A Phenolic Acid Decarboxylase-Based All-Enzyme Hydrogel for Flow Reactor Technology

 ,
,  and
and
Abstract
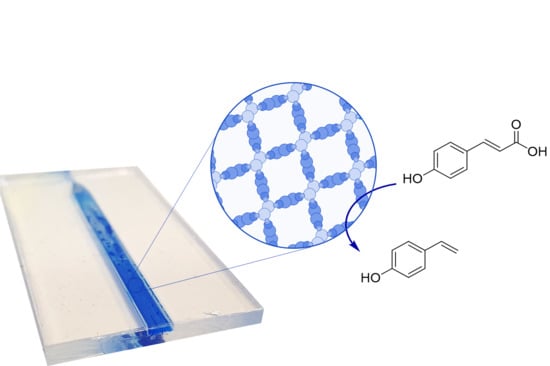
1. Introduction
2. Materials and Methods
2.1. Synthesis of p-Hydroxystyrene Via Wittig Reaction
2.2. Plasmid Construction
2.3. Gene Expression and Protein Production
2.4. Protein Purification
2.5. Sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) Analysis
2.6. Determination of Decarboxylase Activity
2.7. Hydrogel Prepration
2.8. Microrheology Measurements
2.9. High Performance Liquid Chromatography (HPLC) Analysis
2.10. Microfluidic Setup and Analysis of the Hydrogels Under Continuous Flow
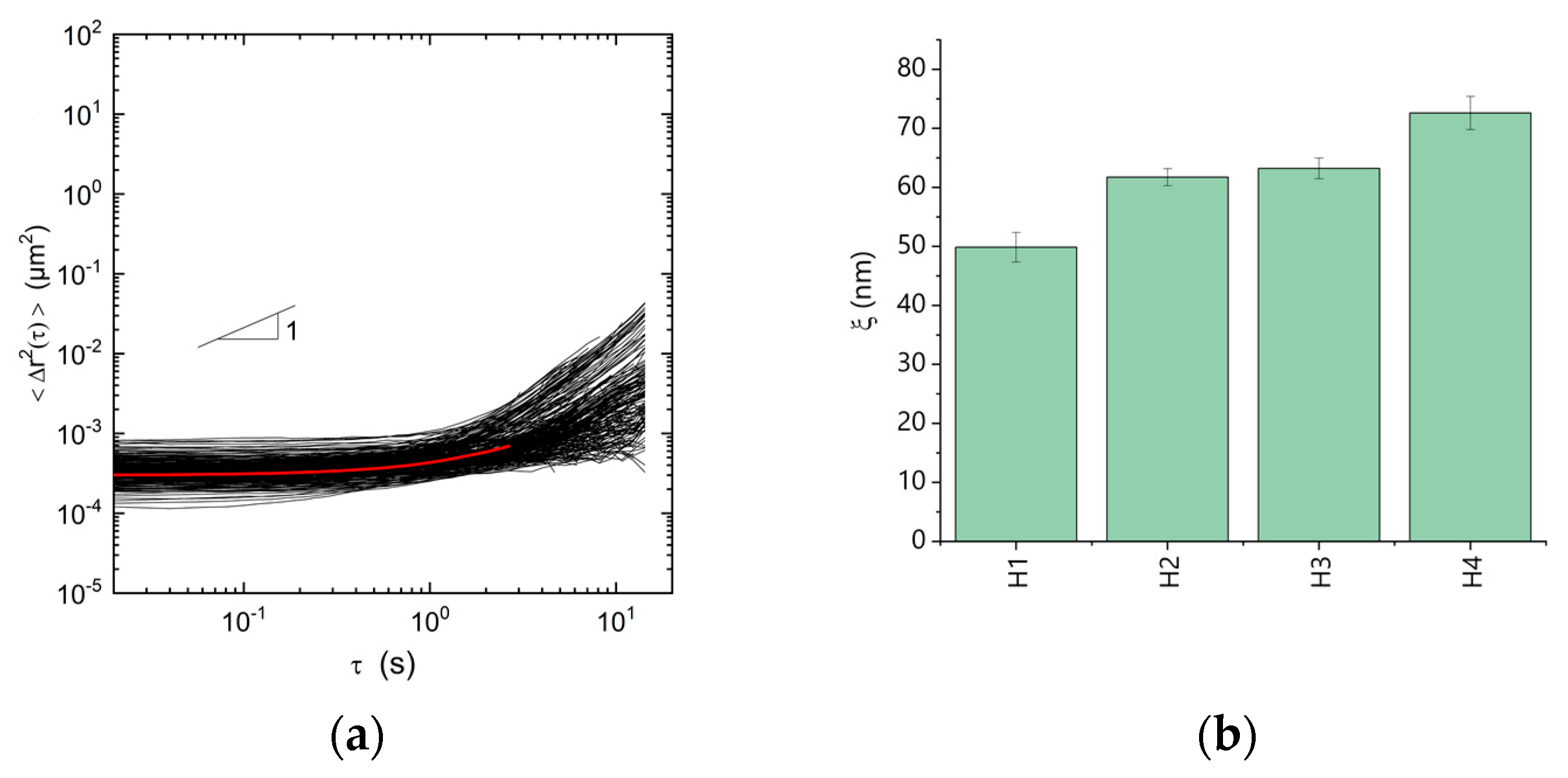
3. Results and Discussion
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Dawes, G.J.S.; Scott, E.L.; Le Notre, J.; Sanders, J.P.M.; Bitter, J.H. Deoxygenation of biobased molecules by decarboxylation and decarbonylation-A review on the role of heterogeneous, homogeneous and bio-catalysis. Green Chem. 2015, 17, 3231–3250. [Google Scholar] [CrossRef]
- Cadot, S.; Rameau, N.; Mangematin, S.; Pinel, C.; Djakovitch, L. Preparation of functional styrenes from biosourced carboxylic acids by copper catalyzed decarboxylation in PEG. Green Chem. 2014, 16, 3089–3097. [Google Scholar] [CrossRef]
- Robinson, A.M.; Hensley, J.E.; Medlin, J.W. Bifunctional Catalysts for Upgrading of Biomass-Derived Oxygenates: A Review. ACS Catal. 2016, 6, 5026–5043. [Google Scholar] [CrossRef]
- Gunawardena, D.A.; Fernando, S.D. Methods and Applications of Deoxygenation for the Conversion of Biomass to Petrochemical Products. In Biomass Now-Cultivation and Utilization; Matovic, M.D., Ed.; InTech: Croatia, Yugoslavia, 2013. [Google Scholar] [CrossRef]
- Xiao, X.; Bergstrom, H.; Saenger, R.; Johnson, B.; Sun, R.C.; Peterson, A. The role of oxygen vacancies in biomass deoxygenation by reducible zinc/zinc oxide catalysts. Catal. Sci. Technol. 2018, 8, 1819–1827. [Google Scholar] [CrossRef]
- Ni, J.; Wu, Y.T.; Tao, F.; Peng, Y.; Xu, P. A Coenzyme-Free Biocatalyst for the Value-Added Utilization of Lignin-Derived Aromatics. J. Am. Chem. Soc. 2018, 140, 16001–16005. [Google Scholar] [CrossRef] [PubMed]
- Leisch, H.; Grosse, S.; Morley, K.; Abokitse, K.; Perrin, F.; Denault, J.; Lau, P.C.K. Chemicals from agricultural biomass: Chemoenzymatic approach for production of vinylphenols and polyvinylphenols from phenolic acids. Green Process. Synth. 2013, 2, 7–17. [Google Scholar] [CrossRef]
