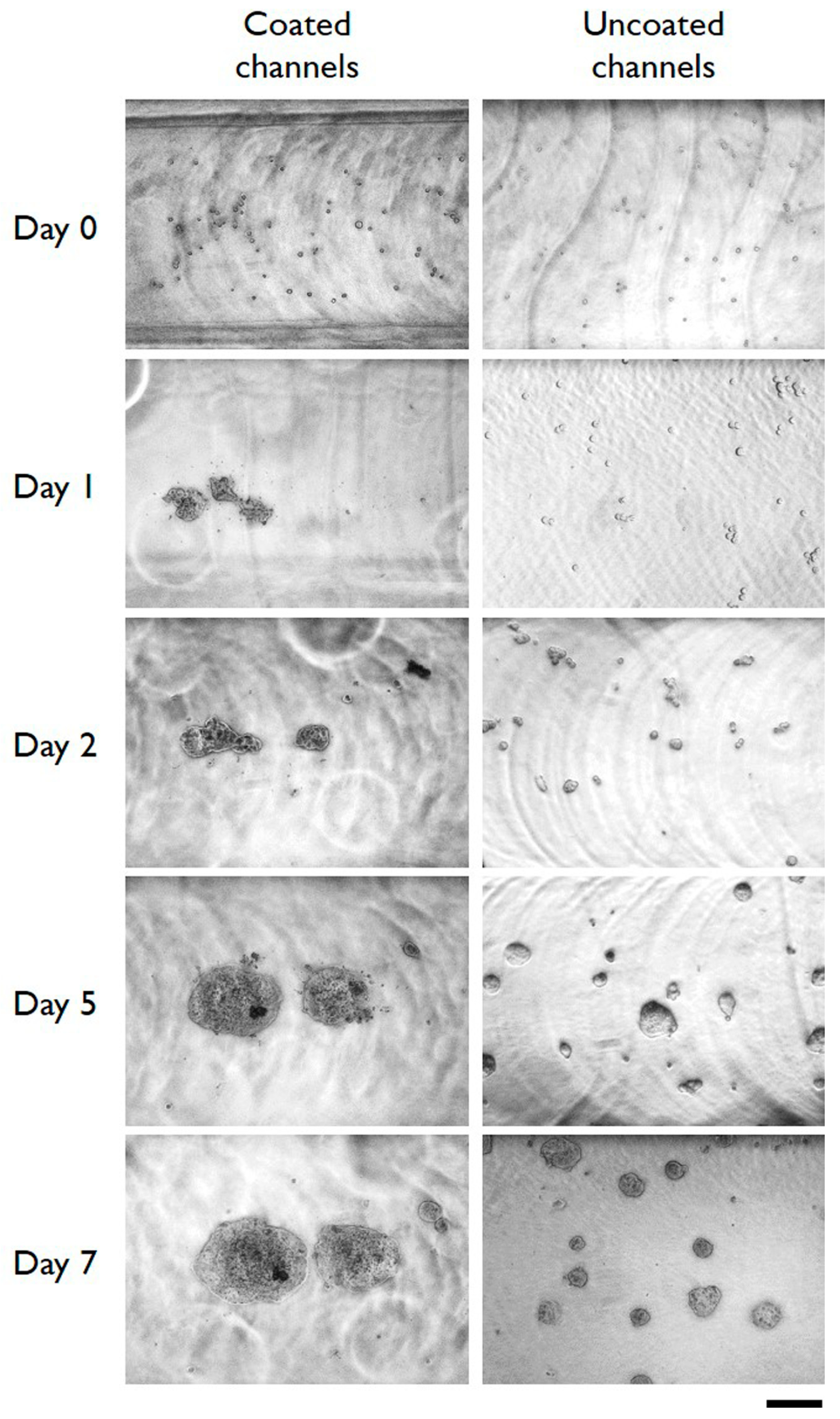
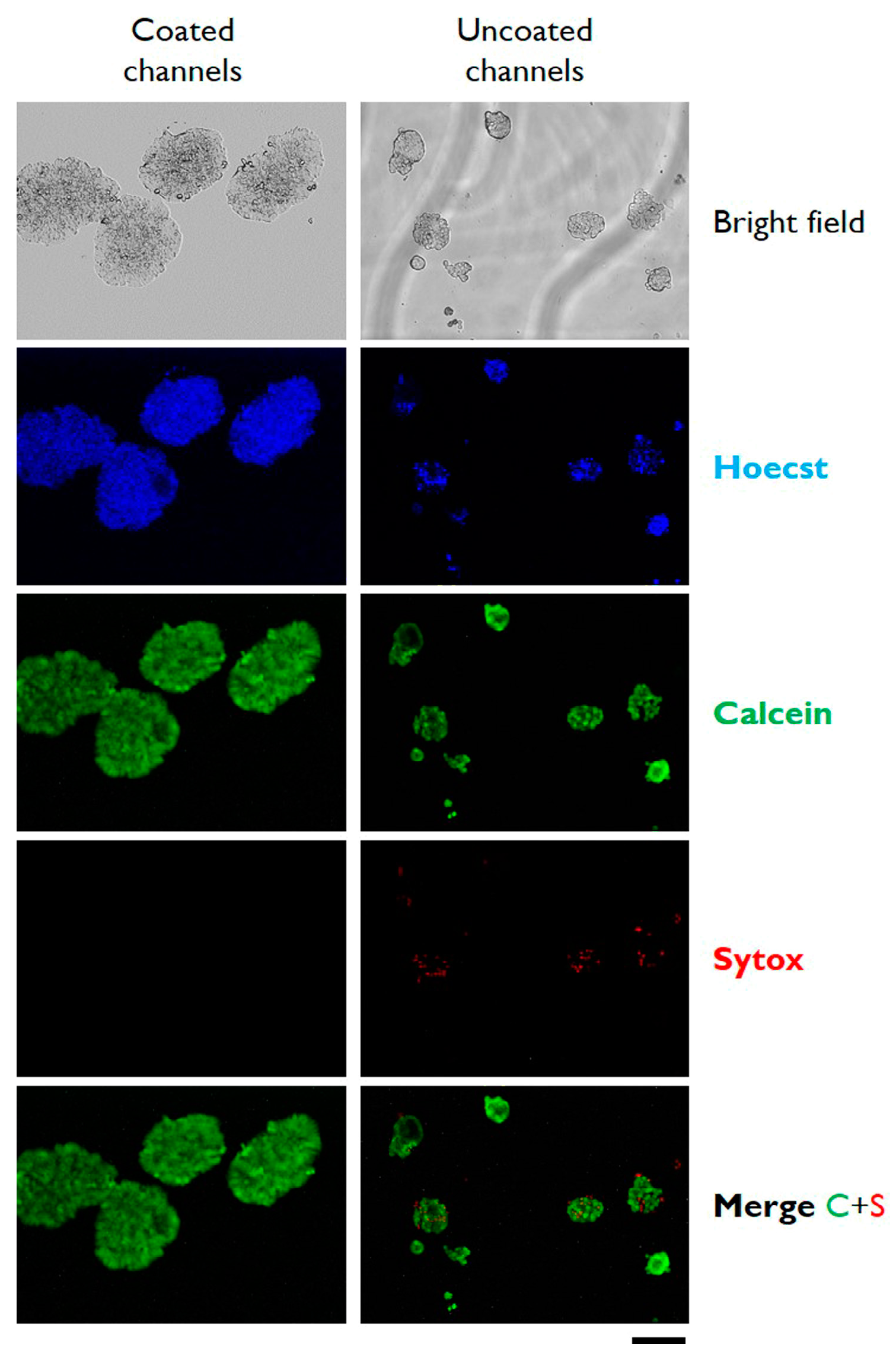
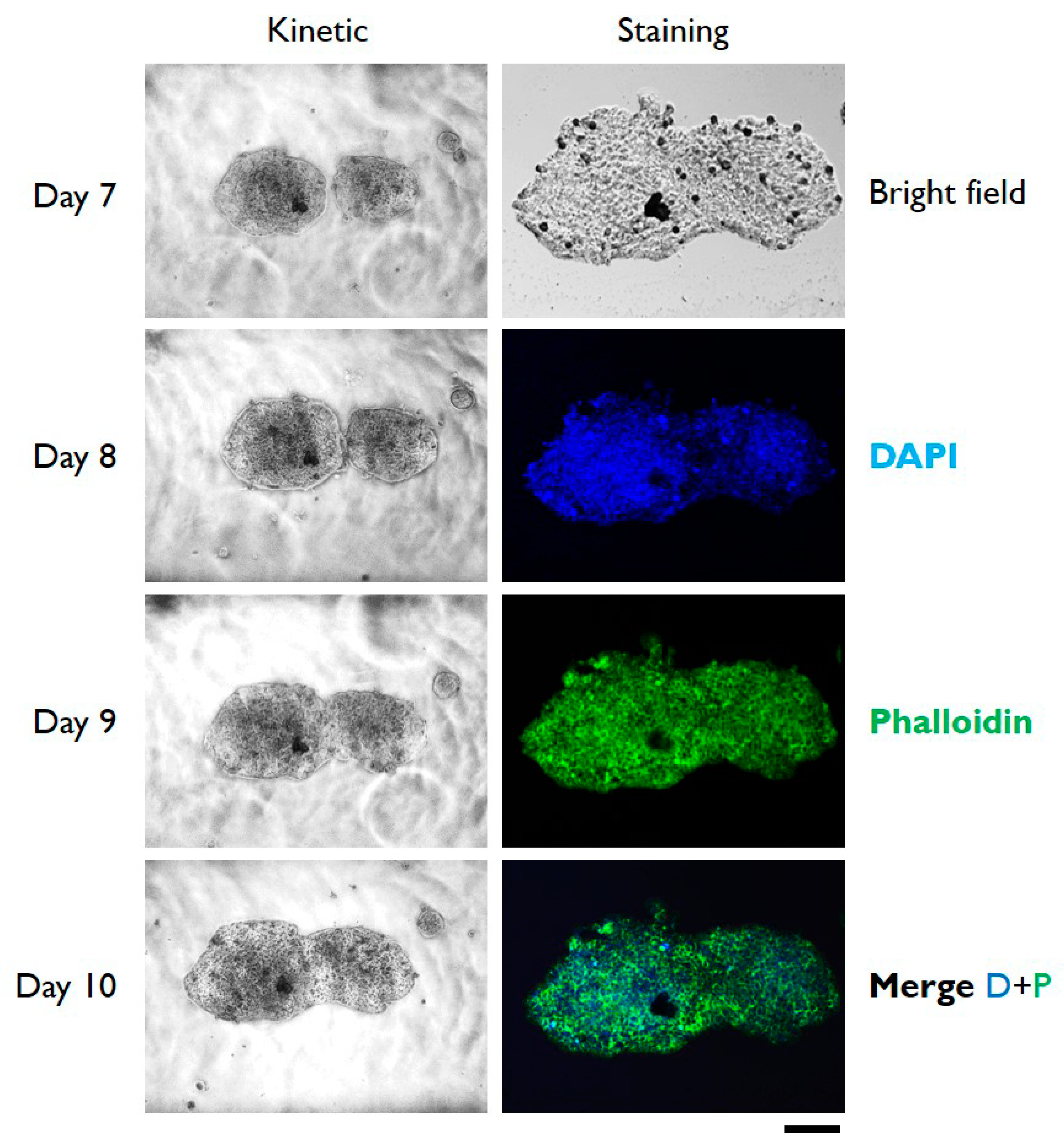
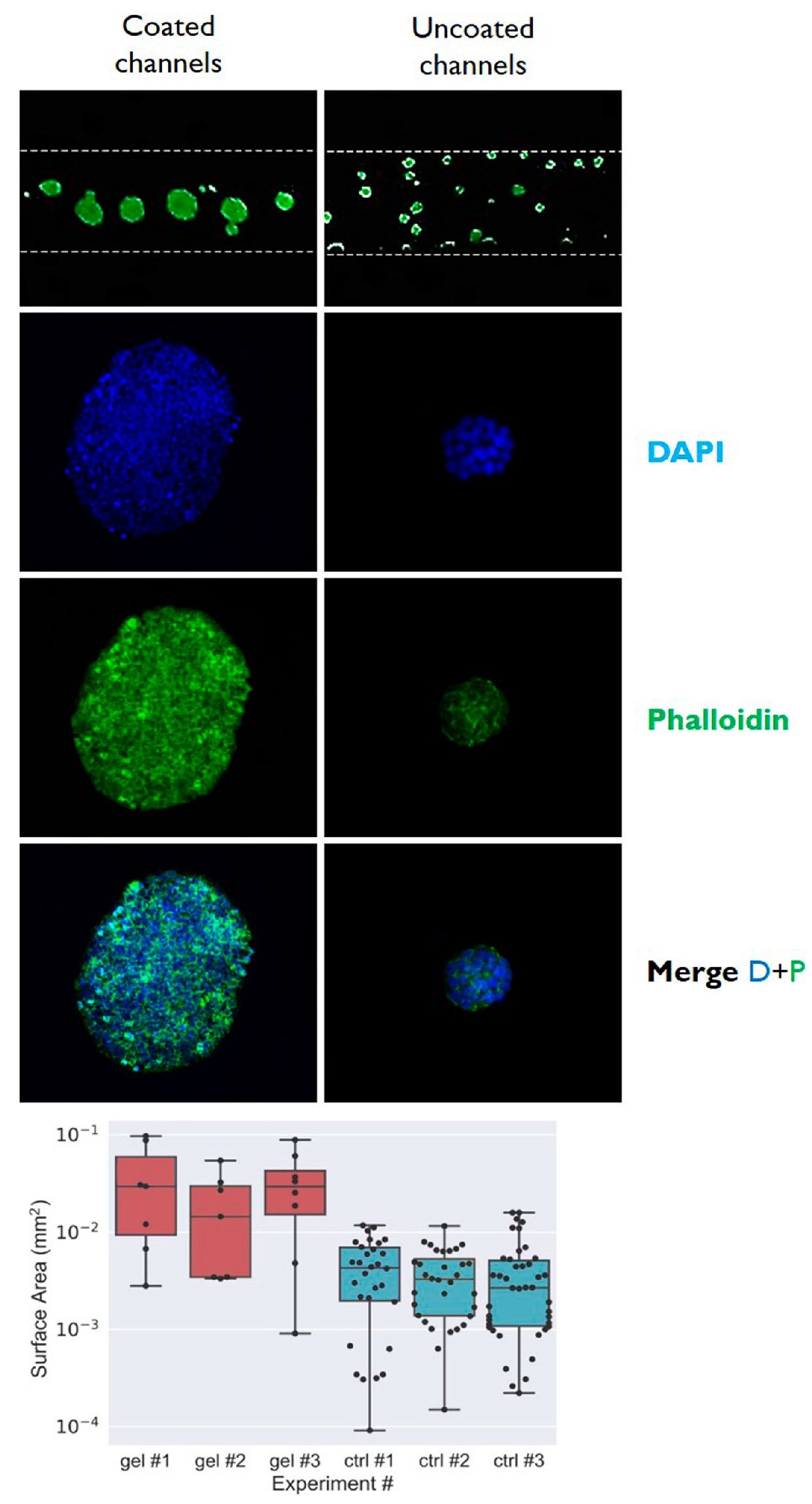
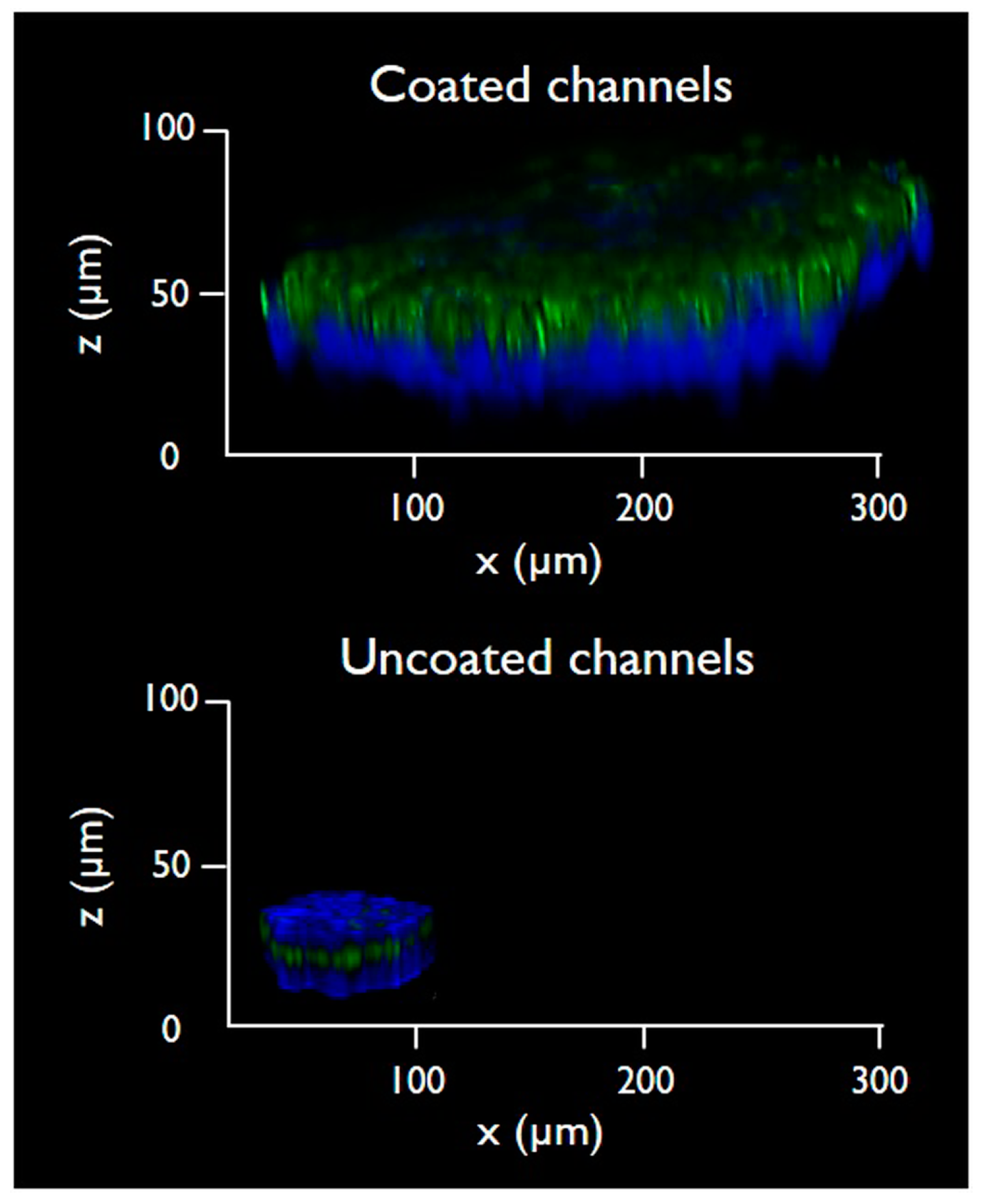
3.2. Cells Aggregation and 3D Microtissue Formation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Pampaloni, F.; Reynaud, E.G.; Stelzer, E.H. The third dimension bridges the gap between cell culture and live tissue. Nat. Rev. Mol. Cell Biol. 2007, 8, 839. [Google Scholar] [CrossRef] [PubMed]
- Huh, D.; Hamilton, G.A.; Ingber, D.E. From 3D cell culture to organs-on-chips. Trends Cell Biol. 2011, 21, 745–754. [Google Scholar] [CrossRef]
- Dolznig, H.; Rupp, C.; Puri, C.; Haslinger, C.; Schweifer, N.; Wieser, E.; Kerjaschki, D.; Garin-Chesa, P. Modeling colon adenocarcinomas in vitro: A 3D co-culture system induces cancer-relevant pathways upon tumor cell and stromal fibroblast interaction. Am. J. Pathol. 2011, 179, 487–501. [Google Scholar] [CrossRef]
- Fischbach, C.; Kong, H.J.; Hsiong, S.X.; Evangelista, M.B.; Yuen, W.; Mooney, D.J. Cancer cell angiogenic capability is regulated by 3D culture and integrin engagement. Proc. Natl. Acad. Sci. USA 2009, 106, 399–404. [Google Scholar] [CrossRef]
