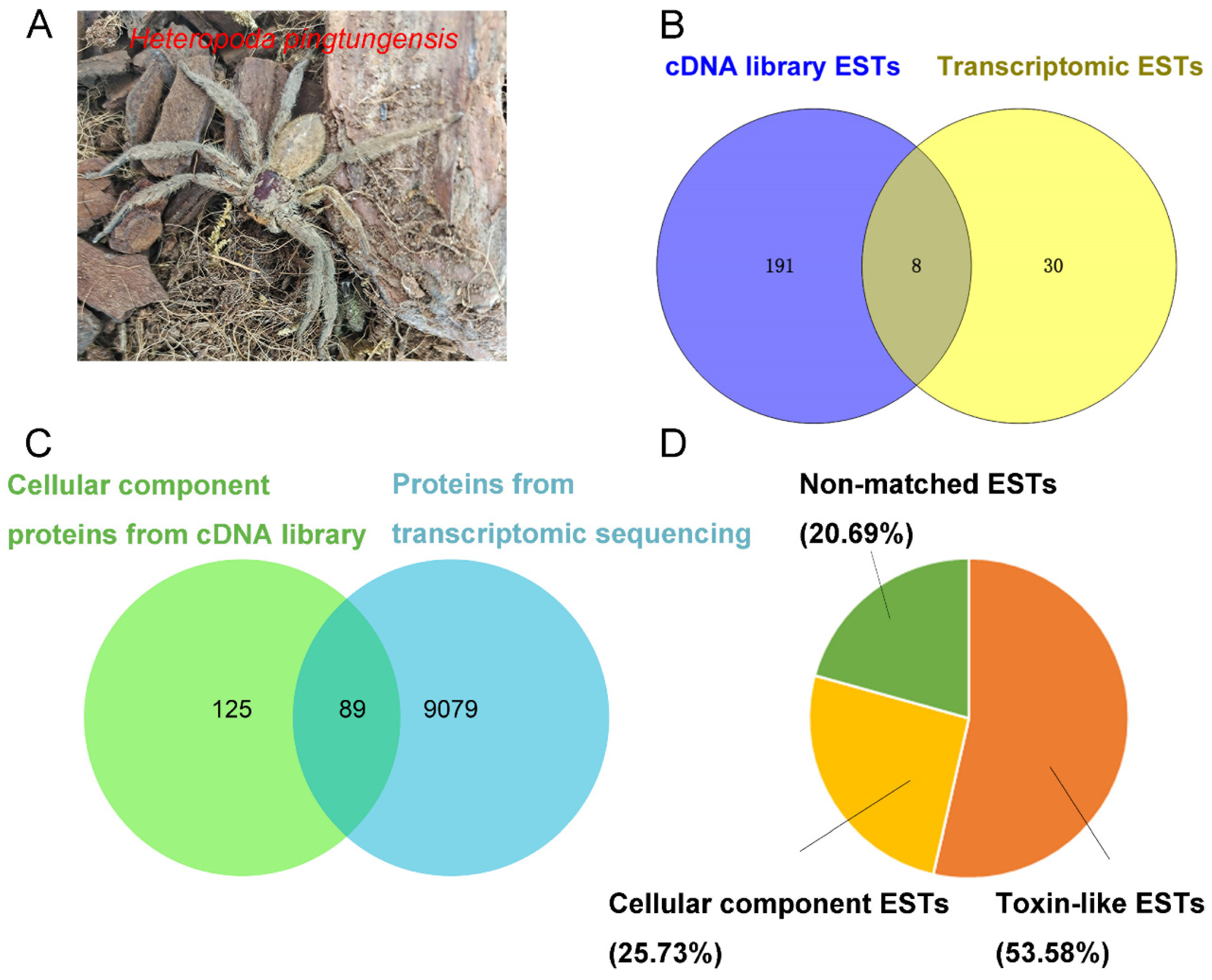
2.2. Cluster Analysis of H. Pingtungensis Venom Gland ESTs
As shown in
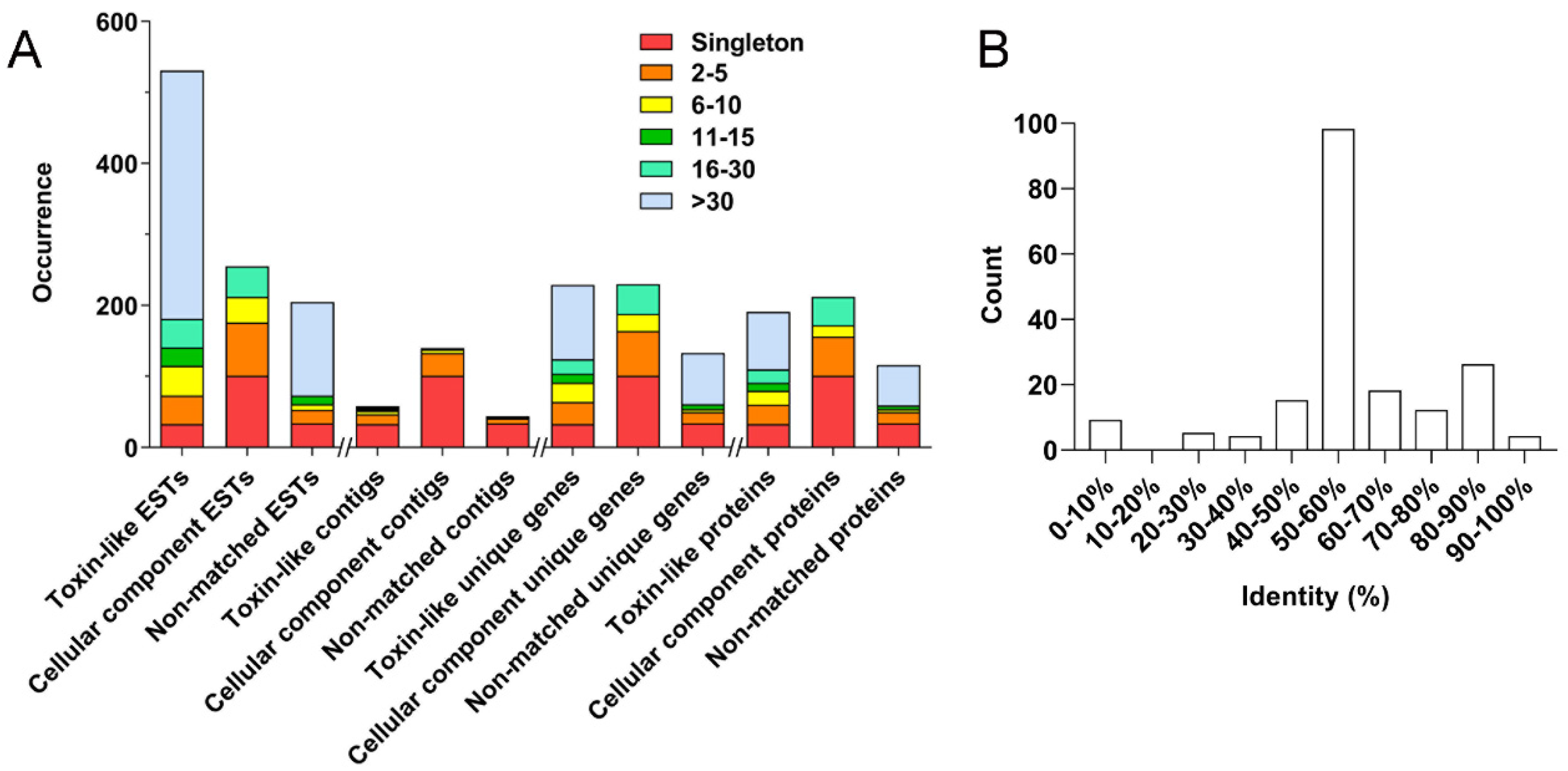
Figure 2A, cluster analysis of the 991 ESTs results in 242 clusters, including 168 singletons and 74 contigs of different sizes (2–5, 6–10, 11–15, 16–30, and >30, which indicates the number of clustered sequences in each contig). Firstly, 531 toxin-like ESTs are grouped into 58 clusters, including 33 singletons and 25 contigs: (1) 14 contigs of size 2–5 contain 40 ESTs, which represent 31 unique genes encoding 27 proteins; (2) 5 contigs of size 6–10 contain 42 ESTs, which represent 27 unique genes encoding 20 proteins; (3) 2 contigs of size 11–15 contain 26 ESTs, which represent 13 unique genes encoding 11 proteins; (4) 2 contigs of size 16–30 contain 40 ESTs, which represent 20 unique genes encoding 19 proteins; (5) 2 contig of size >30 contains 350 ESTs, which represent 105 unique genes encoding 80 proteins.
Secondly, the cellular component ESTs are grouped into 140 clusters, including 101 singletons and 39 contigs: (1) 32 contigs of size 2–5 contain 75 ESTs, which represent 63 unique genes encoding 55 proteins; (2) 5 contigs of size 6–10 contain 36 ESTs, which represent 24 unique genes encoding 16 proteins; (3) 2 contigs of size 16–30 contain 43 ESTs, which represent 42 unique genes encoding 40 proteins. No contigs of size 11–15 and >30 are observed in this category.
Lastly, the non-matched ESTs are grouped into 44 clusters, including 34 singletons and 10 contigs: (1) 7 contigs of size 2–5 contain 19 ESTs, which represent 16 unique genes encoding 16 proteins; (2) 1 contig of size 6–10 contain 8 ESTs, which represent 4 unique genes encoding 4 proteins; (3) 1 contig of size 11–15 contain 12 ESTs, which represent 7 unique genes encoding 5 proteins; (4) 1 contig of size > 30 contain 132 ESTs, which represent 72 unique genes encoding 57 proteins. No contigs of size 16–30 are presented in this category.
From the above analysis, one can find that most toxin-like ESTs (93.79%) are grouped into contigs, which are consistent with the notion that most peptide toxin transcripts are always high-copy in the venom gland. The high-abundance ESTs in the cellular component category, however, mostly encode ribosomal proteins, which is also in line with the active protein synthesis in the venom gland. Accordingly, a total of 518 putative non-redundant proteins were identified in the