- Morley, K.L.; Grosse, S.; Leisch, H.; Lau, P.C.K. Antioxidant canolol production from a renewable feedstock via an engineered decarboxylase. Green Chem. 2013, 15, 3312–3317. [Google Scholar] [CrossRef]
- Zakzeski, J.; Bruijnincx, P.C.; Jongerius, A.L.; Weckhuysen, B.M. The catalytic valorization of lignin for the production of renewable chemicals. Chem. Rev. 2010, 110, 3552–3599. [Google Scholar] [CrossRef]
- Liu, D.J.; Sun, J.; Simmons, B.A.; Singh, S. N-Heterocyclic Carbene Promoted Decarboxylation of Lignin-Derived Aromatic Acids. ACS Sustain. Chem. Eng. 2018, 6, 7232–7238. [Google Scholar] [CrossRef]
- Tinikul, R.; Chenprakhon, P.; Maenpuen, S.; Chaiyen, P. Biotransformation of Plant-Derived Phenolic Acids. Biotechnol. J. 2018, 13, e1700632. [Google Scholar] [CrossRef]
- Liu, J.; Liu, Q.; Yi, H.; Qin, C.; Bai, R.; Qi, X.; Lan, Y.; Lei, A. Visible-light-mediated decarboxylation/oxidative amidation of alpha-keto acids with amines under mild reaction conditions using O(2). Angew. Chem. Int. Ed. Engl. 2014, 53, 502–506. [Google Scholar] [CrossRef] [PubMed]
- Aleku, G.A.; Prause, C.; Bradshaw-Allen, R.T.; Plasch, K.; Glueck, S.M.; Bailey, S.S.; Payne, K.A.P.; Parker, D.A.; Faber, K.; Leys, D. Terminal Alkenes from Acrylic Acid Derivatives via Non-Oxidative Enzymatic Decarboxylation by Ferulic Acid Decarboxylases. ChemCatChem 2018, 10, 3736–3745. [Google Scholar] [CrossRef] [PubMed]
- Payer, S.E.; Faber, K.; Glueck, S.M. Non-Oxidative Enzymatic (De)Carboxylation of (Hetero)Aromatics and Acrylic Acid Derivatives. Adv. Synth Catal. 2019, 361, 2402–2420. [Google Scholar] [CrossRef] [PubMed]
- Rosazza, J.P.; Huang, Z.; Dostal, L.; Volm, T.; Rousseau, B. Review: Biocatalytic transformations of ferulic acid: An abundant aromatic natural product. J. Ind. Microbiol. 1995, 15, 457–471. [Google Scholar] [CrossRef]
- Ferré-Filmon, K.; Delaude, L.; Demonceau, A.; Noels, A.F. Catalytic methods for the synthesis of stilbenes with an emphasis on their phytoalexins. Coordin. Chem. Rev. 2004, 248, 2323–2336. [Google Scholar] [CrossRef]
- Savio, M.; Ferraro, D.; Maccario, C.; Vaccarone, R.; Jensen, L.D.; Corana, F.; Mannucci, B.; Bianchi, L.; Cao, Y.; Stivala, L.A. Resveratrol analogue 4,4′-dihydroxy-trans-stilbene potently inhibits cancer invasion and metastasis. Sci. Rep. 2016, 6, 19973. [Google Scholar] [CrossRef]
- Nasrullah, J.M.; Raja, S.; Vijayakumaran, K.; Dhamodharan, R. A practical route for the preparation of poly(4-hydroxystyrene), a useful photoresist material. J. Polym. Sci. Part A Polym. Chem. 2000, 38, 453–461. [Google Scholar] [CrossRef]