- Pickl, M.; Ries, C. Comparison of 3D and 2D tumor models reveals enhanced HER2 activation in 3D associated with an increased response to trastuzumab. Oncogene 2009, 28, 461. [Google Scholar] [CrossRef] [PubMed]
- Takagi, A.; Watanabe, M.; Ishii, Y.; Morita, J.; Hirokawa, Y.; Matsuzaki, T.; Shiraishi, T. Three-dimensional cellular spheroid formation provides human prostate tumor cells with tissue-like features. Anticancer Res. 2007, 27, 45–53. [Google Scholar]
- Desoize, B.; Jardillier, J.-C. Multicellular resistance: A paradigm for clinical resistance? Crit. Rev. Oncol./Hematol. 2000, 36, 193–207. [Google Scholar] [CrossRef]
- Brancato, V.; Gioiella, F.; Profeta, M.; Imparato, G.; Guarnieri, D.; Urciuolo, F.; Melone, P.; Netti, P.A. 3D tumor microtissues as an in vitro testing platform for microenvironmentally-triggered drug delivery systems. Acta Biomater. 2017, 57, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, R.M.; McCredie, J.A.; Inch, W.R. Growth of Multicell Spheroids in Tissue Culture as a Model of Nodular Carcinomas 2. J. Natl. Cancer Inst. 1971, 46, 113–120. [Google Scholar]
- Gong, X.; Lin, C.; Cheng, J.; Su, J.; Zhao, H.; Liu, T.; Wen, X.; Zhao, P. Generation of multicellular tumor spheroids with microwell-based agarose scaffolds for drug testing. PLoS ONE 2015, 10, e0130348. [Google Scholar] [CrossRef]