H. pingtungensis venom gland, including 190 toxin precursors, 212 cellular component proteins, and 116 non-matched proteins. With BLAST, all these toxin precursors from
H. pingtungensis against the toxins sequence database from the close spider species,
H. venatoria, showed that their toxins are generally of medium homology, with most of the toxins (98 out of a total of 190) from
H. pingtungensis showing 50–60% sequence identity with those from
H. venatoria (
Figure 2B).
2.3. Family Analysis of Putative Toxin Precursors in H. Pingtungensis Venom Gland
We named the toxin-like peptide from
H. pingtungensis as HptTx-n, in which ‘Hpt’, ‘Tx’, and ‘n’ represents
H. pingtungensis, toxin, and the clone number, respectively. In some cases, the capital letter ‘P’ or ‘T’ is added to the end of the toxin name to indicate that toxin is a partial sequence (HptTx-n-P) or derived from transcriptomic data (HptTx-n-T), respectively. BLAST analysis revealed that 176 out of 190 toxin precursors are likely of full-length with the complete signal peptide and mature peptide, for which the signal peptide was predicted using signalP5.0 (
https://services.healthtech.dtu.dk/service.php?SignalP-5.0; accessed on 23 January 2022) [
20], and the cut site between the propeptide and predicted mature peptide was determined by the Processing Quadruplet Motif (PQM) mode [
21,
22]. Sequences of all these novel toxins were deposited in the Genbank database (
https://www.ncbi.nlm.nih.gov/genbank/; accessed on 24 January 2022) (Genbank accession numbers: OM362623—OM362812). As shown in
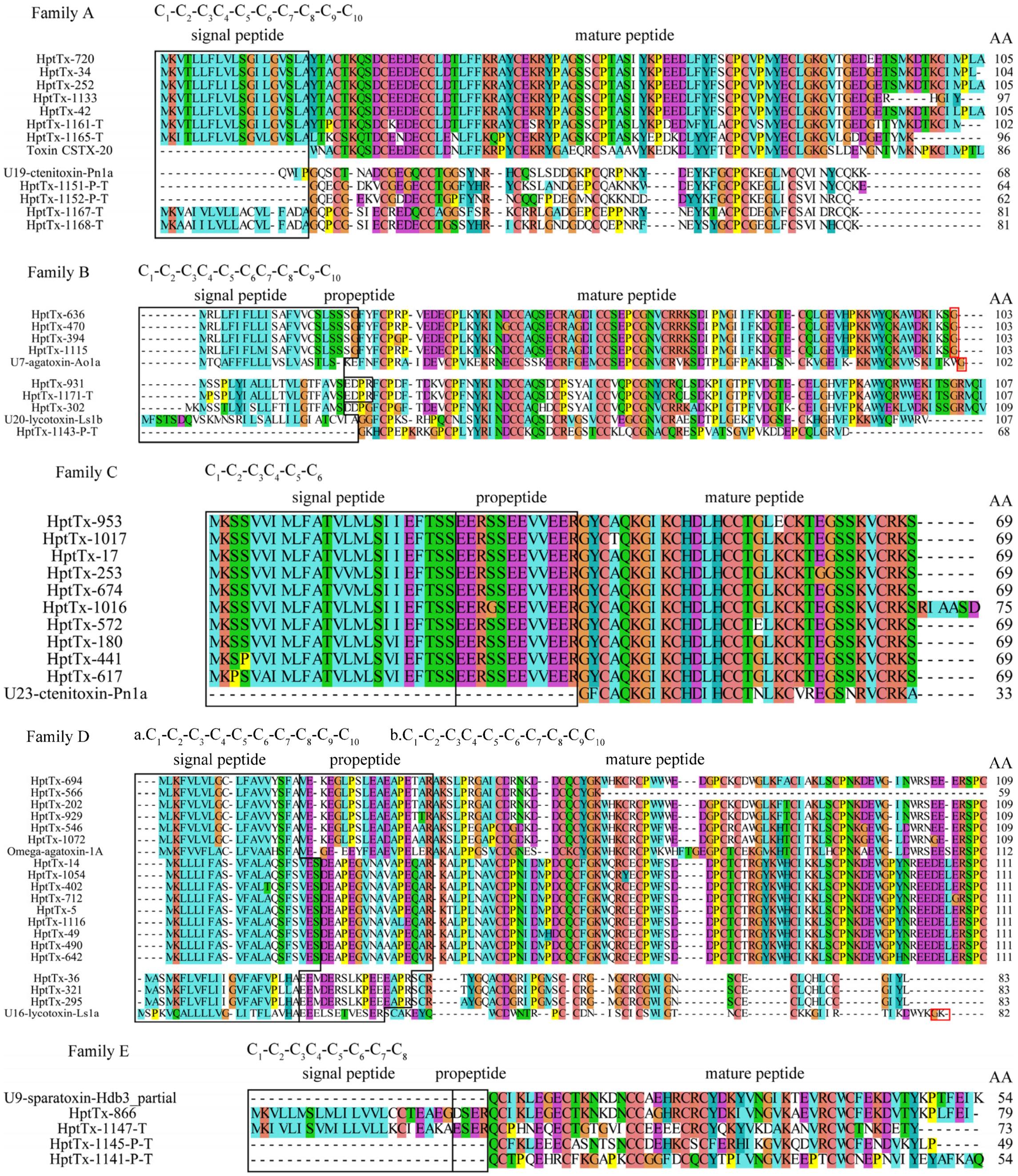
Figure 3, these toxins were grouped into 13 families based on their sequence homology and cysteine frameworks.
2.3.1. Family A
Family A contains 11 precursor toxins, all of which lack a typical propeptide cutting site. (1) HptTx-720, HptTx-34, HptTx-252, HptTx-1133, HptTx-42, HptTx-1161-T, and HptTx-1165-T are a subset of toxins showing the highest homology with toxin CSTX-20 (64–70% identity) from the spider
Cupiennius salei (
American wandering spider) and U4-sparatoxin-Hv1a (75–91% identity) from the spider
Heteropoda venatoria, when blasted against the Uniprot and NR databases, respectively. Both these two hit toxins’ biological functions are unknown. HpTx-1161-T and HpTx-1165-T are derived from transcriptome sequencing and thus might represent the artificially C-terminus truncated peptides. Interestingly, HptTx-1133 lacks the most C-terminal cysteine residue, whereas its cDNA contains a complete 3′ poly-A tail, suggesting it is a full-length peptide with its last cysteine residue mutated. (2) HptTx-1151-P-T, HptTx-1152-P-T, HptTx-1167-T, and HptTx-1168-T are derived from transcriptomic data and make another cluster of toxins in this family. Their predicted mature peptides showed the highest homology with U19-ctenitoxin-Pn1a (54–70% identity) from the spider
