Botulinum Toxin and Neuronal Regeneration after Traumatic Injury of Central and Peripheral Nervous System


{kind=link}
{kind=link}
Abstract
1. Introduction
2. Peripheral Nervous System
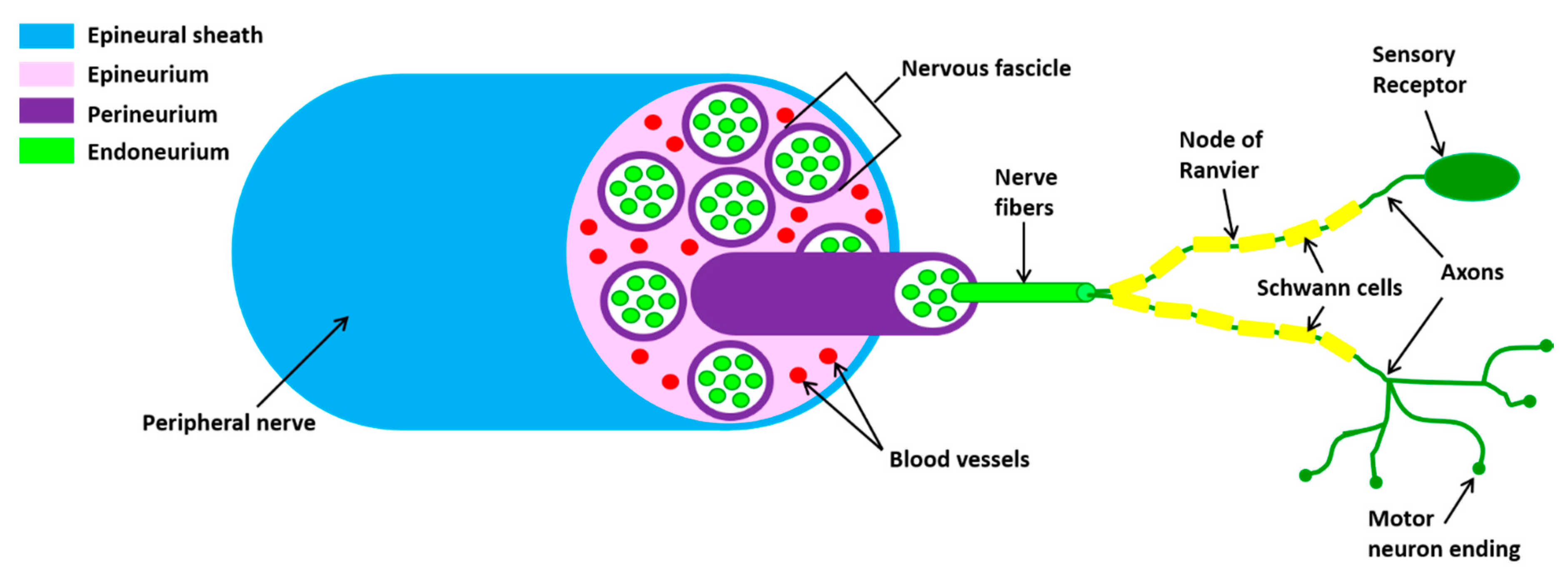
2.1. Outline of Peripheral Nerve Anatomy
2.2. Pathophysiology of Traumatic Injuries of Peripheral Nerve
2.3. Mechanisms of Nerve Regeneration after Peripheral Injuries
2.4. Botulinum Toxin and Nerve Regeneration after Peripheral Injuries
3. Central Nervous System
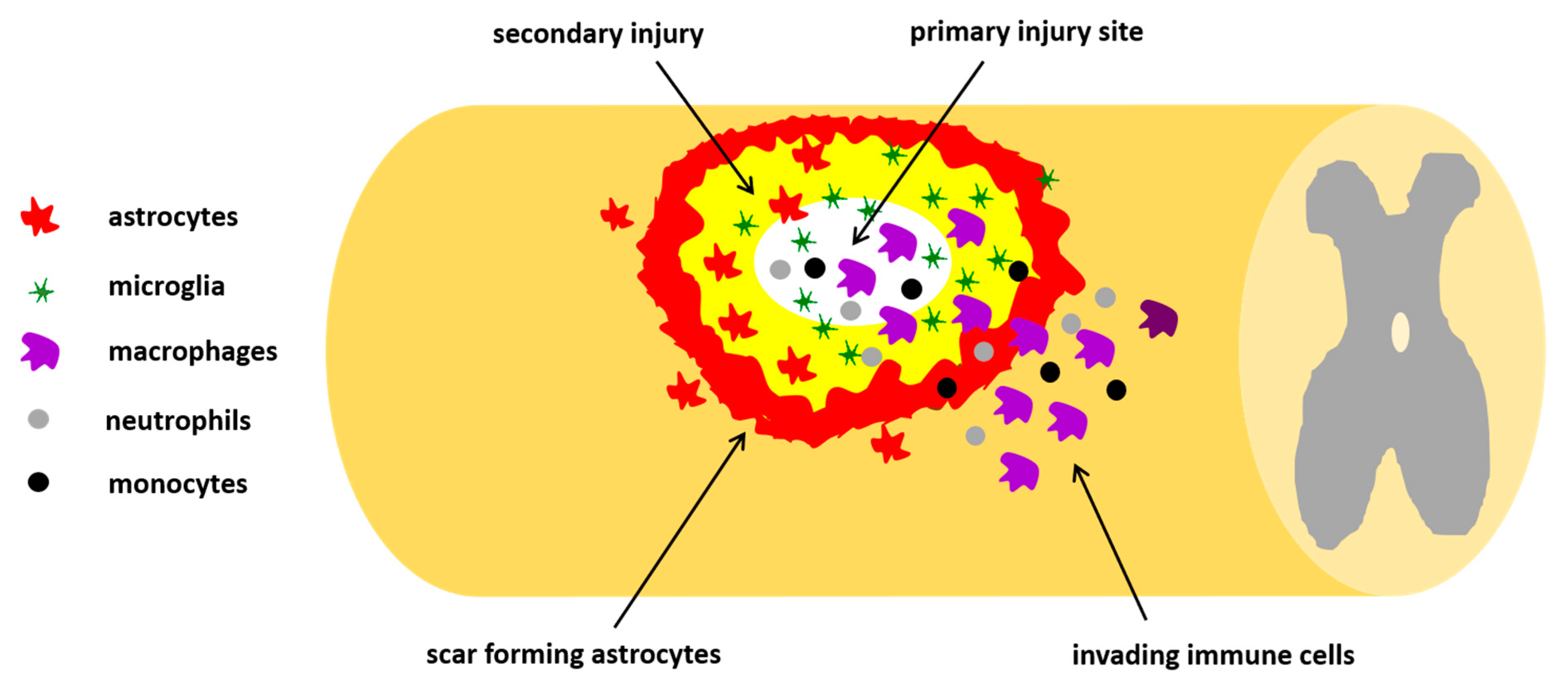
3.1. Pathophysiology of Traumatic SCI
3.2. Mechanisms of Regeneration after SCI
3.3. Botulinum Toxin and Spinal Cord Injuries
4. Conclusions
5. Patents
Funding
Conflicts of Interest
References
- Káradóttir, R.T.; Kuo, C.T. Neuronal Activity-Dependent Control of Postnatal Neurogenesis and Gliogenesis. Annu. Rev. Neurosci. 2018, 41, 139–161. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Belin, S.; He, Z. Signaling regulations of neuronal regenerative ability. Curr. Opin. Neurobiol. 2014, 27, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, T.C.; Geoffroy, C.G. The Influence of Neuron-Extrinsic Factors and Aging on Injury Progression and Axonal Repair in the Central Nervous System. Front. Cell Dev. Biol. 2020, 8, 190. [Google Scholar] [CrossRef]
- Hu, X.; De Silva, T.M.; Chen, J.; Faraci, F.M. Cerebral Vascular Disease and Neurovascular Injury in Ischemic Stroke. Circ. Res. 2017, 120, 449–471. [Google Scholar] [CrossRef]
- Vargas, M.E.; Barres, B.A. Why is wallerian degeneration in the CNS so slow? Annu. Rev. Neurosci. 2007, 30, 153–179. [Google Scholar] [CrossRef]
- Putatunda, R.; Bethea, J.R.; Hu, W.-H. Potential immunotherapies for traumatic brain and spinal cord injury. Chin. J. Traumatol. 2018, 21, 125–136. [Google Scholar] [CrossRef] [PubMed]
- Shah, M.; Peterson, C.; Yilmaz, E.; Halalmeh, D.R.; Moisi, M. Current advancements in the management of spinal cord injury: A comprehensive review of literature. Surg. Neurol. Int. 2020, 11, 2. [Google Scholar] [CrossRef]
- Robinson, L.R. Traumatic injury to peripheral nerves. Muscle Nerve 2000, 23, 863–873. [Google Scholar] [CrossRef]
- He, Z.; Jin, Y. Intrinsic control of axon regeneration. Neuron 2016, 90, 437–451. [Google Scholar] [CrossRef]
- Navarro, X.; Vivó, M.; Valero-Cabré, A. Neural plasticity after peripheral nerve injury and regeneration. Prog. Neurobiol. 2007, 82, 163–201. [Google Scholar] [CrossRef]