- Santo, V.E.; Estrada, M.F.; Rebelo, S.P.; Abreu, S.; Silva, I.; Pinto, C.; Veloso, S.C.; Serra, A.T.; Boghaert, E.; Alves, P.M.; et al. Adaptable stirred-tank culture strategies for large scale production of multicellular spheroid-based tumor cell models. J. Biotechnol. 2016, 221, 118–129. [Google Scholar] [CrossRef] [PubMed]
- Timmins, N.E.; Nielsen, L.K. Generation of multicellular tumor spheroids by the hanging-drop method. Tissue Eng. 2007, 140, 141–151. [Google Scholar]
- Hirschhaeuser, F.; Menne, H.; Dittfeld, C.; West, J.; Mueller-Klieser, W.; Kunz-Schughart, L.A. Multicellular tumor spheroids: An underestimated tool is catching up again. J. Biotechnol. 2010, 148, 3–15. [Google Scholar] [CrossRef]
- Vadivelu, R.K.; Kamble, H.; Shiddiky, M.J.; Nguyen, N.-T. Microfluidic Technology for the Generation of Cell Spheroids and Their Applications. Micromachines 2017, 8, 94. [Google Scholar] [CrossRef]
- Pitingolo, G.; Nizard, P.; Riaud, A.; Taly, V. Beyond the on/off Chip Trade-off: A Reversibly Sealed Microfluidic Platform for 3D Tumor Microtissue Analysis. Sens. Actuators B 2018, 274, 393–401. [Google Scholar] [CrossRef]