- Landete, J.M.; Rodriguez, H.; Curiel, J.A.; de las Rivas, B.; Mancheno, J.M.; Munoz, R. Gene cloning, expression, and characterization of phenolic acid decarboxylase from Lactobacillus brevis RM84. J. Ind. Microbiol. Biotechnol. 2010, 37, 617–624. [Google Scholar] [CrossRef]
- Rodriguez, H.; Landete, J.M.; Curiel, J.A.; de Las Rivas, B.; Mancheno, J.M.; Munoz, R. Characterization of the p-coumaric acid decarboxylase from Lactobacillus plantarum CECT 748(T). J. Agric. Food Chem. 2008, 56, 3068–3072. [Google Scholar] [CrossRef]
- Sheng, X.; Lind, M.E.; Himo, F. Theoretical study of the reaction mechanism of phenolic acid decarboxylase. FEBS J. 2015, 282, 4703–4713. [Google Scholar] [CrossRef]
- Rodriguez, H.; Angulo, I.; de Las Rivas, B.; Campillo, N.; Paez, J.A.; Munoz, R.; Mancheno, J.M. p-Coumaric acid decarboxylase from Lactobacillus plantarum: Structural insights into the active site and decarboxylation catalytic mechanism. Proteins 2010, 78, 1662–1676. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.; Yu, H.N.; Wu, Y.F.; Liu, X.Y.; Cheng, A.X.; Lou, H.X. Cloning and functional characterization of a phenolic acid decarboxylase from the liverwort Conocephalum japonicum. Biochem. Biophys. Res. Commun. 2016, 481, 239–244. [Google Scholar] [CrossRef] [PubMed]
- Sheng, X.; Himo, F. Theoretical Study of Enzyme Promiscuity: Mechanisms of Hydration and Carboxylation Activities of Phenolic Acid Decarboxylase. ACS Catal. 2017, 7, 1733–1741. [Google Scholar] [CrossRef]
- Gu, W.; Yang, J.; Lou, Z.; Liang, L.; Sun, Y.; Huang, J.; Li, X.; Cao, Y.; Meng, Z.; Zhang, K.Q. Structural basis of enzymatic activity for the ferulic acid decarboxylase (FADase) from Enterobacter sp. Px6-4. PLoS ONE 2011, 6, e16262. [Google Scholar] [CrossRef] [PubMed]
- Schweiger, A.K.; Rios-Lombardia, N.; Winkler, C.K.; Schmidt, S.; Moris, F.; Kroutil, W.; Gonzalez-Sabin, J.; Kourist, R. Using Deep Eutectic Solvents to Overcome Limited Substrate Solubility in the Enzymatic Decarboxylation of Bio-Based Phenolic Acids. ACS Sustain. Chem. Eng. 2019, 7, 16364–16370. [Google Scholar] [CrossRef]
- Busto, E.; Simon, R.C.; Kroutil, W. Vinylation of Unprotected Phenols Using a Biocatalytic System. Angew. Chem. Int. Ed. Engl. 2015, 54, 10899–10902. [Google Scholar] [CrossRef] [PubMed]
- Pesci, L.; Baydar, M.; Glueck, S.; Faber, K.; Liese, A.; Kara, S. Development and Scaling-Up of the Fragrance Compound 4-Ethylguaiacol Synthesis via a Two-Step Chemo-Enzymatic Reaction Sequence. Org. Process Res. Dev. 2017, 21, 85–93. [Google Scholar] [CrossRef]