- Cattin, A.L.; Lloyd, A.C. The multicellular complexity of peripheral nerve regeneration. Curr. Opin. Neurobiol. 2016, 39, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Seddon, H. Three types of nerve injury. Brain 1943, 66, 237–288. [Google Scholar] [CrossRef]
- Dubový, P. Wallerian degeneration and peripheral nerve conditions for both axonal regeneration and neuropathic pain induction. Ann. Anat. 2011, 193, 267–275. [Google Scholar] [CrossRef] [PubMed]
- Sunderland, S. A classification of peripheral nerve injuries producing loss of function. Brain 1951, 74, 491–516. [Google Scholar] [CrossRef] [PubMed]
- Mackinnon, S.; Dellon, A.L. Surgery of the peripheral nerve. In Diagnosis of Nerve Injury; Thieme: New York, NY, USA, 1988; pp. 74–78. [Google Scholar]
- Fu, S.Y.; Gordon, T. The cellular and molecular basis of peripheral nerve regeneration. Mol. Neurobiol. 1997, 14, 67–116. [Google Scholar] [CrossRef]
- Jonsson, S.; Wiberg, R.; McGrath, A.M.; Novikov, L.N.; Wiberg, M.; Novikova, L.N.; Kingham, P.J. Effect of delayed peripheral nerve repair on nerve regeneration, Schwann cell function and target muscle recovery. PLoS ONE 2013, 8, e56484. [Google Scholar] [CrossRef]
- Langert, K.A.; Brey, E.M. Strategies for Targeted Delivery to the Peripheral Nerve. Front. Neurosci. 2018, 12, 887. [Google Scholar] [CrossRef]
- Hussain, G.; Wang, J.; Rasul, A.; Anwar, H.; Qasim, M.; Zafar, S.; Aziz, N.; Razzaq, A.; Hussain, R.; Gonzalez de Aguilar, J.-L.; et al. Current Status of Therapeutic Approaches against Peripheral Nerve Injuries: A Detailed Story from Injury to Recovery. Int. J. Biol. Sci. 2020, 16, 116–134. [Google Scholar] [CrossRef] [PubMed]
- Deumens, R.; Bozkurt, A.; Meek, M.F.; Marcus, M.A.; Joosten, E.A.; Weis, J.; Brook, G.A. Repairing injured peripheral nerves: Bridging the gap. Prog. Neurobiol. 2010, 92, 245–276. [Google Scholar] [CrossRef]
- Doolabh, V.; Hertl, M.; Mackinnon, S. The role of conduits in nerve repair: A review. Rev. Neurosci. 1996, 7, 47–84. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Perez, F.; Cobianchi, S.; Geuna, S.; Barwig, C.; Freier, T.; Udina, E.; Navarro, X. Tubulization with chitosan guides for the repair of long gap peripheral nerve injury in the rat. Microsurgery 2015, 35, 300–308. [Google Scholar] [CrossRef]
- Gu, X.; Ding, F.; Yang, Y.; Liu, J. Construction of tissue engineered nerve grafts and their application in peripheral nerve regeneration. Prog. Neurobiol. 2011, 93, 204–230. [Google Scholar] [CrossRef]
