Exploring the Utility of ssDNA Aptamers Directed against Snake Venom Toxins as New Therapeutics for Snakebite Envenoming


 , , and
, , and
Abstract
:1. Introduction
2. Results
2.1. ssDNA Aptamer Selection
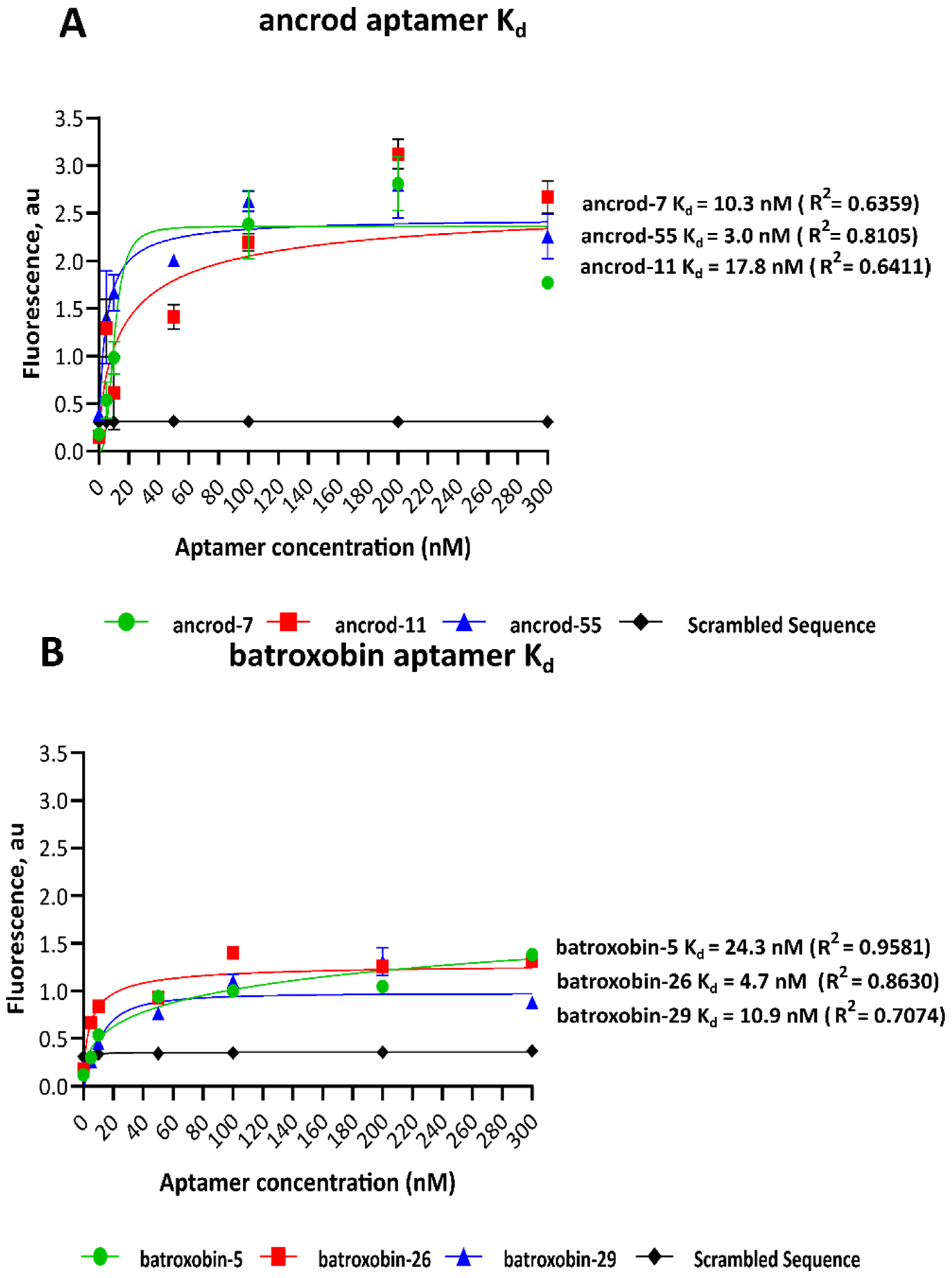
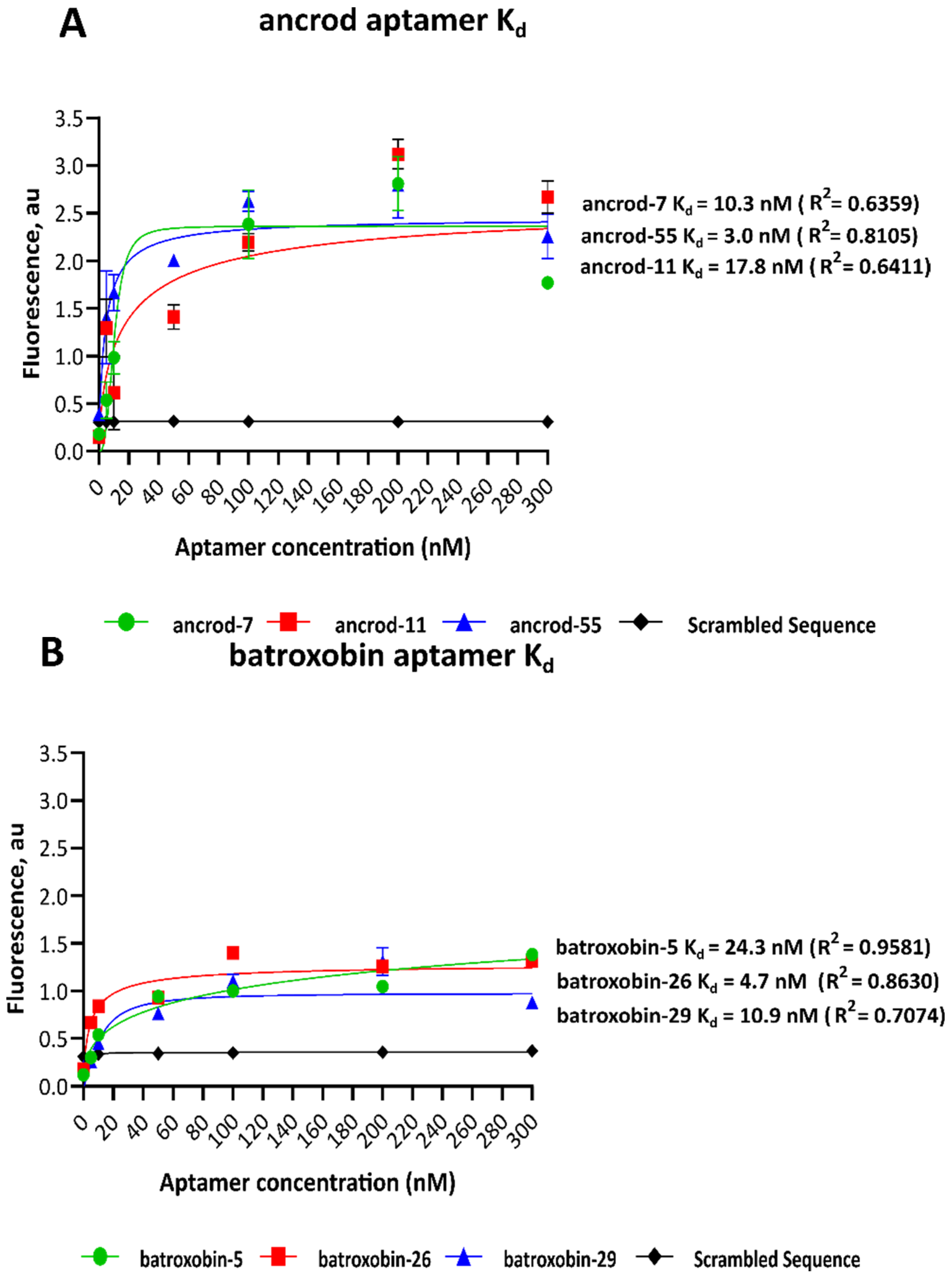
2.2. Dissociation Constants of Identified Aptamers
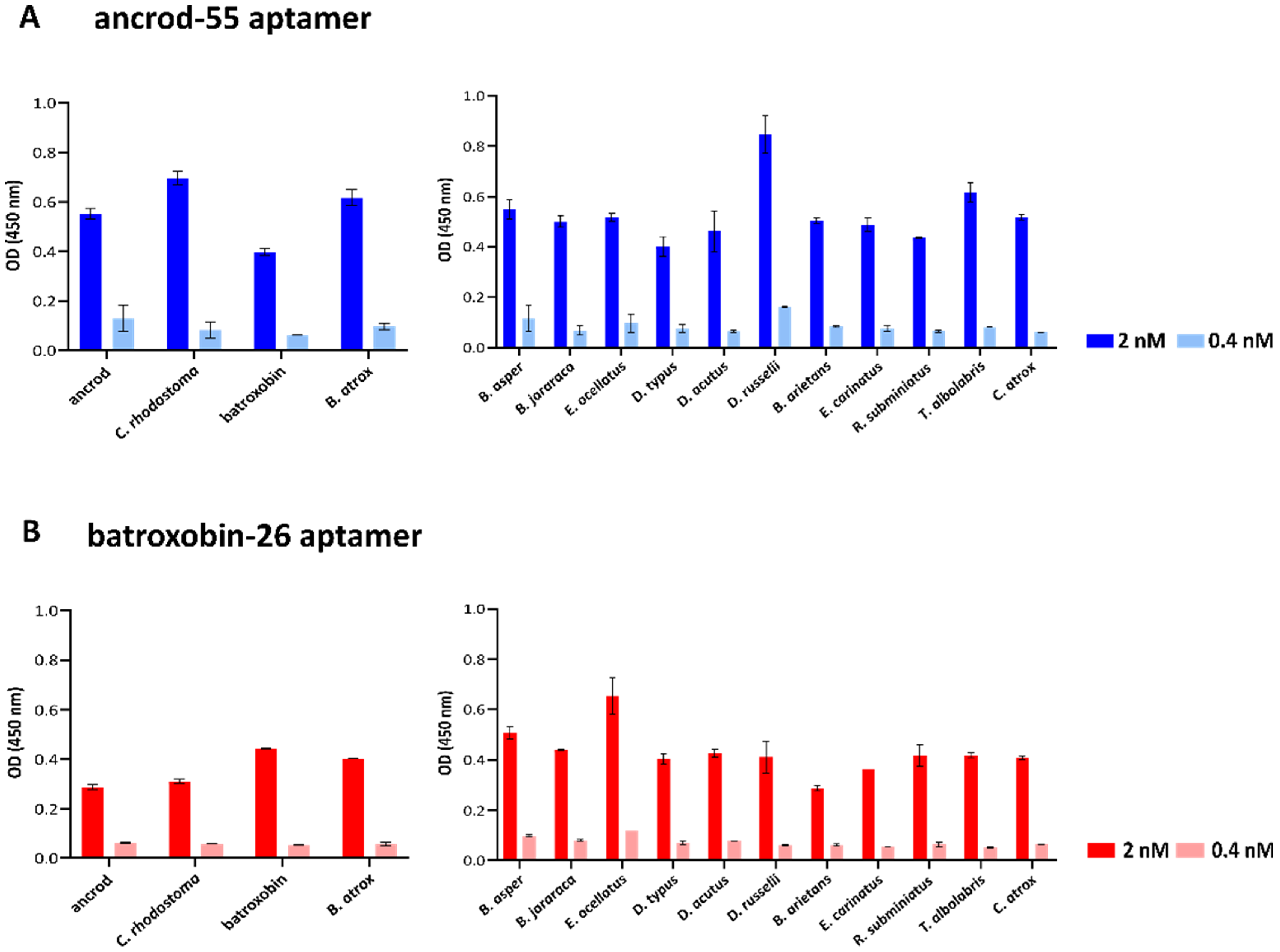
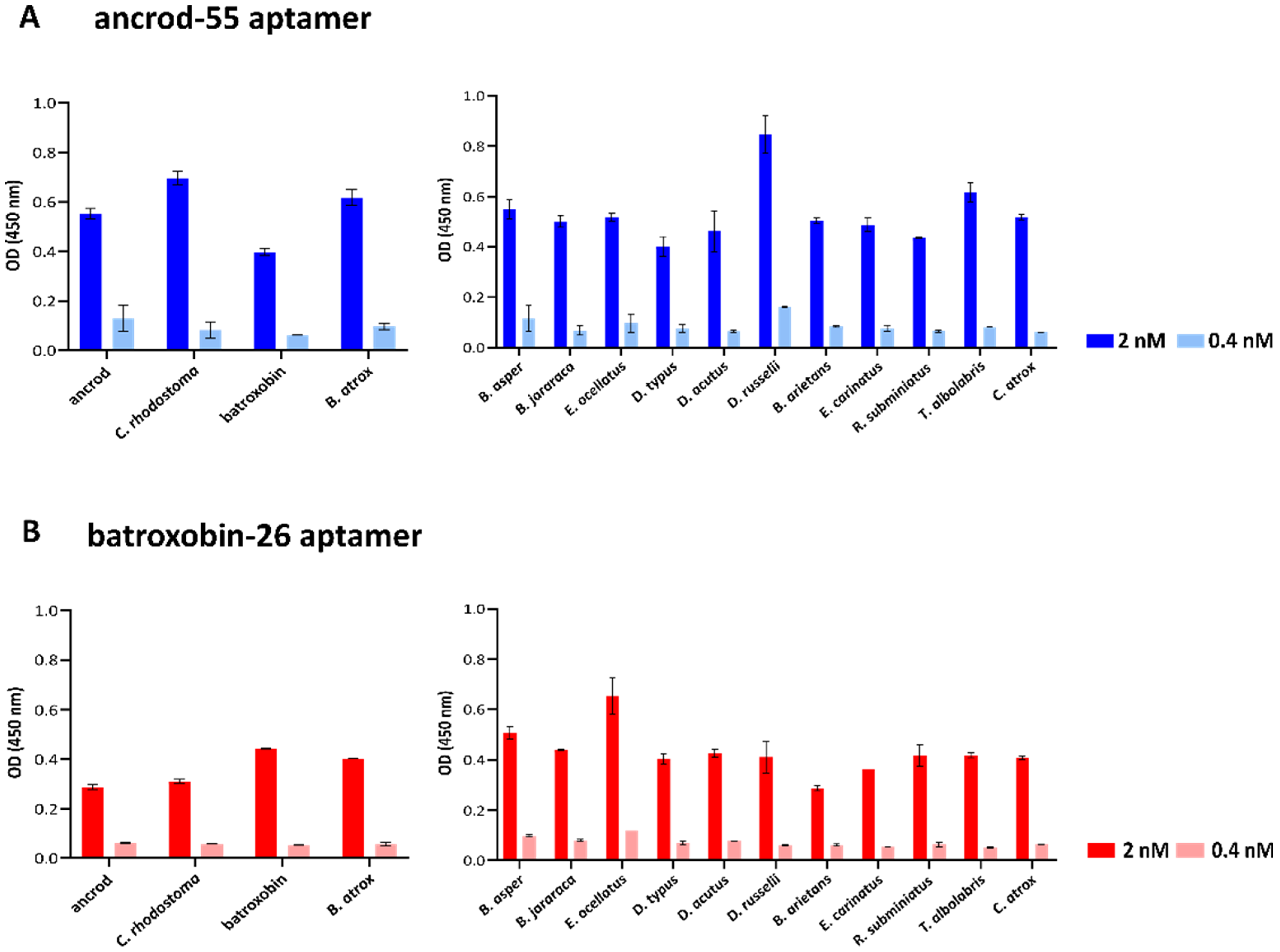
2.3. Quantifying Aptamer Binding to Recombinant Toxins and Venom by ALISA
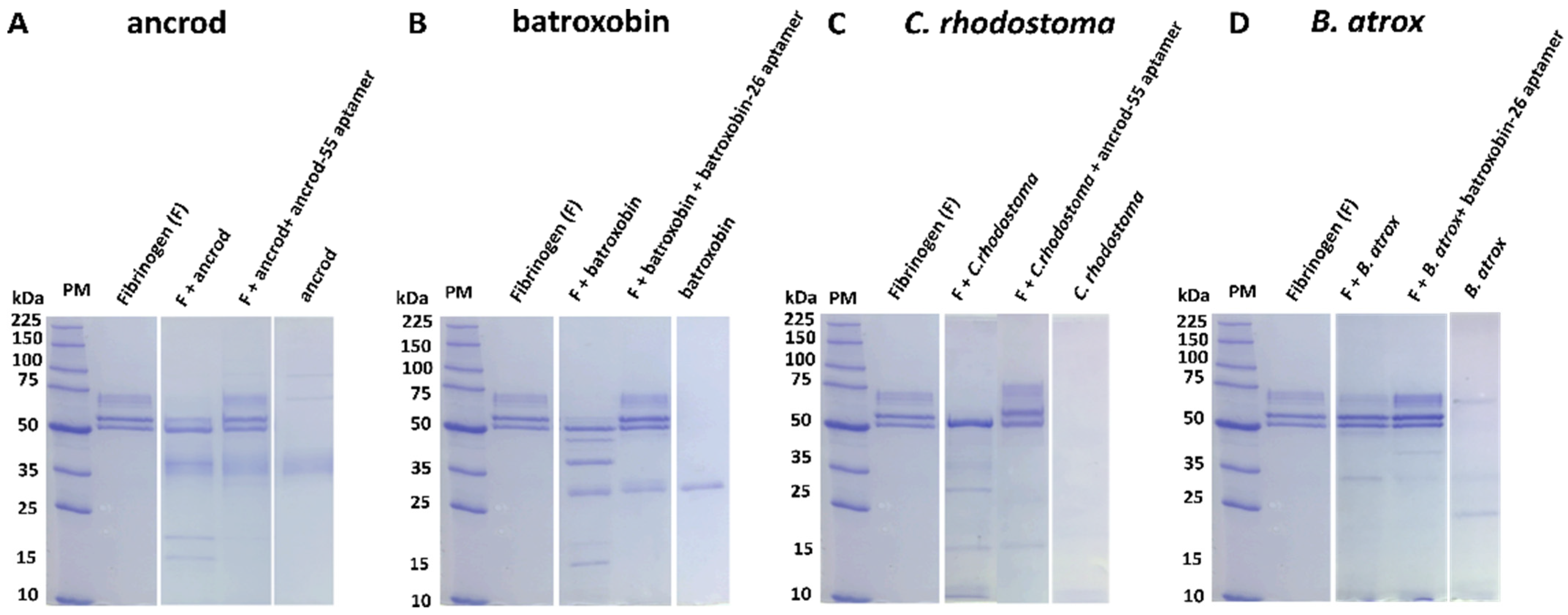
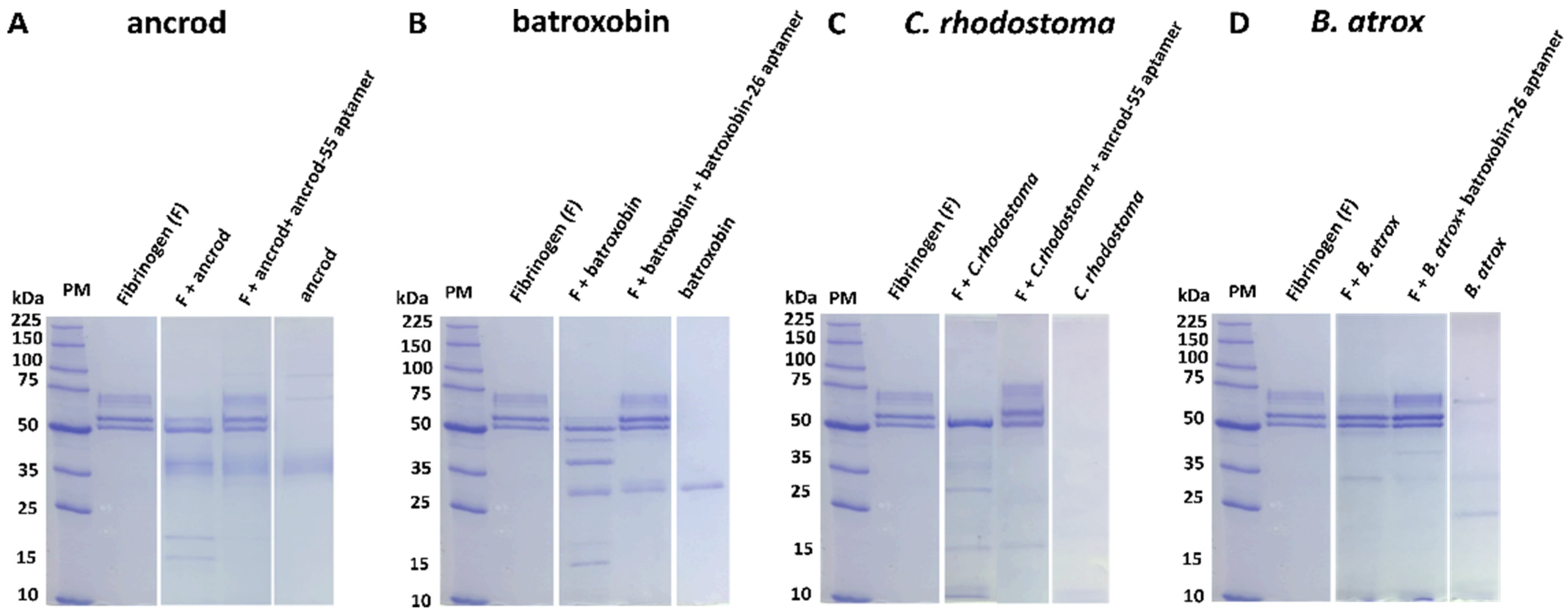
2.4. Aptamers Inhibit Toxin-Induced Fibrinogenolysis and Fibrinogen Depletion
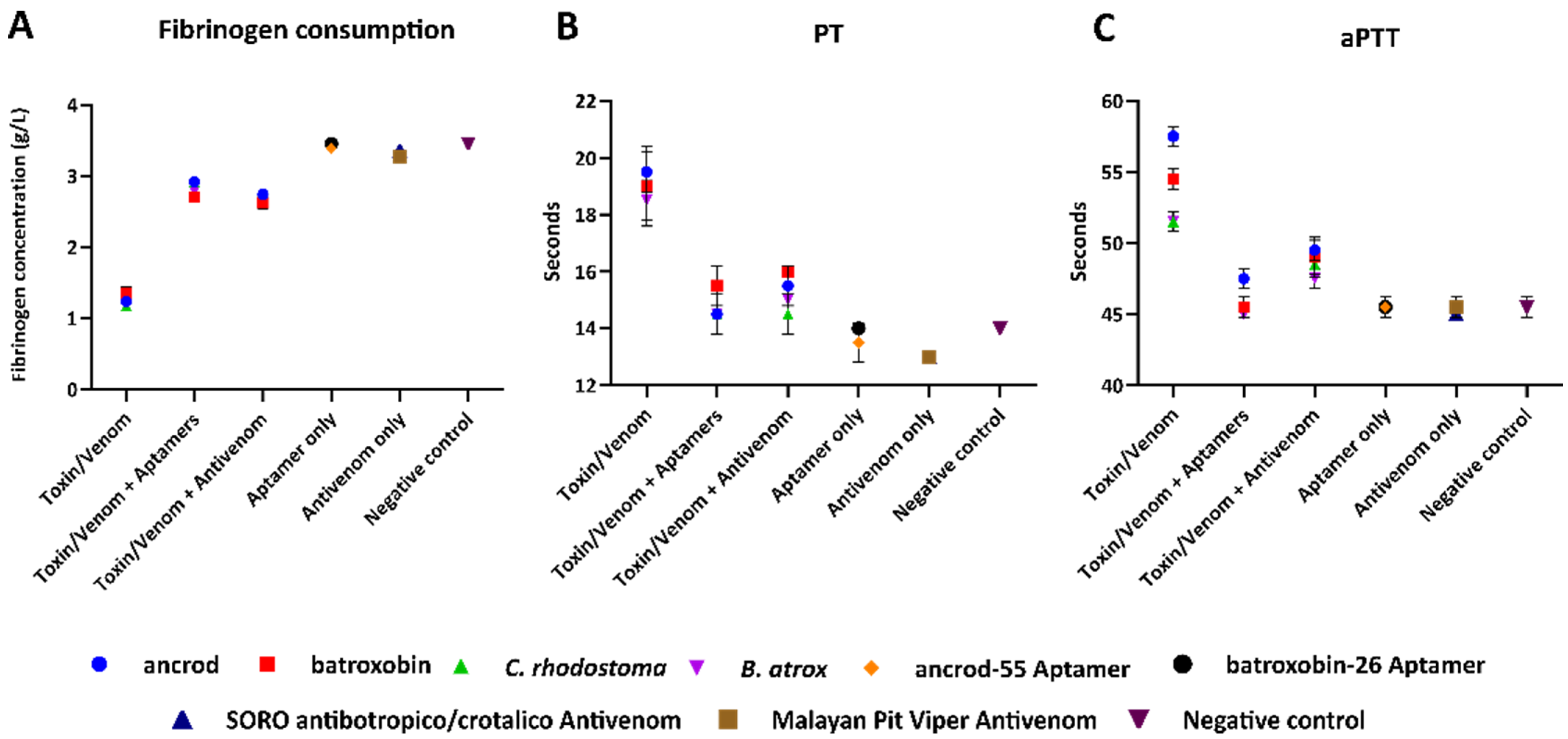
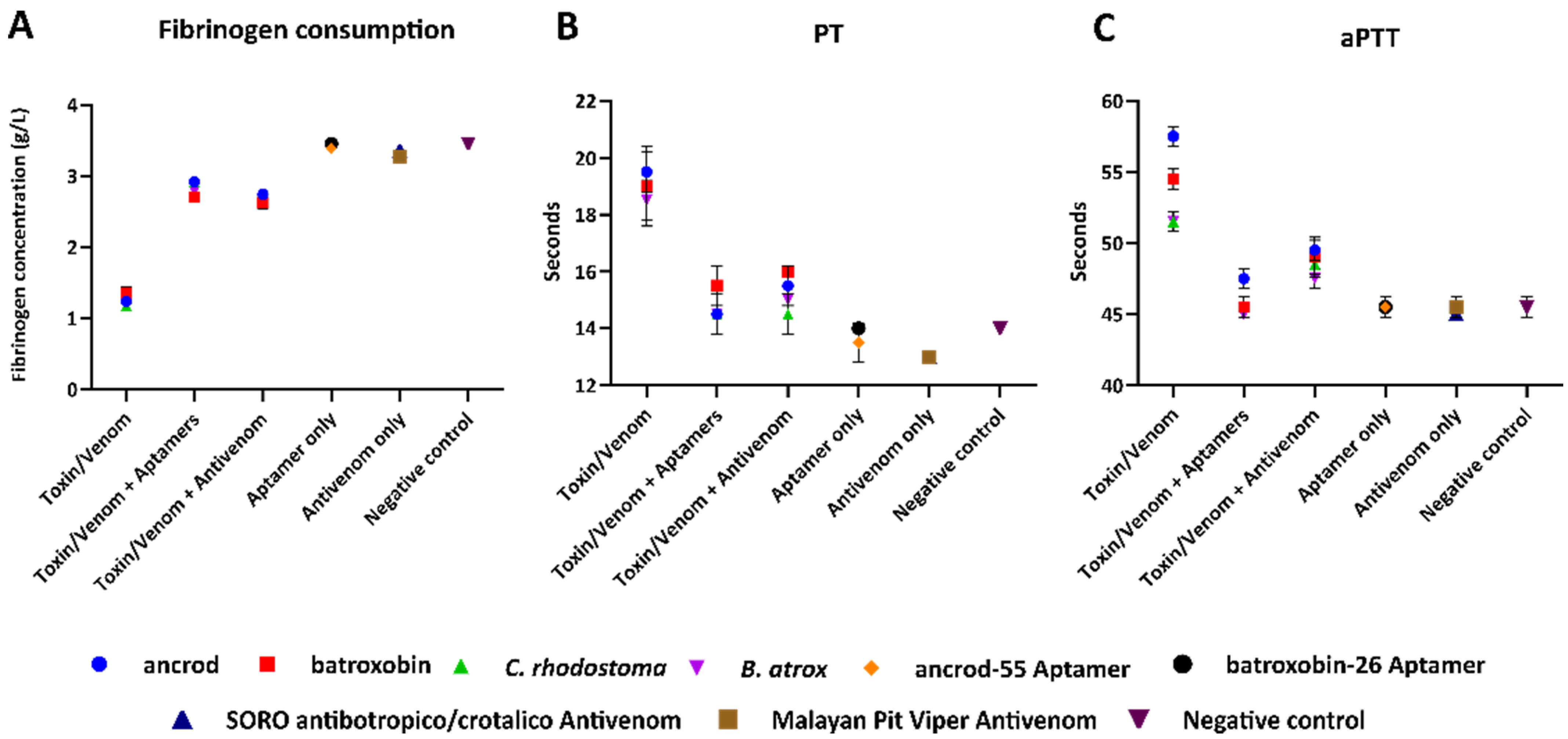
2.5. Aptamers Reduce Toxin- and Venom-Induced Prolongations of Clotting Times
3. Discussion
4. Materials and Methods
4.1. DNA Library and Primer Design
4.2. Target Conjugation
4.3. PCR Amplification
4.4. Separation of ssDNA by Denaturing PAGE
4.5. Counter-Selection
4.6. Cloning and Sequencing ssDNA Aptamers
4.7. Dissociation Constants
4.8. Binding by ALISA
4.9. Visualisation of Fibrinogenolysis via SDS-PAGE Gel Electrophoresis
4.10. Sample Preparation for Clotting Profiling Experiments
4.11. Fibrinogen Consumption via the Clauss Method
4.12. Prothrombin Time (PT)
4.13. Activated Partial Thromboplastin Time (aPTT)
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Alirol, E.; Sharma, S.K.; Bawaskar, H.S.; Kuch, U.; Chappuis, F. Snake bite in South Asia: A review. PLoS Negl. Trop. Dis. 2010, 4, e603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gutiérrez, J.M.; Calvete, J.J.; Habib, A.G.; Harrison, R.A.; Williams, D.J.; Warrell, D.A. Snakebite envenoming. Nat. Rev. Dis. Primers 2017, 3, 17063. [Google Scholar] [CrossRef] [PubMed]
- Kasturiratne, A.; Wickremasinghe, A.R.; de Silva, N.; Gunawardena, N.K.; Pathmeswaran, A.; Premaratna, R.; Savioli, L.; Lalloo, D.G.; de Silva, H.J. The global burden of snakebite: A literature analysis and modelling based on regional estimates of envenoming and deaths. PLoS Med. 2008, 5, e218. [Google Scholar] [CrossRef] [Green Version]
- Chippaux, J.-P. Snakebite envenomation turns again into a neglected tropical disease! J. Venom. Anim. Toxins Incl. Trop. Dis. 2017, 23, 38. [Google Scholar] [CrossRef] [PubMed]
- Laxme, R.S.; Khochare, S.; de Souza, H.F.; Ahuja, B.; Suranse, V.; Martin, G.; Whitaker, R.; Sunagar, K. Beyond the ‘big four’: Venom profiling of the medically important yet neglected Indian snakes reveals disturbing antivenom deficiencies. PLoS Negl. Trop. Dis. 2019, 13, e0007899. [Google Scholar]
- Casewell, N.R.; Jackson, T.N.W.; Laustsen, A.H.; Sunagar, K. Causes and consequences of snake venom variation. Trends Pharmacol. Sci. 2020, 41, 570–581. [Google Scholar] [CrossRef]
- Slagboom, J.; Kool, J.; Harrison, R.A.; Casewell, N.R. Haemotoxic snake venoms: Their functional activity, impact on snakebite victims and pharmaceutical promise. Br. J. Haematol. 2017, 177, 947–959. [Google Scholar] [CrossRef] [Green Version]