- Verhulsel, M.; Vignes, M.; Descroix, S.; Malaquin, L.; Vignjevic, D.M.; Viovy, J.-L. A review of microfabrication and hydrogel engineering for micro-organs on chips. Biomaterials 2014, 35, 1816–1832. [Google Scholar] [CrossRef] [PubMed]
- Tibbitt, M.W.; Anseth, K.S. Hydrogels as extracellular matrix mimics for 3D cell culture. Biotechnol. Bioeng. 2009, 103, 655–663. [Google Scholar] [CrossRef]
- Loebel, C.; Szczesny, S.E.; Cosgrove, B.D.; Alini, M.; Zenobi-Wong, M.; Mauck, R.L.; Eglin, D. Cross-linking chemistry of tyramine-modified hyaluronan hydrogels alters mesenchymal stem cell early attachment and behavior. Biomacromolecules 2017, 18, 855–864. [Google Scholar] [CrossRef] [PubMed]
- Khademhosseini, A.; Langer, R. Microengineered hydrogels for tissue engineering. Biomaterials 2007, 28, 5087–5092. [Google Scholar] [CrossRef]
- Wang, L.; Lu, G.; Lu, Q.; Kaplan, D.L. Controlling Cell Behavior on Silk Nanofiber Hydrogels with Tunable Anisotropic Structures. ACS Biomater. Sci. Eng. 2018, 4, 933–941. [Google Scholar] [CrossRef]
- Chung, B.G.; Lee, K.-H.; Khademhosseini, A.; Lee, S.-H. Microfluidic fabrication of microengineered hydrogels and their application in tissue engineering. Lab Chip 2012, 12, 45–59. [Google Scholar] [CrossRef] [PubMed]