- Gomez Baraibar, A.; Reichert, D.; Mugge, C.; Seger, S.; Groger, H.; Kourist, R. A One-Pot Cascade Reaction Combining an Encapsulated Decarboxylase with a Metathesis Catalyst for the Synthesis of Bio-Based Antioxidants. Angew. Chem. Int. Ed. Engl. 2016, 55, 14823–14827. [Google Scholar] [CrossRef]
- Peng, M.; Mittmann, E.; Wenger, L.; Hubbuch, J.; Engqvist, M.K.M.; Niemeyer, C.M.; Rabe, K.S. 3D-Printed phenacrylate decarboxylase flow reactors for the chemoenzymatic synthesis of 4-hydroxystilbene. Chemistry 2019. [Google Scholar] [CrossRef]
- Lindeque, R.M.; Woodley, J.M. Reactor Selection for Effective Continuous Biocatalytic Production of Pharmaceuticals. Catalysts 2019, 9, 262. [Google Scholar] [CrossRef]
- Rabe, K.S.; Muller, J.; Skoupi, M.; Niemeyer, C.M. Cascades in Compartments: En Route to Machine-Assisted Biotechnology. Angew. Chem. Int. Ed. Engl. 2017, 56, 13574–13589. [Google Scholar] [CrossRef] [PubMed]
- Sheldon, R.A.; Pereira, P.C. Biocatalysis engineering: The big picture. Chem. Soc. Rev. 2017, 46, 2678–2691. [Google Scholar] [CrossRef] [PubMed]
- Tamborini, L.; Fernandes, P.; Paradisi, F.; Molinari, F. Flow Bioreactors as Complementary Tools for Biocatalytic Process Intensification. Trends Biotechnol. 2018, 36, 73–88. [Google Scholar] [CrossRef] [PubMed]
- Peschke, T.; Skoupi, M.; Burgahn, T.; Gallus, S.; Ahmed, I.; Rabe, K.S.; Niemeyer, C.M. Self-Immobilizing Fusion Enzymes for Compartmentalized Biocatalysis. ACS Catal. 2017, 7, 7866–7872. [Google Scholar] [CrossRef]
- Woodley, J.M. New frontiers in biocatalysis for sustainable synthesis. Curr. Opin. Green Sustain. Chem. 2020, 21, 22–26. [Google Scholar] [CrossRef]
- Britton, J.; Majumdar, S.; Weiss, G.A. Continuous flow biocatalysis. Chem. Soc. Rev. 2018, 47, 5891–5918. [Google Scholar] [CrossRef]
- Zdarta, J.; Meyer, A.S.; Jesionowski, T.; Pinelo, M. Developments in support materials for immobilization of oxidoreductases: A comprehensive review. Adv. Colloid Interface Sci. 2018, 258, 1–20. [Google Scholar] [CrossRef]
- Sheldon, R.A. CLEAs, Combi-CLEAs and ‘Smart’ Magnetic CLEAs: Biocatalysis in a Bio-Based Economy. Catalysts 2019, 9, 261. [Google Scholar] [CrossRef]
- Cui, J.D.; Jia, S.R. Optimization protocols and improved strategies of cross-linked enzyme aggregates technology: Current development and future challenges. Crit. Rev. Biotechnol. 2015, 35, 15–28. [Google Scholar] [CrossRef]