- Lu, Q.; Navdaev, A.; Clemetson, J.M.; Clemetson, K.J. Snake venom C-type lectins interacting with platelet receptors. Structure–function relationships and effects on haemostasis. Toxicon 2005, 45, 1089–1098. [Google Scholar] [CrossRef]
- White, J. Snake venoms and coagulopathy. Toxicon 2005, 45, 951–967. [Google Scholar] [CrossRef]
- Warrell, D.A. Snake bite. Lancet 2010, 375, 77–88. [Google Scholar] [CrossRef]
- Gutiérrez, J.M.; Vargas, M.; Segura, A.; Herrera, M.; Villalta, M.; Solano, G.; Sánchez, A.; Herrera, C.; León, G. In vitro tests for assessing the neutralizing ability of snake antivenoms: Toward the 3Rs principles. Front. Immunol. 2021, 11, 617429. [Google Scholar] [CrossRef] [PubMed]
- Isbister, G.K. (Ed.) Snakebite doesn’t cause disseminated intravascular coagulation: Coagulopathy and thrombotic microangiopathy in snake envenoming. In Seminars in Thrombosis and Hemostasis; Thieme Medical Publishers: New York, NY, USA, 2010. [Google Scholar]
- Ainsworth, S.; Slagboom, J.; Alomran, N.; Pla, D.; Alhamdi, Y.; King, S.I.; Bolton, F.; Gutiérrez, J.M.; Vonk, F.J.; Toh, C.H.; et al. The paraspecific neutralisation of snake venom induced coagulopathy by antivenoms. Commun. Biol. 2018, 1, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phillips, D.J.; Swenson, S.D.; Francis, S.; Markland, J.; Mackessy, S. Thrombin-like snake venom serine proteinases. Handb. Venoms Toxins Rep. 2010, 139, 154. [Google Scholar]
- Maduwage, K.; Isbister, G.K. Current treatment for venom-induced consumption coagulopathy resulting from snakebite. PLoS Negl. Trop. Dis. 2014, 8, e3220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joseph, J.S.; Kini, R.M. Snake venom prothrombin activators homologous to blood coagulation factor Xa. Pathophysiol. Haemost. Thromb. 2001, 31, 234–240. [Google Scholar] [CrossRef]
- Isbister, G.K. (Ed.) Procoagulant snake toxins: Laboratory studies, diagnosis, and understanding snakebite coagulopathy. In Seminars in Thrombosis Hemostasis; Thieme Medical Publishers: New York, NY, USA, 2009. [Google Scholar]
- Xie, C.; Slagboom, J.; Albulescu, L.O.; Bruyneel, B.; Still, K.; Vonk, F.J.; Somsen, G.W.; Casewell, N.R.; Kool, J. Antivenom neutralization of coagulopathic snake venom toxins assessed by bioactivity profiling using nanofractionation analytics. Toxins 2020, 12, 53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gutiérrez, J.M.; Escalante, T.; Rucavado, A.; Herrera, C. Hemorrhage caused by snake venom metalloproteinases: A journey of discovery and understanding. Toxins 2016, 8, 93. [Google Scholar] [CrossRef] [Green Version]
- Gutiérrez, J.M.; Escalante, T.; Rucavado, A.; Herrera, C.; Fox, J.W. A comprehensive view of the structural and functional alterations of extracellular matrix by snake venom metalloproteinases (SVMPs): Novel perspectives on the pathophysiology of envenoming. Toxins 2016, 8, 304. [Google Scholar] [CrossRef] [Green Version]