- Campbell, J.J.; Husmann, A.; Hume, R.D.; Watson, C.J.; Cameron, R.E. Development of three-dimensional collagen scaffolds with controlled architecture for cell migration studies using breast cancer cell lines. Biomaterials 2017, 114, 34–43. [Google Scholar] [CrossRef] [PubMed]
- Lam, J.; Truong, N.F.; Segura, T. Design of cell–matrix interactions in hyaluronic acid hydrogel scaffolds. Acta Biomater. 2014, 10, 1571–1580. [Google Scholar] [CrossRef]
- Kleinman, H.K.; Martin, G.R. Matrigel: Basement membrane matrix with biological activity. Semin. Cancer Biol. 2005, 15, 378–386. [Google Scholar] [CrossRef] [PubMed]
- Camci-Unal, G.; Cuttica, D.; Annabi, N.; Demarchi, D.; Khademhosseini, A. Synthesis and characterization of hybrid hyaluronic acid-gelatin hydrogels. Biomacromolecules 2013, 14, 1085–1092. [Google Scholar] [CrossRef]
- Rose, J.; Pacelli, S.; Haj, A.; Dua, H.; Hopkinson, A.; White, L.; Rose, F. Gelatin-based materials in ocular tissue engineering. Materials 2014, 7, 3106–3135. [Google Scholar] [CrossRef] [PubMed]
- Speer, D.P.; Chvapil, M.; Eskelson, C.; Ulreich, J. Biological effects of residual glutaraldehyde in glutaraldehyde-tanned collagen biomaterials. J. Biomed. Mater. Res. 1980, 14, 753–764. [Google Scholar] [CrossRef]
- Benton, J.A.; DeForest, C.A.; Vivekanandan, V.; Anseth, K.S. Photocrosslinking of gelatin macromers to synthesize porous hydrogels that promote valvular interstitial cell function. Tissue Eng. Part A 2009, 15, 3221–3230. [Google Scholar] [CrossRef]