- Arslantunali, D.; Dursun, T.; Yucel, D.; Hasirci, H. Peripheral nerve conduits: Technology update. Med. Devices (Auckl. N.Z.) 2014, 7, 405–424. [Google Scholar]
- Raza, C.; Riaz, H.A.; Anjum, R.; Shakeel, N.U.A. Repair strategies for injured peripheral nerve: Review. Life Sci. 2020, 243, 117308. [Google Scholar] [CrossRef]
- Alvites, R.D.; Caseiro, A.R.; Pedrosa, S.S.; Branquinho, M.E.; Varejão, A.S.P.; Maurício, A.C. The Nasal Cavity of the Rat and Mouse-Source of Mesenchymal Stem Cells for Treatment of Peripheral Nerve Injury. Anat. Rec. (Hoboken) 2018, 301, 1678–1689. [Google Scholar] [CrossRef]
- Asensio-Pinilla, E.; Udina, E.; Jaramillo, J.; Navarro, X. Electrical stimulation combined with exercise increase axonal regeneration after peripheral nerve injury. Exp. Neurol. 2009, 219, 258–265. [Google Scholar] [CrossRef] [PubMed]
- Ewald, S.G.; Beckmann-Fries, V. Rehabilitation Following Peripheral Nerve Injury. In Modern Concepts of Peripheral Nerve Repair; Haastert-Talini, K., Assmus, H., Antoniadis, G., Eds.; Springer: Cham, Switzerland, 2007; pp. 109–125. [Google Scholar]
- E Udina, E.; Cobianchi, S.; Allodi, I.; Navarro, X. Effects of activity-dependent strategies on regeneration and plasticity after peripheral nerve injuries. Ann. Anat. 2011, 193, 347–353. [Google Scholar] [CrossRef]
- Armada-da-Silva, P.A.; Pereira, C.; Amado, S.; Veloso, A.P. Role of physical exercise for improving posttraumatic nerve regeneration. Int. Rev. Neurobiol. 2013, 109, 125–149. [Google Scholar]
- Montecucco, C.; Rasotto, M.B. On botulinum neurotoxin variability. mBio 2015, 6, e02131. [Google Scholar] [CrossRef]
- Dover, N.; Barash, J.R.; Hill, K.K.; Xie, G.; Arnon, S.S. Molecular characterization of a novel botulinum neurotoxin type H gene. J. Infect. Dis. 2014, 209, 192–202. [Google Scholar] [CrossRef]
- Zhang, S.; Masuyer, G.; Zhang, J.; Shen, Y.; Lundin, D.; Henriksson, L.; Miyashita, S.I.; Martínez-Carranza, M.; Dong, M.; Stenmark, P. Identification and characterization of a novel botulinum neurotoxin. Nat. Commun. 2017, 8, 14130. [Google Scholar] [CrossRef] [PubMed]
- Brunt, J.; Carter, A.T.; Stringer, S.C.; Peck, M.W. Identification of a novel botulinum neurotoxin gene cluster in Enterococcus. FEBS Lett. 2018, 592, 310–317. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Barash, J.R.; Conrad, F.; Lou, J.; Tam, C.; Cheng, L.W.; Arnon, S.S.; Marks, J.D. The Novel Clostridial Neurotoxin Produced by Strain IBCA10-7060 Is Immunologically Equivalent to BoNT/HA. Toxins 2019, 12, 9. [Google Scholar] [CrossRef] [PubMed]