Phoneutria nigriventer (
Brazilian armed spider), which is non-toxic to mice and insects. HptTx-1167-T and HptTx-1168-T contain the complete signal peptide, whereas HptTx-1151-P-T and HptTx-1152-P-T are likely just the mature peptides. It is unknown whether their mature peptides are complete. All the predicted mature peptides for toxins in this family share a consensus cysteine framework as C
1-C
2-C
3C
4-C
5-C
6-C
7-C
8-C
9-C
10. This cysteine pattern is also seen in the snake toxin, Toxin MIT1, in which the cysteine connecting mode is determined as C
1-4, C
2-5, C
3-7, C
6-9, and C
8-10 [
23]. We supposed that these toxins have the same disulfide mode as Toxin MIT1.
2.3.2. Family B
Family B contains eight precursor sequences. Except for HptTx-1143-P-T, which is derived from transcriptome sequencing and has the lowest homology with other toxins in this family, all the other eight toxins are of full-length. HptTx-931, HptTx-1171-T, and HptTx-302 cluster into a subset showing the highest homology with U20-lycotoxin-Ls1b (51–52% identity) from the spider Lycosa Singoriensis and U6-sparatoxin-Hv1h (80–84% identity) from the spider Heteropoda venatoria, in the Uniprot and NR databases, respectively. The former was predicted to have antibacterial activity. Compared with HptTx-931 and HptTx-1171-T, HptTx-302 is supposed to lack the propeptide due to an R to G mutation at the PQM region. HptTx-636, HptTx-470, HptTx-394, and HptTx-1115 cluster into another group, with the signal peptide cutting site being determined as SSG/FY. No propetide is presented in their sequences. Blasting analysis showed they share the highest homology with U7-agatoxin-Ao1a from the spider Agelena orientalis (52–53% identity, Uniprot database) and U7-sparatoxin-Hv1a_1 from the spider Heteropoda venatoria (87–88% identity, NR database), both of whose biological functions are unknown. Predicted mature peptides for toxin precursors in this family have a consensus cysteine framework as C1-C2-C3C4-C5-C6C7-C8-C9-C10, and the putative disulfide mode is C1-C7, C2-C8, C3-C6, C4-C10, and C5-C9 based on similarity analysis.