- Gutierrez JLeón, G.; Lomonte, B.; Angulo, Y. Antivenoms for snakebite envenomings. Inflamm. Allergy-Drug Targets 2011, 10, 369–380. [Google Scholar] [CrossRef] [Green Version]
- Gutierrez, J.M.; Lomonte, B.; León, G.; Rucavado, A.; Chaves, F.; Angulo, Y. Trends in snakebite envenomation therapy: Scientific, technological and public health considerations. Curr. Pharm. Des. 2007, 13, 2935–2950. [Google Scholar] [CrossRef]
- Lalloo, D.G.; Theakston, R.D.G. Snake antivenoms. J. Toxicol. Clin. Toxicol. 2003, 41, 277–290. [Google Scholar] [CrossRef] [PubMed]
- de Silva, H.A.; Pathmeswaran, A.; Ranasinha, C.D.; Jayamanne, S.; Samarakoon, S.B.; Hittharage, A.; Kalupahana, R.; Ratnatilaka, G.A.; Uluwatthage, W.; Aronson, J.K.; et al. Low-dose adrenaline, promethazine, and hydrocortisone in the prevention of acute adverse reactions to antivenom following snakebite: A randomised, double-blind, placebo-controlled trial. PLoS Med. 2011, 8, e1000435. [Google Scholar] [CrossRef] [PubMed]
- Kularatne, S.A.; Gawarammana, I.B.; Kumarasiri, P.V.; Senanayake, N.; Dissanayake, W.P.; Ariyasena, H. Safety and efficacy of subcutaneous adrenaline as a treatment for anaphylactic reactions to polyvalent antivenom. Ceylon Med. J. 2003, 48, 148–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casewell, N.R.; Cook, D.A.; Wagstaff, S.C.; Nasidi, A.; Durfa, N.; Wüster, W.; Harrison, R.A. Pre-clinical assays predict pan-African Echis viper efficacy for a species-specific antivenom. PLoS Negl. Trop. Dis. 2010, 4, e851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, D.J.; Gutiérrez, J.M.; Calvete, J.J.; Wüster, W.; Ratanabanangkoon, K.; Paiva, O.; Brown, N.I.; Casewell, N.R.; Harrison, R.A.; Rowley, P.D.; et al. Ending the drought: New strategies for improving the flow of affordable, effective antivenoms in Asia and Africa. J. Proteom. 2011, 74, 1735–1767. [Google Scholar] [CrossRef]
- Abubakar, I.S.; Abubakar, S.B.; Habib, A.G.; Nasidi, A.; Durfa, N.; Yusuf, P.O.; Larnyang, S.; Garnvwa, J.; Sokomba, E.; Salako, L.; et al. Randomised controlled double-blind non-inferiority trial of two antivenoms for saw-scaled or carpet viper (Echis ocellatus) envenoming in Nigeria. PLoS Negl. Trop. Dis. 2010, 4, e767. [Google Scholar] [CrossRef] [Green Version]
- Brown, N.I. Consequences of neglect: Analysis of the sub-Saharan African snake antivenom market and the global context. PLoS Negl. Trop. Dis. 2012, 6, e1670. [Google Scholar] [CrossRef] [Green Version]
- Habib, A.G.; Brown, N.I. The snakebite problem and antivenom crisis from a health-economic perspective. Toxicon 2018, 150, 115–123. [Google Scholar] [CrossRef]
- Harrison, R.A.; Hargreaves, A.; Wagstaff, S.C.; Faragher, B.; Lalloo, D.G. Snake envenoming: A disease of poverty. PLoS Negl. Trop. Dis. 2009, 3, e569. [Google Scholar] [CrossRef] [Green Version]
- Laustsen, A.H. Guiding recombinant antivenom development by omics technologies. New Biotechnol. 2018, 45, 19–27. [Google Scholar] [CrossRef] [Green Version]