- Bae, H.; Ahari, A.F.; Shin, H.; Nichol, J.W.; Hutson, C.B.; Masaeli, M.; Kim, S.H.; Aubin, H.; Yamanlar, S.; Khademhosseini, A. Cell-laden microengineered pullulan methacrylate hydrogels promote cell proliferation and 3D cluster formation. Soft Matter 2011, 7, 1903–1911. [Google Scholar] [CrossRef] [PubMed]
- Zuo, Y.; Liu, X.; Wei, D.; Sun, J.; Xiao, W.; Zhao, H.; Guo, L.; Wei, Q.; Fan, H.; Zhang, X. Photo-cross-linkable methacrylated gelatin and hydroxyapatite hybrid hydrogel for modularly engineering biomimetic osteon. ACS Appl. Mater. Interfaces 2015, 7, 10386–10394. [Google Scholar] [CrossRef]
- Zhao, X.; Lang, Q.; Yildirimer, L.; Lin, Z.Y.; Cui, W.; Annabi, N.; Ng, K.W.; Dokmeci, M.R.; Ghaemmaghami, A.M.; Khademhosseini, A. Photocrosslinkable gelatin hydrogel for epidermal tissue engineering. Adv. Healthc. Mater. 2016, 5, 108–118. [Google Scholar] [CrossRef] [PubMed]
- Yannas, A.; Tobolsky, A. Cross-Linking of gelatine by dehydration. Nature 1967, 215, 509. [Google Scholar] [CrossRef] [PubMed]
- Vecchione, R.; Pitingolo, G.; Falanga, A.P.; Guarnieri, D.; Netti, P.A. Confined gelatin dehydration as a viable route to go beyond micromilling resolution and miniaturize biological assays. ACS Appl. Mater. Interfaces 2016, 8, 12075–12081. [Google Scholar] [CrossRef]