2.3.3. Family C
The ten peptide precursors in family C showed great homology with each other, with a variation of residues being only present in the signal peptide and/or propeptide regions in several members. All of them are from cDNA library sequencing and are of full-length. The predicted mature peptides of this family are best aligned to the toxin U23-ctenitoxin-Pn1a from the spider Phoneutria nigriventer (73–76% identity, Uniprot database) and U22-sparatoxin-Hv1a from the spider Heteropoda venatoria (79–82% identity, NR database). Both these two toxins’ biological functions are unknown, but the former was shown to be non-toxic to mice. Interestingly, HptTx-1016 has a longer mature peptide than the others, which might be caused by a single nucleotide mutation in the stop codon, as revealed by analyzing their cDNA sequences (the first stop codon TGA in several other toxins is mutated to CGA in HptTx-1016). It is unknown whether such a mature peptide extension would render new function to this toxin. Furthermore, their classic cysteine framework, C1-C2-C3C4-C5-C6, as observed in most inhibitor cysteine knot (ICK) motif spider toxins, strongly suggests the mature peptides in this family are also knotting toxins (disulfide mode: C1-C4, C2-C5, and C3-C6).
2.3.4. Family D
There are three clusters of peptide precursors in this family, which all contain a signal peptide and a propeptide. Among them, the first cluster of toxins (HptTx-694, HptTx-566, HptTx-202, HptTx-929, HptTx-546, and HptTx-1072) match the best with toxin omega-agatoxin-1A from the spider
Agelenopsis aperta in the Uniprot database (58–65% identity and 71–75% similarity). This toxin is previously determined to be a blocker of presynaptic calcium channels at the insect neuromuscular junction [
24,
25]. The high sequence homology of their mature peptides with that of omega-agatoxin-1A suggests that they may share the same biological targets and play a key role for
H. pingtungensis to prey on insects. The second group of toxins, including HptTx-14, HptTx-1054, HptTx-402, HptTx-712, HptTx-5, HptTx-1116, HptTx-49, HptTx-490, and HptTx-642, are also best aligned with omega-agatoxin-1A but with lower homology than the first cluster of toxins (47–49% identity and 57–59% similarity). Both these two classes of toxins have a consensus cysteine framework as C
1-C
2-C
3-C
4-C
5-C
6-C
7-C
8-C
9-C
10, the same as omega-agatoxin-1A. The other three toxins in this family (HptTx-36, HptTx-321, and HptTx-295), however, have another different cysteine framework with their last two cysteines directly connected (C
1-C
2-C
3-C
4-C
5-C
6-C
7-C
8-C
9C
10). Moreover, blasting analysis revealed that they are best aligned to, although with relatively low homology, U16-lycotoxin-Ls1a (35–36% identity), which was predicted to have ion channel inhibitor activity but is currently not experimentally verified.
2.3.5. Family E
Family E contains four toxin precursors, with HptTx-866 being derived from the cDNA library and the other three from transcriptome sequencing. Consequently, it remains elusive whether the mature peptides in the latter three toxins are complete. Among them, HptTx-1147-T has the signal peptide and propeptide, whereas HptTx-1145-P-T and HptTx-1141-P-T seem to be just the mature peptides. The overall sequence homology between these four toxins is relatively low but they share a consensus cysteine framework as C1-C2-C3C4-C5-C6-C7-C8. Blasting analysis revealed they all have moderate sequence homology with U6-lycotoxin-Ls1g from the spider Lycosa singoriensis (40–54% identity, Uniprot database), which has an unknown biological function. Moreover, blasting against the NR database showed that HptTx-1141-P-T has the same mature peptide as U9-sparatoxin-Hdb4, partial (100% identity), and HptTx-866 has extremely high homology with U9-sparatoxin-Hdb3, partial (91% identity), from the spider Heteropoda davidbowie.
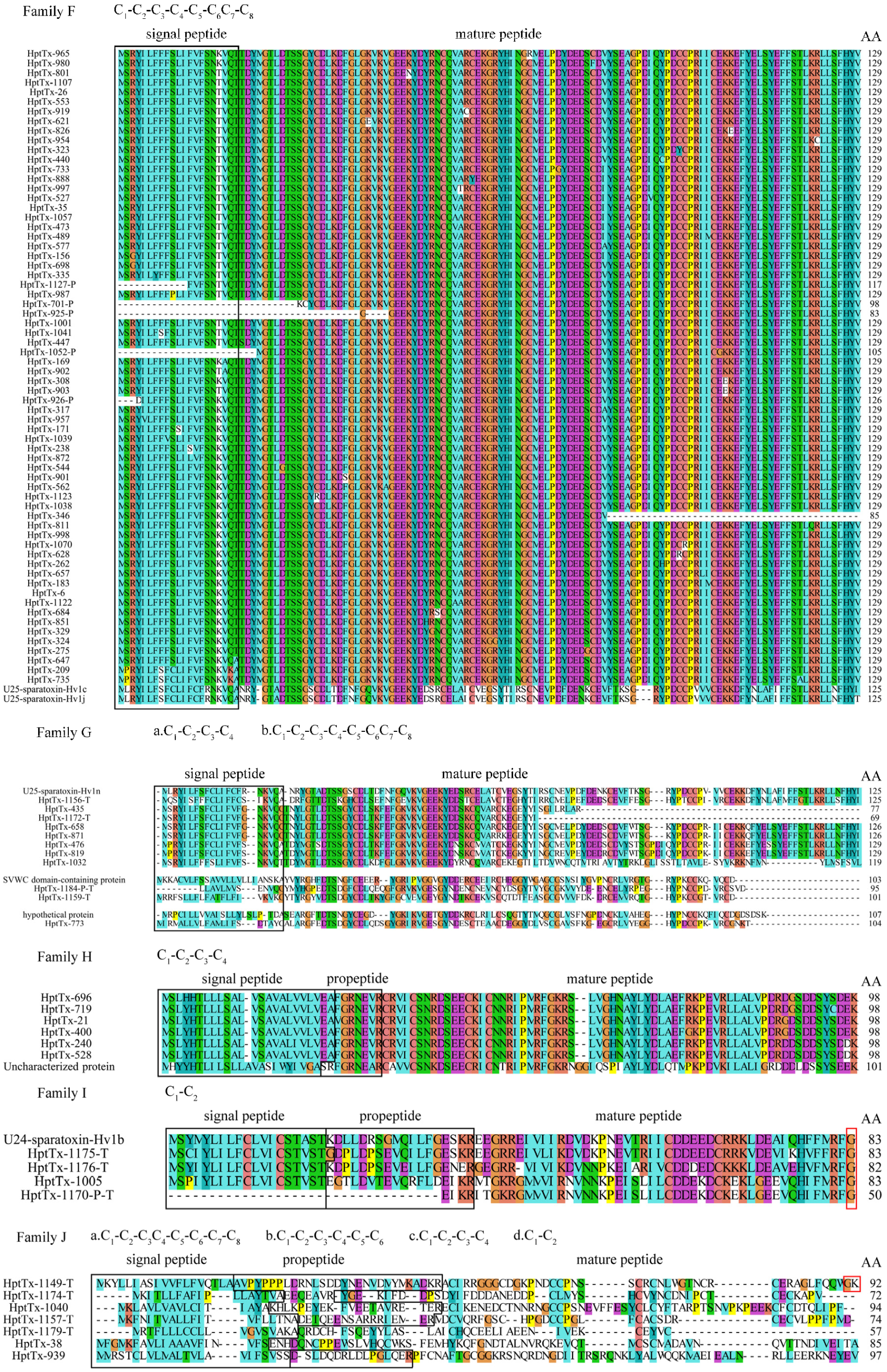
2.3.6. Family F
There are 66 toxin precursors in family F, which represents the most abundant cluster in the cDNA library. Five toxins (HptTx-1127-P, HptTx-701-P, HptTx-925-P, HptTx-926-P, and HptTx-1052-P) in this family with a truncated N-terminus should be caused by mRNA fragmentation during cDNA library construction, whereas the toxin HptTx-346 with a truncated C-terminus is generated by a tyrosine codon (UAC) mutation to the stop codon (UAG). These toxins are of extremely high homology and are made of short signal peptides and rather long predicted mature peptides, which have a consensus cysteine framework of C