- Clare, R.H.; Hall, S.R.; Patel, R.N.; Casewell, N.R. Small molecule drug discovery for neglected tropical snakebite. Trends Pharmacol. Sci. 2021, 42, 340–353. [Google Scholar] [CrossRef] [PubMed]
- Albulescu, L.O.; Kazandjian, T.; Slagboom, J.; Bruyneel, B.; Ainsworth, S.; Alsolaiss, J.; Wagstaff, S.C.; Whiteley, G.; Harrison, R.A.; Ulens, C.; et al. A decoy-receptor approach using nicotinic acetylcholine receptor mimics reveals their potential as novel therapeutics against neurotoxic snakebite. Front. Pharmacol. 2019, 10, 848. [Google Scholar] [CrossRef] [Green Version]
- Ye, F.; Zheng, Y.; Wang, X.; Tan, X.; Zhang, T.; Xin, W.; Wang, J.; Huang, Y.; Fan, Q.; Wang, J. Recognition of Bungarus multicinctus venom by a DNA aptamer against β-bungarotoxin. PLoS ONE 2014, 9, e105404. [Google Scholar] [CrossRef] [PubMed]
- Savchik, E.Y.; Kalinina, T.B.; Drozd, N.N.; Makarov, V.A.; Zav’Yalova, E.G.; Lapsheva, E.N.; Mudrik, N.N.; Babij, A.V.; Pavlova, G.V.; Golovin, A.V.; et al. Aptamer RA36 inhibits of human, rabbit, and rat plasma coagulation activated with thrombin or snake venom coagulases. Bull. Exp. Biol. Med. 2013, 156, 44–48. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-J.; Tsai, C.-Y.; Hu, W.-P.; Chang, L.-S. DNA aptamers against Taiwan banded krait α-bungarotoxin recognize Taiwan cobra cardiotoxins. Toxins 2016, 8, 66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thiviyanathan, V.; Gorenstein, D.G. Aptamers and the next generation of diagnostic reagents. PROTEOMICS—Clin. Appl. 2012, 6, 563–573. [Google Scholar] [CrossRef] [Green Version]
- Gao, S.; Zheng, X.; Hu, B.; Sun, M.; Wu, J.; Jiao, B.; Wang, L. Enzyme-linked, aptamer-based, competitive biolayer interferometry biosensor for palytoxin. Biosens. Bioelectron. 2017, 89, 952–958. [Google Scholar] [CrossRef]
- Gao, S.; Hu, B.; Zheng, X.; Cao, Y.; Liu, D.; Sun, M.; Jiao, B.; Wang, L. Gonyautoxin 1/4 aptamers with high-affinity and high-specificity: From efficient selection to aptasensor application. Biosens. Bioelectron. 2016, 79, 938–944. [Google Scholar] [CrossRef]
- Marrazza, G. Aptamer sensors; Multidisciplinary Digital Publishing Institute: Basel, Switzerland, 2017. [Google Scholar]
- Ruscito, A.; DeRosa, M.C. Small-molecule binding aptamers: Selection strategies, characterization, and applications. Front. Chem. 2016, 4, 14. [Google Scholar] [CrossRef]
- Tuerk, C.; Gold, L. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science 1990, 249, 505–510. [Google Scholar] [CrossRef]
- Ellington, A.D.; Szostak, J.W. In vitro selection of RNA molecules that bind specific ligands. Nature 1990, 346, 818–822. [Google Scholar] [CrossRef] [PubMed]
- Groff, K.; Brown, J.; Clippinger, A.J. Modern affinity reagents: Recombinant antibodies and aptamers. Biotechnol. Adv. 2015, 33, 1787–1798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nezlin, R. Aptamers in immunological research. Immunol. Lett. 2014, 162, 252–255. [Google Scholar] [CrossRef]
- Ahmadvand, D.; Rahbarizadeh, F.; Moghimi, S.M. Biological targeting and innovative therapeutic interventions with phage-displayed peptides and structured nucleic acids (aptamers). Curr. Opin. Biotechnol. 2011, 22, 832–838. [Google Scholar] [CrossRef] [PubMed]