- Pitingolo, G.; Taly, V.; Nastruzzi, C. A Second Life for Old Electronic Parts: A Spin Coater for Microfluidic Applications; Royal Soc Chemistry: Cambridge, UK, 2018. [Google Scholar]
- Tsao, C.-W.; DeVoe, D.L. Bonding of thermoplastic polymer microfluidics. Microfluid. Nanofluid. 2009, 6, 1–16. [Google Scholar] [CrossRef]
- Brouzes, E.; Medkova, M.; Savenelli, N.; Marran, D.; Twardowski, M.; Hutchison, J.B.; Rothberg, J.M.; Link, D.R.; Perrimon, N.; Samuels, M.L. Droplet microfluidic technology for single-cell high-throughput screening. Proc. Natl. Acad. Sci. USA 2009, 106, 14195–14200. [Google Scholar] [CrossRef] [PubMed]
- Davidenko, N.; Schuster, C.F.; Bax, D.V.; Farndale, R.W.; Hamaia, S.; Best, S.M.; Cameron, R.E. Evaluation of cell binding to collagen and gelatin: A study of the effect of 2D and 3D architecture and surface chemistry. J. Mater. Sci. Mater. Med. 2016, 27, 148. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Day | Semi-Major Axis Coated (µm) | Semi-Minor Axis Coated (µm) | Semi-Major Axis Uncoated (µm) | Semi-Minor Axis Uncoated (µm) | Total Area Coated (mm2) | Total Area Uncoated (mm2) |