1-C
2-C
3-C
4-C
5-C
6C
7-C
8. However, there are some toxins with mutations at the conserved cysteine sites, including HptTx-980, HptTx-965, HptTx-1123, HptTx-323, HptTx-628, HptTx-888, HptTx-925-P, HptTx-1052-P, and HptTx-1070. On the other hand, HptTx-701-P, HptTx-440, HptTx-919, and HptTx-954 have an additional cysteine in their sequences except for the common cysteine framework. Blasting analysis of their sequences against the UniProt database results in no significant hits, which makes it impossible to predict their biological functions. However, they showed moderate homology with U25-sparatoxin-Hv1c and U25-sparatoxin-Hv1j from the spider
Heteropoda venatoria in the NR database (58–62% identity and 75–80% similarity). Interestingly, the transcripts for these two hit toxins are also the top two most abundant in the venom cDNA library of
H. Venatoria [
11], raising the possibility that this class of toxins plays an important function in prey and defense for these two spider species.
2.3.7. Family G
The eleven toxin precursors in this family can be divided into three groups, with no typical propeptide cutting sites found in these toxins. HptTx-1156-T, HptTx-658, HptTx-476, and HptTx-819 have a consensus cysteine framework of C1-C2-C3-C4-C5-C6C7-C8, as those toxins in family F. HptTx-871 have high sequence homology with HP-658 but its third cysteine (C3) is mutated to arginine. The sequence of HptTx-435 is much shorter than the others because of a premature termination codon in its sequence, and HptTx-1172-T derived from the transcriptomic data might represent an artificial C-terminus truncated peptide. These toxins showed the highest homology with U25-sparatoxin-Hv1n from the spider Heteropoda venatoria (67–71% identity). Unlike other U25-sparatoxin-Hv1n related peptides in this subset, HptTx-1032 has a different cysteine framework of C1-C2-C3-C4. HptTx-773, HptTx-1184-P-T, and HptTx-1159-T also have a cysteine pattern of C1-C2-C3-C4-C5-C6C7-C8, with HptTx-773 being best aligned with hypothetical protein AVEN_267295-1 from spider Araneus ventricosus (44% identity), and the latter two toxins have moderate sequence homology with the SVWC domain-containing protein from the spider Caerostris darwini (42–45% identity). Furthermore, the predicted mature peptides in HptTx-1184-P-T and HptTx-1159-T might not be complete. We are unable to predict the functions of toxins in family G based on sequence homology analysis.
2.3.8. Family H
There are six toxin precursor peptides in this family, all of which are derived from cDNA libraries. Except for HptTx-719, which has an extra cysteine residue at its C-terminus, all other toxins in this family have a cysteine pattern of C1-C2-C3-C4. A short propeptide is presented between their signal and the predicted mature peptides. The overall sequence homology between family H members is extremely high, and blasting analysis showed they are best aligned with uncharacterized protein TNCT_234811 from the spider Trichonephila clavate (55–57% identity and 70–71% similarity, NR database), which has an unknown biological function.
2.3.9. Family I
This family has four toxin precursor sequences, whose predicted mature peptides have only two cysteines (cysteine framework: C
1-C
2). HptTx-1005 is derived from cDNA library sequencing, and HptTx-1175-T, HptTx-1176-T, and HptTx-1170-P-T are from transcriptomic data. These four toxin precursors have completed mature peptides, with an amidation signal at their C-termini (the C-terminal glycine residue), which would result in amidation of its upstream phenylalanine residue at the carboxyl group. All these toxins are bested aligned with U24-sparatoxin-Hv1b from the spider
Heteropoda venatoria (52–82% identity, NR database), which has an unknown biological function. Interestingly, the predicted mature toxins from family I members all have an -RFamide sequence at their C-termini, which is a characteristic feature of FMRFamide-related peptides (FaRPs), therefore, these toxins are also likely to activate respective GPCRs(G-protein-coupled receptors). Actually, FaRPs are also identified in other toxic animals, such as the solitary wasp and cone snail [
26,
27].