- Bouchard, P.R.; Hutabarat, R.M.; Thompson, K.M. Discovery and development of therapeutic aptamers. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 237–257. [Google Scholar] [CrossRef] [PubMed]
- Bock, L.C.; Griffin, L.C.; Latham, J.A.; Vermaas, E.H.; Toole, J.J. Selection of single-stranded DNA molecules that bind and inhibit human thrombin. Nature 1992, 355, 564–566. [Google Scholar] [CrossRef]
- Bagheri, E.; Abnous, K.; Alibolandi, M.; Ramezani, M.; Taghdisi, S.M. Triple-helix molecular switch-based aptasensors and DNA sensors. Biosens. Bioelectron. 2018, 111, 1–9. [Google Scholar] [CrossRef]
- Razmi, N.; Baradaran, B.; Hejazi, M.; Hasanzadeh, M.; Mosafer, J.; Mokhtarzadeh, A.; de la Guardia, M. Recent advances on aptamer-based biosensors to detection of platelet-derived growth factor. Biosens. Bioelectron. 2018, 113, 58–71. [Google Scholar] [CrossRef]
- Kim, Y.S.; Raston, N.H.A.; Gu, M.B. Aptamer-based nanobiosensors. Biosens. Bioelectron. 2016, 76, 2–19. [Google Scholar]
- Ronkainen, N.J.; Halsall, H.B.; Heineman, W.R. Electrochemical biosensors. Chem. Soc. Rev. 2010, 39, 1747–1763. [Google Scholar] [CrossRef]
- Chen, A.; Yang, S. Replacing antibodies with aptamers in lateral flow immunoassay. Biosens. Bioelectron. 2015, 71, 230–242. [Google Scholar] [CrossRef] [PubMed]
- Radom, F.; Jurek, P.M.; Mazurek, M.P.; Otlewski, J.; Jeleń, F. Aptamers: Molecules of great potential. Biotechnol. Adv. 2013, 31, 1260–1274. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Yang, J.; Liu, M.; Wu, Y.; Shen, Z.; Li, G. Sensitive detection of human breast cancer cells based on aptamer–cell–aptamer sandwich architecture. Anal. Chim. Acta 2013, 764, 59–63. [Google Scholar] [CrossRef] [PubMed]
- Keefe, A.D.; Pai, S.; Ellington, A. Aptamers as therapeutics. Nat. Rev. Drug Discov. 2010, 9, 537–550. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.H.; Elsherbiny, M.E.; Emara, M. Updates on aptamer research. Int. J. Mol. Sci. 2019, 20, 2511. [Google Scholar] [CrossRef] [Green Version]
- Vater, A.; Klussmann, S. Turning mirror-image oligonucleotides into drugs: The evolution of Spiegelmer® therapeutics. Drug Discov. Today 2015, 20, 147–155. [Google Scholar] [CrossRef] [Green Version]
- Nolte, A.; Klußmann, S.; Bald, R.; Erdmann, V.A.; Fürste, J.P. Mirror-design of L-oligonucleotide ligands binding to L-arginine. Nat. Biotechnol. 1996, 14, 1116–1119. [Google Scholar] [CrossRef]
- Odeh, F.; Nsairat, H.; Alshaer, W.; Ismail, M.A.; Esawi, E.; Qaqish, B.; Bawab, A.A.; Ismail, S.I. Aptamers chemistry: Chemical modifications and conjugation strategies. Molecules 2019, 25, 3. [Google Scholar] [CrossRef] [Green Version]
- Sundaram, P.; Kurniawan, H.; Byrne, M.E.; Wower, J. Therapeutic RNA aptamers in clinical trials. Eur. J. Pharm. Sci. 2013, 48, 259–271. [Google Scholar] [CrossRef]
- Bunka, D.H.J.; Platonova, O.; Stockley, P.G. Development of aptamer therapeutics. Curr. Opin. Pharmacol. 2010, 10, 557–562. [Google Scholar] [CrossRef]