- Velasco-Lozano, S.; López-Gallego, F.; Mateos-Díaz, J.C.; Favela-Torres, E. Cross-linked enzyme aggregates (CLEA) in enzyme improvement—A review. Biocatalysis 2016, 1, 7626. [Google Scholar] [CrossRef]
- Yan, E.K.; Cao, H.L.; Zhang, C.Y.; Lu, Q.Q.; Ye, Y.J.; He, J.; Huang, L.J.; Yin, D.C. Cross-linked protein crystals by glutaraldehyde and their applications. RSC Adv. 2015, 5, 26163–26174. [Google Scholar] [CrossRef]
- Honda, T.; Miyazaki, M.; Nakamura, H.; Maeda, H. Facile preparation of an enzyme-immobilized microreactor using a cross-linking enzyme membrane on a microchannel surface. Adv. Synth. Catal. 2006, 348, 2163–2171. [Google Scholar] [CrossRef]
- Peschke, T.; Bitterwolf, P.; Gallus, S.; Hu, Y.; Oelschlaeger, C.; Willenbacher, N.; Rabe, K.S.; Niemeyer, C.M. Self-Assembling All-Enzyme Hydrogels for Flow Biocatalysis. Angew. Chem. Int. Ed. Engl. 2018, 57, 17028–17032. [Google Scholar] [CrossRef] [PubMed]
- Peschke, T.; Bitterwolf, P.; Hansen, S.; Gasmi, J.; Rabe, K.S.; Niemeyer, C.M. Self-Immobilizing Biocatalysts Maximize Space-Time Yields in Flow Reactors. Catalysts 2019, 9, 164. [Google Scholar] [CrossRef]
- Bitterwolf, P.; Gallus, S.; Peschke, T.; Mittmann, E.; Oelschlaeger, C.; Willenbacher, N.; Rabe, K.S.; Niemeyer, C.M. Valency engineering of monomeric enzymes for self-assembling biocatalytic hydrogels. Chem. Sci. 2019, 10, 9752–9757. [Google Scholar] [CrossRef]
- Zakeri, B.; Fierer, J.O.; Celik, E.; Chittock, E.C.; Schwarz-Linek, U.; Moy, V.T.; Howarth, M. Peptide tag forming a rapid covalent bond to a protein, through engineering a bacterial adhesin. Proc. Natl. Acad. Sci. USA 2012, 109, E690–E697. [Google Scholar] [CrossRef]
- Sutherland, A.R.; Alam, M.K.; Geyer, C.R. Post-translational Assembly of Protein Parts into Complex Devices by Using SpyTag/SpyCatcher Protein Ligase. Chembiochem 2019, 20, 319–328. [Google Scholar] [CrossRef]
- Hatlem, D.; Trunk, T.; Linke, D.; Leo, J.C. Catching a SPY: Using the SpyCatcher-SpyTag and Related Systems for Labeling and Localizing Bacterial Proteins. Int. J. Mol. Sci. 2019, 20, 2129. [Google Scholar] [CrossRef]
- Peschke, T.; Rabe, K.S.; Niemeyer, C.M. Orthogonal Surface Tags for Whole-Cell Biocatalysis. Angew. Chem. Int. Ed. Engl. 2017, 56, 2183–2186. [Google Scholar] [CrossRef]
- Gu, W.; Li, X.; Huang, J.; Duan, Y.; Meng, Z.; Zhang, K.Q.; Yang, J. Cloning, sequencing, and overexpression in Escherichia coli of the Enterobacter sp. Px6-4 gene for ferulic acid decarboxylase. Appl. Microbiol. Biotechnol. 2011, 89, 1797–1805. [Google Scholar] [CrossRef]