2.3.10. Family J
Family J contains seven toxin precursors that are of very low sequence homology. Among them, HptTx-1149-T, HptTx-1174-T, HptTx-1157-T, and HptTx-1179-T are derived from transcriptomic data. HptTx-1149-T has a complete mature peptide, as revealed by a typical amidation motif (GK) at its C-terminus, whereas the mature peptide of HptTx-1179-T might be incomplete, as only five cysteines are presented in its sequence. It is unknown whether HptTx-1174-T and HptTx-1157-T have complete mature peptides. (1) HptTx-1149-T matches the best with the U8-agatoxin-Ao1a toxin from the spider Stegodyphus dumicola (37% identity) and U8-agatoxin-Ao1a-like toxin from the spider Stegodyphus dumicola (76% identity), in the Uniprot and NR database, respectively. (2) HptTx-1174-T has a cysteine pattern of C1-C2-C3-C4-C5-C6. Blasting analysis revealed its high homology with U1-sparatoxin-Hv1a from the spider Heteropoda venatoria (77% identity and 82% similarity). (3) HptTx-1040 is best aligned with U1-lycotoxin-Ls1b (33.7% identity, Uniprot database) from the spider Lycosa singoriensis and U3-sparatoxin-Hdb1, partial from the spider Heteropoda davidbowie (81% identity, NR database). (4) HptTx-1157-T is best aligned with U29-sparatoxin-Hv1a (65% identity, NR database) from the spider Heteropoda venatoria. Notably, HptTx-1149-T, HptTx-1040, and HptTx-1157-T share a consensus cysteine framework as C1-C2-C3C4-C5-C6-C7-C8, which is also a typical pattern in ICK motif toxins. The disulfide mode in them is speculated to be C1-4, C2-5, C3-8, C6-7. (5) HptTx-1179-T and HptTx-38 have no propeptides in their sequences. Blasting analysis showed HptTx-1179-T has the highest homology with U27-sparatoxin-Hv1c (79% identity) from the spider Heteropoda venatoria. (6) HptTx-38 has a cysteine framework of C1-C2-C3-C4 and is best aligned with hypothetical protein X975_08641, partial from the spider Stegodyphus mimosarum (64% identity). (7) HptTx-939 only has two cysteines in sequence (C1-C2). Blasting analysis showed it matches uncharacterized protein TNCT_72511 from the spider Trichonephila clavate (66% identity). We are unable to predict the biological functions of these toxins in family J as the functions of all their hit toxins in the database are unknown.
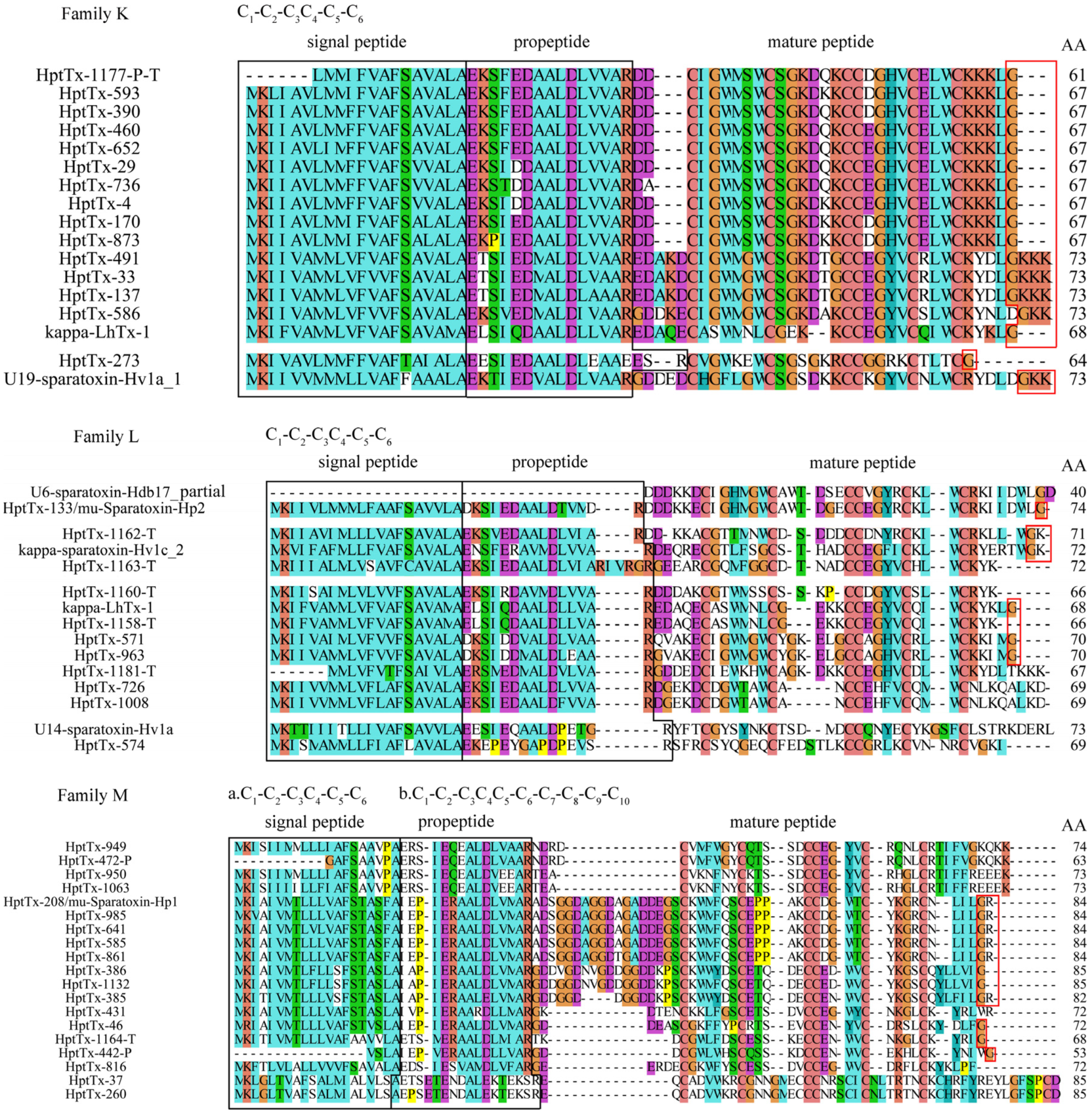
2.3.11. Family K
There are 15 toxin precursors in family K, all of which have a propeptide in their sequences. Their predicted mature peptides share a consensus cysteine framework popularly found in ICK motif toxins (C
1-C
2-C
3C
4-C
5-C
6), suggesting the disulfide mode is C1-C
4, C
2-C
5, and C
3-C
6. The C-terminal amidation signals are observed in family K members (red-boxed region), which implies the C-termini of these toxins are amidated when expressed in the venom. These toxins can be grouped into three subsets based on their sequence homology: cluster 1 (HptTx-1177-P, HptTx-460, HptTx-652, HptTx-390, HptTx-29, HptTx-4, HptTx-736, HptTx-170, HptTx-873, and HptTx-593), cluster 2 (HptTx-491, HptTx-33, HptTx-137, and HptTx-586), and cluster 3 (HptTx-273 alone). Blasting analysis revealed that most toxins, except for HptTx-273, in this family are best aligned with kappa-LhTx-1 from the spider
Pandercetes sp, with cluster 2 toxins showing slightly higher sequence homology (65–70% identity and 75–80% similarity) than that of cluster 1 toxins (57–61% identity and 72–81% similarity). This spider toxin was proved in our previous study to be a Kv4 channels’ specific antagonist, which inhibits Kv4.1-4.3 subtypes with different voltage dependence [
28]. HptTx-273, however, has the highest homology with U19-sparatoxin-Hv1a from the spider
Heteropoda venatoria (57% identity and 69% similarity), whose biological function is not experimentally determined yet.