- Toh, S.Y.; Citartan, M.; Gopinath, S.C.B.; Tang, T.-H. Aptamers as a replacement for antibodies in enzyme-linked immunosorbent assay. Biosens. Bioelectron. 2015, 64, 392–403. [Google Scholar] [CrossRef] [PubMed]
- Ni, S.; Yao, H.; Wang, L.; Lu, J.; Jiang, F.; Lu, A.; Zhang, G. Chemical modifications of nucleic acid aptamers for therapeutic purposes. Int. J. Mol. Sci. 2017, 18, 1683. [Google Scholar] [CrossRef] [PubMed]
- Kong, H.Y.; Byun, J. Nucleic acid aptamers: New methods for selection, stabilization, and application in biomedical science. Biomol. Therapeut. 2013, 21, 423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischer, M.J.M.; Schmidt, J.; Koulchitsky, S.; Klussmann, S.; Vater, A.; Messlinger, K. Effect of a calcitonin gene-related peptide-binding L-RNA aptamer on neuronal activity in the rat spinal trigeminal nucleus. J. Headache Pain 2018, 19, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruno, J.G.; Carrillo, M.P.; Phillips, T.; Vail, N.K.; Hanson, D. Competitive FRET-aptamer-based detection of methylphosphonic acid, a common nerve agent metabolite. J. Fluoresc. 2008, 18, 867–876. [Google Scholar] [CrossRef]
- Sun, W.; Du, L.; Li, M. Aptamer-based carbohydrate recognition. Curr. Pharm. Des. 2010, 16, 2269–2278. [Google Scholar] [CrossRef]
- Ma, L.-H.; Wang, H.-B.; Fang, B.-Y.; Tan, F.; Cao, Y.-C.; Zhao, Y.-D. Visual detection of trace lead ion based on aptamer and silver staining nano-metal composite. Colloids Surf. B Biointerfaces 2018, 162, 415–419. [Google Scholar] [CrossRef]
- Emahi, I.; Gruenke, P.R.; Baum, D.A. Effect of aptamer binding on the electron-transfer properties of redox cofactors. J. Mol. Evol. 2015, 81, 186–193. [Google Scholar] [CrossRef]
- Chabata, C.V.; Frederiksen, J.W.; Sullenger, B.A.; Gunaratne, R. Emerging applications of aptamers for anticoagulation and hemostasis. Curr. Opin. Hematol. 2018, 25, 382–388. [Google Scholar] [CrossRef]
- Song, K.-M.; Lee, S.; Ban, C. Aptamers and their biological applications. Sensors 2012, 12, 612–631. [Google Scholar] [CrossRef] [Green Version]
- Lauridsen, L.H.; Shamaileh, H.A.; Edwards, S.L.; Taran, E.; Veedu, R.N. Rapid one-step selection method for generating nucleic acid aptamers: Development of a DNA aptamer against α-bungarotoxin. PLoS ONE 2012, 7, e41702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anand, A.; Chatterjee, B.; Dhiman, A.; Goel, R.; Khan, E.; Malhotra, A.; Santra, V.; Salvi, N.; Khadilkar, M.V.; Bhatnagar, I.; et al. Complex target SELEX-based identification of DNA aptamers against Bungarus caeruleus venom for the detection of envenomation using a paper-based device. Biosens. Bioelectron. 2021, 193, 113523. [Google Scholar] [CrossRef] [PubMed]
- Alomran, N.; Blundell, P.; Alsolaiss, J.; Crittenden, E.; Ainsworth, S.; Dawson, C.A.; Edge, R.J.; Hall, S.R.; Harrison, R.A.; Wilkinson, M.C.; et al. Exploring the utility of recombinant snake venom serine protease toxins as immunogens for generating experimental snakebite antivenoms. Toxins 2022, 14, 443. [Google Scholar] [CrossRef]