- Demidoff, F.C.; de Souza, F.P.; Netto, C.D. Synthesis of Stilbene-Quinone Hybrids through Heck Reactions in PEG-400. Synthesis-Stuttgart 2017, 49, 5217–5223. [Google Scholar] [CrossRef]
- Gibson, D.G.; Young, L.; Chuang, R.Y.; Venter, J.C.; Hutchison, C.A., 3rd; Smith, H.O. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 2009, 6, 343–345. [Google Scholar] [CrossRef] [PubMed]
- Crocker, J.C.; Grier, D.G. Methods of digital video microscopy for colloidal studies. J. Colloid Interface Sci. 1996, 179, 298–310. [Google Scholar] [CrossRef]
- Van Hove, L. Correlations in Space and Time and Born Approximation Scattering in Systems of Interacting Particles. Phys. Rev. 1954, 95, 249–262. [Google Scholar] [CrossRef]
- Weeks, E.R.; Crocker, J.C.; Levitt, A.C.; Schofield, A.; Weitz, D.A. Three-dimensional direct imaging of structural relaxation near the colloidal glass transition. Science 2000, 287, 627–631. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer | DNA-Sequence |
|---|---|
| EM01 | GGTCATCATCACCATCATCATTAAGATCC |
| EM02 | GCTACCACCACCACCTTTCAGATTATC |
| EM03 | CCGGATAATCTGAAAGGTGGTGGTGGTAGCGTTGATACCCTGAGCGGTCTGAG |
| EM04 | CGGATCTTAATGATGATGGTGATGATGACCAATATGTGCATCACCTTTGGTTGCTTTACC |
| EM05 | TGGTTGATGCATATAAACCGACCAAAGGTCATCATCACCATCATCATTAAGATCC |
| EM06 | CGGTTTATATGCATCAACCATAACAATATGTGCGCTACCACCACCACCTTTCAGATTATC |
| EM07 | CACAATTCCCCTATAGTGAGTCGTATTAATTTC |
| EM08 | GTGAAAGCATCTATAAAATCAGCTGGACC |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mittmann, E.; Gallus, S.; Bitterwolf, P.; Oelschlaeger, C.; Willenbacher, N.; Niemeyer, C.M.; Rabe, K.S. A Phenolic Acid Decarboxylase-Based All-Enzyme Hydrogel for Flow Reactor Technology. Micromachines 2019, 10, 795. https://doi.org/10.3390/mi10120795
Mittmann E, Gallus S, Bitterwolf P, Oelschlaeger C, Willenbacher N, Niemeyer CM, Rabe KS. A Phenolic Acid Decarboxylase-Based All-Enzyme Hydrogel for Flow Reactor Technology. Micromachines. 2019; 10(12):795. https://doi.org/10.3390/mi10120795
Chicago/Turabian StyleMittmann, Esther, Sabrina Gallus, Patrick Bitterwolf, Claude Oelschlaeger, Norbert Willenbacher, Christof M. Niemeyer, and Kersten S. Rabe. 2019. "A Phenolic Acid Decarboxylase-Based All-Enzyme Hydrogel for Flow Reactor Technology" Micromachines 10, no. 12: 795. https://doi.org/10.3390/mi10120795
APA StyleMittmann, E., Gallus, S., Bitterwolf, P., Oelschlaeger, C., Willenbacher, N., Niemeyer, C. M., & Rabe, K. S. (2019). A Phenolic Acid Decarboxylase-Based All-Enzyme Hydrogel for Flow Reactor Technology. Micromachines, 10(12), 795. https://doi.org/10.3390/mi10120795