2.3.12. Family L
This family has 11 toxin precursors, all of which have a propeptide and contain 6 cysteines arranged as C1-C2-C3C4-C5-C6. Based on this cysteine scaffold, typically observed in ICK motif toxins, their disulfide bond was predicted to be C1-4, C2-5, and C3-6. Toxins in this family are diverse and are divided into four subsets: (1) HptTx-571, HptTx-963, HptTx-1181-T, HptTx-1158-T, HptTx-726, HptTx-1008, and HptTx-1160-T are best aligned with kappa-LhTx-1 from the spider Pandercetes sp. Among them, HptTx-1158-T showed 100% sequence identity with kappa-LhTx-1 but its mature peptide is one residue shorter than the latter, suggesting HptTx-1158-T is not a complete sequence. The other toxins have moderate homology with kappa-LhTx-1 (59–61% identity and 71–78% similarity). HptTx-571 and HptTx-963 are homologous peptides with an amidation signal (the C-terminal glycine residue) at their C-termini, and HptTx-726 and HptTx-1008 only differ from each other by a single residue in the propeptide region. (2) HptTx-1162-T and HptTx-1163-T are derived from the transcriptomic data, and the mature peptide of HptTx-1162-T should be complete due to an amidation signal (GK) at its C-terminus. BLAST analysis showed that they have the highest homology with Kappa-sparatoxin-Hv1c_2 (56% identity and 72% similarity) and Kappa-sparatoxin-Hv1e_2 (56% identity and 59% similarity), from the spider Heteropoda venatoria, respectively. (3) HptTx-133 has an amidation signal at its C-terminus. Its predicted mature peptide has extremely high homology with the toxin U6-sparatoxin-Hdb17, partial from the spider Heteropoda davidbowie (92% identity and 94% similarity; (4) HptTx-574 is best aligned with U14-sparatoxin-Hv1a from the spider Heteropoda venatoria (42% identity and 55% similarity). It is worth noting that most toxins in family K and L are related to kappa-LhTx-1, which raises the possibility that they also act on Kv channels.
2.3.13. Family M
This family has 19 precursor peptides, all of which contain propeptides. Except for HptTx-37 and HptTx-260 which have a novel cysteine framework of C
1-C
2-C
3C
4C
5-C
6-C
7-C
8-C
9-C
10, the other members are likely ICK motif toxins that have a consensus cysteines arrangement of C
1-C
2-C
3C
4-C
5-C
6, and the disulfide mode should be C
1-4, C
2-5, and C
3-6. These toxins are relatively diverse and can be grouped as follows: (1) HptTx-949 and HptTx-472-P have the same predicted mature peptide and propeptide, but possibly different signal peptides. HptTx-950 and HptTx-1063 also differ from each other by only two residues in their signal peptide region. These four toxins are best aligned with μ-sparatoxin-Hv2 from the spider
Heteropoda venatoria (43–46% identity and 62–65% similarity) and U20-sparatoxin-Hv1g from the spider
Heteropoda venatoria (52–60% identity and 65–68% similarity), in the Uniprot and NR database, respectively. In our previous study, μ-sparatoxin-Hv2 was identified as an insecticidal toxin functioning by potently and irreversibly inhibiting the insect voltage-gated sodium channel [
19], whereas the biological function of U20-sparatoxin-Hv1g is unknown; (2) HptTx-208, HptTx-985, HptTx-641, HptTx-585, and HptTx-861 are another homologous subset, and their predicted mature peptides are likely amidated at the C-terminus, as indicated by a GR amidation signal. BLAST analysis showed they are best aligned with μ-sparatoxin-Hv2 (40–41% identity and 52% similarity, Uniprot database); (3) HptTx-386, HptTx-1132, and HptTx-385 have high sequence homology with each other, their predicted mature peptides are also presumably amidated. These three peptides also have the highest homology with μ-sparatoxin-Hv2 (42–43% identity and 54–56% similarity, Uniprot database); (4) HptTx-431, HptTx-46, HptTx-1164-T, HptTx-442-P, and HptTx-816 have higher sequence homology with μ-sparatoxin-Hv2 than other toxins in this family (51–58% identity and 58–69% similarity). Except for HptTx-431 and HptTx-816, the predicted mature peptides of the other three toxins are likely amidated; (5) HptTx-37 and HP-260 differ by a residue in the propeptide region. They are best aligned with Kappa-ctenitoxin-Pn1a from the spider
Phoneutria nigriventer in the Uniprot database, although with low sequence homology (35–36% identity and 51–53% similarity). However, they have high homology with U8-sparatoxin-Hdb1, partial from the spider
Heteropoda davidbowie (73% identity and 84% similarity) in the NR database.