- Alomran, N.; Alsolaiss, J.; Albulescu, L.O.; Crittenden, E.; Harrison, R.A.; Ainsworth, S.; Casewell, N.R. Pathology-specific experimental antivenoms for haemotoxic snakebite: The impact of immunogen diversity on the in vitro cross-reactivity and in vivo neutralisation of geographically diverse snake venoms. PLoS Negl. Trop. Dis. 2021, 15, e0009659. [Google Scholar] [CrossRef]
- Castro, H.C.; Zingali, R.B.; Albuquerque, M.G.; Pujol-Luz, M.; Rodrigues, C.R. Snake venom thrombin-like enzymes: From reptilase to now. Cell. Mol. Life Sci. CMLS 2004, 61, 843–856. [Google Scholar] [CrossRef]
- Serrano, S.M.T.; Maroun, R.C. Snake venom serine proteinases: Sequence homology vs. substrate specificity, a paradox to be solved. Toxicon 2005, 45, 1115–1132. [Google Scholar] [CrossRef] [PubMed]
- Clauss, A. Gennungsphysiologishe schnellmethode sur Bestimmung des Fibrinogens. Acta Haematol. 1957, 17, 237–246. [Google Scholar] [CrossRef]
- Knudsen, C.; Ledsgaard, L.; Dehli, R.I.; Ahmadi, S.; Sørensen, C.V.; Laustsen, A.H. Engineering and design considerations for next-generation snakebite antivenoms. Toxicon 2019, 167, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Liu, Q.X.; Guo, Z.H.; Lin, J.S. Practical application of aptamer-based biosensors in detection of low molecular weight pollutants in water sources. Molecules 2018, 23, 344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eissa, S.; Ng, A.; Siaj, M.; Tavares, A.C.; Zourob, M. Selection and identification of DNA aptamers against okadaic acid for biosensing application. Anal. Chem. 2013, 85, 11794–11801. [Google Scholar] [CrossRef]
- Eissa, S.; Ng, A.; Siaj, M.; Zourob, M. Label-free voltammetric aptasensor for the sensitive detection of microcystin-LR using graphene-modified electrodes. Anal. Chem. 2014, 86, 7551–7557. [Google Scholar] [CrossRef] [PubMed]
- Eissa, S.; Siaj, M.; Zourob, M. Aptamer-based competitive electrochemical biosensor for brevetoxin-2. Biosens. Bioelectron. 2015, 69, 148–154. [Google Scholar] [CrossRef] [PubMed]
- El-Aziz, T.M.; Ravelet, C.; Molgo, J.; Fiore, E.; Pale, S.; Amar, M.; Al-Khoury, S.; Dejeu, J.; Fadl, M.; Ronjat, M.; et al. Efficient functional neutralization of lethal peptide toxins in vivo by oligonucleotides. Sci. Rep. 2017, 7, 7202. [Google Scholar] [CrossRef] [Green Version]
- Kini, R.M. Serine proteases affecting blood coagulation and fibrinolysis from snake venoms. Pathophysiol. Haemost. Thrombos. 2005, 34, 200–204. [Google Scholar] [CrossRef] [PubMed]
- Brody, E.N.; Gold, L. Aptamers as therapeutic and diagnostic agents. Rev. Mol. Biotechnol. 2000, 74, 5–13. [Google Scholar] [CrossRef]
- Ravelet, C.; Grosset, C.; Peyrin, E. Liquid chromatography, electrochromatography and capillary electrophoresis applications of DNA and RNA aptamers. J. Chromatogr. A 2006, 1117, 1–10. [Google Scholar] [CrossRef]
- Cao, Z.; Tan, W. Molecular Aptamers for Real-Time Protein-Protein Interaction Study. Chem. Eur. J. 2005, 11, 4502–4508. [Google Scholar] [CrossRef]