|---|---|---|---|---|---|---|
| 7 | 201 ± 20 | 153 ± 12 | 50 ± 10 | 41 ± 7 | 0.965 | 0.064 |
| 8 | 216 ± 24 | 162 ± 18 | 53 ± 8 | 43 ± 4 | 1.098 | 0.071 |
| 9 | 249 ± 18 | 165 ± 26 | 60 ± 12 | 47 ± 9 | 1.290 | 0.088 |
| 10 | 262 ± 24 | 172 ± 9 | 72 ± 9 | 54 ± 11 | 1.415 | 0.122 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pitingolo, G.; Riaud, A.; Nastruzzi, C.; Taly, V. Gelatin-Coated Microfluidic Channels for 3D Microtissue Formation: On-Chip Production and Characterization. Micromachines 2019, 10, 265. https://doi.org/10.3390/mi10040265
Pitingolo G, Riaud A, Nastruzzi C, Taly V. Gelatin-Coated Microfluidic Channels for 3D Microtissue Formation: On-Chip Production and Characterization. Micromachines. 2019; 10(4):265. https://doi.org/10.3390/mi10040265
Chicago/Turabian StylePitingolo, Gabriele, Antoine Riaud, Claudio Nastruzzi, and Valerie Taly. 2019. "Gelatin-Coated Microfluidic Channels for 3D Microtissue Formation: On-Chip Production and Characterization" Micromachines 10, no. 4: 265. https://doi.org/10.3390/mi10040265
APA StylePitingolo, G., Riaud, A., Nastruzzi, C., & Taly, V. (2019). Gelatin-Coated Microfluidic Channels for 3D Microtissue Formation: On-Chip Production and Characterization. Micromachines, 10(4), 265. https://doi.org/10.3390/mi10040265