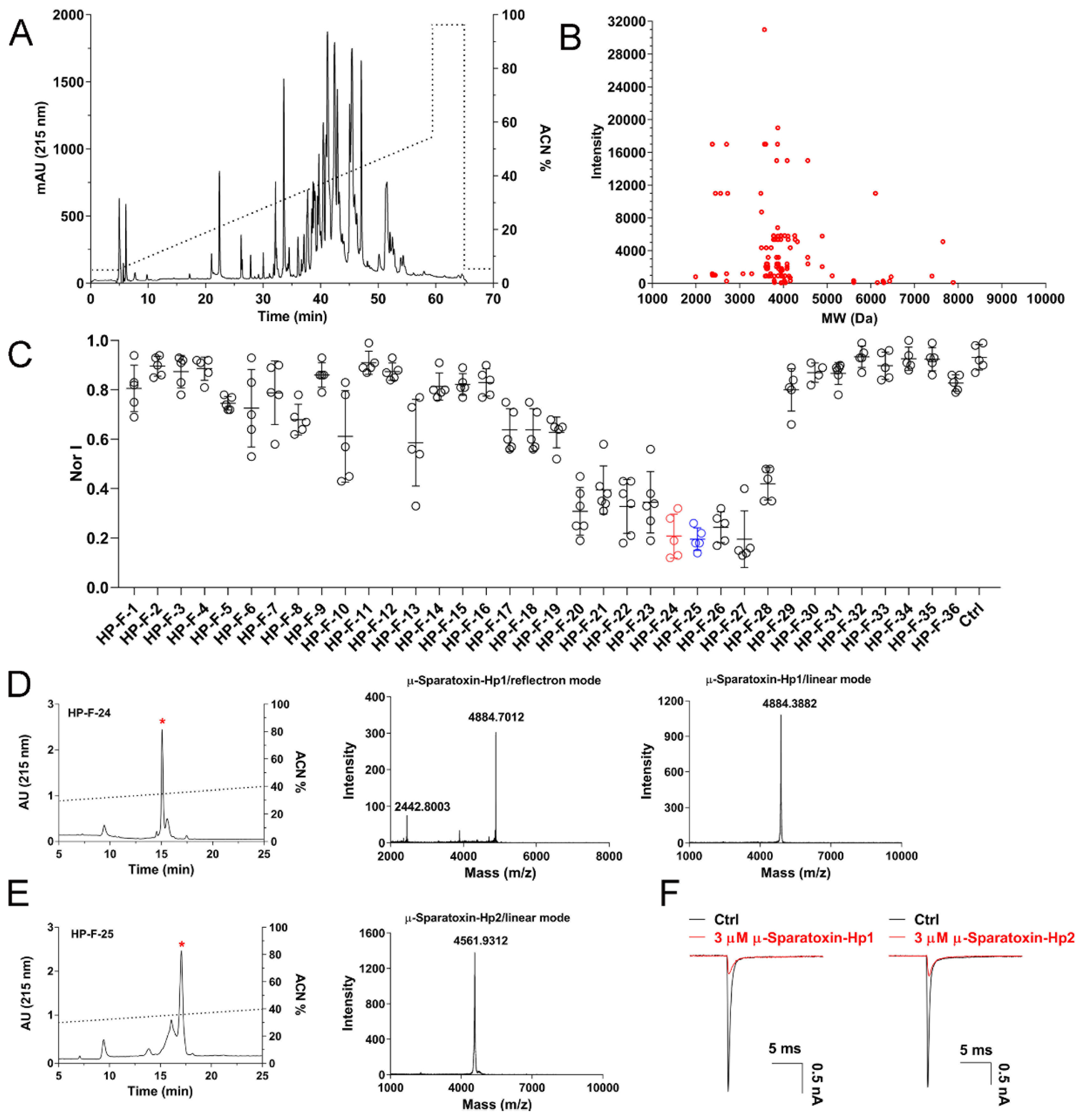
2.4. RP-HPLC Profile of H. Pingtungensis Venom and Screening of NaV1.7 Inhibitory Peptides
To explore the molecular diversity of venom peptide entities in the venom of
H. pingtungensis, we conducted semi-preparative RP-HPLC purification of the venom and offline MALDI-TOF analysis of eluted fractions. As shown in
Figure 4A, the venom is fractionated into 36 fractions in RP-HPLC, for which most fractions are eluted with a retention time between 30 min to 50 min (30–50% acetonitrile gradient). MALDI-TOF MS analysis identified a total of 110 different molecular weights (MWs) in these fractions, with each MW representing a venom peptide [
29]. Most venom peptides have an MW between 3500 Da and 4500 Da, which are clustered near the MW of 4000 Da (
Figure 4B). We next tested the activities of these fractions on NaV1.7 channels heterologously expressed in HEK293-T cells at a final concentration of 2–3 µM (note, it is an estimated molar concentration which is calculated by dividing the mass concentration of each fraction by its most abundant peptide’s MW, as determined by mass spectrometry analysis). Excitingly, nine fractions showed dramatic inhibition on the NaV1.7 currents (>50% inhibition), with HP-F-24, HP-F-25, HP-F-26, and HP-F-27 exhibiting the most potent activity (approximately 80% inhibition) (
Figure 4C,
n = 5–6). We further purified HP-F-24 and HP-F-25 by analytical RP-HPLC with a much slower acetonitrile gradient and evaluated the inhibitory activity of the finely purified components on the NaV1.7 channel. Finally, two peptide toxins with potent NaV1.7 inhibition were purified to homogeneity, and following the nomenclature rules proposed by King et al. [
12], these two toxins were named as µ-Sparatoxin-Hp1 and µ-Sparatoxin-Hp2, respectively (
Figure 4D,E). MALDI-TOF MS analysis showed that the MW of µ-Sparatoxin-Hp1 and µ-Sparatoxin-Hp2 is 4884.7012 Da and 4561.9312 Da (M + H
+), respectively (
Figure 4D,E, middle and right panels). Moreover, combining EDMAN degradation and cDNA library analysis revealed the full sequence of these two toxins, with µ-Sparatoxin-Hp1 and µ-Sparatoxin-Hp2 being the cDNA library clones HptTx-208 and HptTx-133, respectively (µ-Sparatoxin-Hp1: H
2N-ADSGGDAGGDAGADDEGSCKWMFQSCEPPAKCCDGWTCYKGRCNLIL-amide; µ-Sparatoxin-Hp2: H2N-DDDKKECIGHMGWCAWTDGECCEGYRCKLWCRKIIDWL-amide). The identities of these two toxins were also cross-checked by matching their experimentally determined MWs with those derived from their sequences (theoretical MW of µ-Sparatoxin-Hp1 and µ-Sparatoxin-Hp2 is 4883.39 Da and 4560.26 Da, respectively; note that 3 disulfide and the C-terminus amidation in toxin reduces the MW of linear peptide by 7 Da). Furthermore, 3 μM µ-Sparatoxin-Hp1 and µ-Sparatoxin-Hp2 inhibited NaV1.7 currents by 79 ± 1.8% and 80 ± 0.9%, respectively (
Figure 4F,
n = 5–6). These two toxins are novel pioneer molecules for developing analgesics. In summary, these data suggest that
H. pingtungensis venom contains a great diversity of peptides, which are valuable sources for identifying novel ion channel modulators.

 and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}