- Chen, Z.; Ruffner, D.E. Modified crush-and-soak method for recovering oligodeoxynucleotides from polyacrylamide gel. Biotechniques 1996, 21, 820–822. [Google Scholar] [CrossRef]
- Simossis, V.A.; Heringa, J. The PRALINE online server: Optimising progressive multiple alignment on the web. Comput. Biol. Chem. 2003, 27, 511–519. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target | Aptamer Identifier | The Sequence of the Random Region (5′-3′) |
|---|---|---|
| ancrod | ancrod-1 | TGCTCACACGTCCTGTGTGATTATGTCAGGCATTCACATG |
| ancrod-2 | TGCTGGGAAATCCTCCCATTATGTCAGTATGTCTCGACAT | |
| ancrod-5 | ACGCTTGATCCTCCGAAATGTCCTGATCCTCGGCCTGTCA | |
| ancrod-7 | TAGCATGGGTGGTCAATTTAAGTACAGTGTCGTGCTCACT | |
| ancrod-11 | TGGTCTAAGGACTGCTTAGGATTGCGATATGGTCCAGATG | |
| ancrod-12 | GTAAATTGTACAGGTGTATGGATTGCTAGGTCTGCTGGTT | |
| ancrod-18 | TGTCTGGTTTGCAAAGGACTGCTGTACTGTTAGCTTTTGT | |
| ancrod-25 | GGTGCGTTTCACCTCGAGTTTACGATAAATCACCTTCGAG | |
| ancrod-30 | TATTAAGGGACTGCTCGGGATTGCGGATATAGGTATGAGC | |
| ancrod-31 | GTGTATTGTGATAGTCGGTAATTCCCTGACTACGCCGTAT | |
| ancrod-32 | TTGGGCCCTCTAGTGATGGATATCTGCAGAATTCGCCCTT | |
| ncrod-48 | TAGTAACAGGTCTGCTTAGGCTTGCGAGGAATACTAGTAC | |
| ancrod-55 | GGACCGACCCTTTAGCATTTATGACCCTTGTCATCGGGCT | |
| batroxobin | batroxobin-4 | AGGTGGTCAGCTTTATCCTTTATGACCTTAACCCGTCATG |
| batroxobin-5 | TAGTAACAGGTCTGCTTAGGCTTGCGAGGAGTACTAGTAT | |
| batroxobin-6 | AGGTGGATATCAAGATAGGTTTGGTTAGGTAGCGTTCTTG | |
| batroxobin-14 | GGACCGACCCTTTAGCATTTATGACCCTTGTCATCGGGCT | |
| batroxobin-18 | TGCTGGGAAATCCTCCCATTATGTCAGTATGTCTCAACAT | |
| batroxobin-21 | AGGGGGCGACCTTTAATGCTTGTGATCCTTATCCGTCATC | |
| batroxobin-26 | TGTCTGGTATGCAAAGGACTGCTGTACTGTTAGCTTTTGT | |
| batroxobin-28 | TAGCATGGGTGGTCAATTTAAGTACAGTGTCGTGCTCACT | |
| batroxobin-29 | AGGTCCTATTGTATACAGGGAGCCCTCGGTCTTGCTGTGA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alomran, N.; Chinnappan, R.; Alsolaiss, J.; Casewell, N.R.; Zourob, M. Exploring the Utility of ssDNA Aptamers Directed against Snake Venom Toxins as New Therapeutics for Snakebite Envenoming. Toxins 2022, 14, 469. https://doi.org/10.3390/toxins14070469
Alomran N, Chinnappan R, Alsolaiss J, Casewell NR, Zourob M. Exploring the Utility of ssDNA Aptamers Directed against Snake Venom Toxins as New Therapeutics for Snakebite Envenoming. Toxins. 2022; 14(7):469. https://doi.org/10.3390/toxins14070469
Chicago/Turabian StyleAlomran, Nessrin, Raja Chinnappan, Jaffer Alsolaiss, Nicholas R. Casewell, and Mohammed Zourob. 2022. "Exploring the Utility of ssDNA Aptamers Directed against Snake Venom Toxins as New Therapeutics for Snakebite Envenoming" Toxins 14, no. 7: 469. https://doi.org/10.3390/toxins14070469
APA StyleAlomran, N., Chinnappan, R., Alsolaiss, J., Casewell, N. R., & Zourob, M. (2022). Exploring the Utility of ssDNA Aptamers Directed against Snake Venom Toxins as New Therapeutics for Snakebite Envenoming. Toxins, 14(7), 469. https://doi.org/10.3390/toxins14070469