- Rossetto, O.; Pirazzini, M.; Montecucco, C. Botulinum neurotoxins: Genetic, structural and mechanistic insights. Nat. Rev. Microbiol. 2014, 12, 535–549. [Google Scholar] [CrossRef] [PubMed]
- Pirazzini, M.; Rossetto, O.; Eleopra, R.; Montecucco, C. Botulinum Neurotoxins: Biology, Pharmacology, and Toxicology. Pharmacol. Rev. 2017, 69, 200–235. [Google Scholar] [CrossRef]
- Rasetti-Escargueil, C.; Lemichez, E.; Popoff, M.R. Variability of Botulinum Toxins: Challenges and Opportunities for the Future. Toxins 2018, 10, 374. [Google Scholar] [CrossRef] [PubMed]
- Jankovic, J. Botulinum toxin: State of the Art. Mov. Disord. 2017, 32, 1131–1138. [Google Scholar] [CrossRef] [PubMed]
- Pavone, F.; Luvisetto, S. Botulinum neurotoxin for pain management: Insights from animal models. Toxins 2010, 2, 2890–2913. [Google Scholar] [CrossRef]
- Ramachandran, R.; Yaksh, T.L. Therapeutic use of botulinum toxin in migraine: Mechanisms of action. Br. J. Pharmacol. 2014, 171, 4177–4192. [Google Scholar] [CrossRef]
- Matak, I.; Lacković, Z. Botulinum toxin A, brain and pain. Prog. Neurobiol. 2015, 119–120, 39–59. [Google Scholar] [CrossRef]
- Matak, I.; Bölcskei, K.; Bach-Rojecky, L.; Helyes, Z. Mechanisms of Botulinum Toxin Type A Action on Pain. Toxins 2019, 11, 459. [Google Scholar] [CrossRef] [PubMed]
- Lacković, Z. Pain. Handb. Exp. Pharmacol. 2020. [Google Scholar] [CrossRef]
- Yeh, T.C.; Chen, P.C.; Su, Y.R.; Kuo, H.C. Effect of Botulinum Toxin A on Bladder Pain-Molecular Evidence and Animal Studies. Toxins 2020, 12, 98. [Google Scholar] [CrossRef] [PubMed]
- Jabbari, B. Botulinum neurotoxins in the treatment of refractory pain. Nat. Clin. Pract. Neurol. 2008, 4, 676–685. [Google Scholar] [CrossRef] [PubMed]
- Luvisetto, S.; Gazerani, P.; Cianchetti, C.; Pavone, F. Botulinum Toxin Type A as a Therapeutic Agent against Headache and Related Disorders. Toxins 2015, 7, 3818–3844. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Park, H.J. Botulinum Toxin for the Treatment of Neuropathic Pain. Toxins 2017, 9, 260. [Google Scholar] [CrossRef] [PubMed]
- Safarpour, Y.; Jabbari, B. Botulinum toxin treatment of pain syndromes-an evidence based review. Toxicon 2018, 147, 120–128. [Google Scholar] [CrossRef]
- Lin, Y.H.; Chiang, B.J.; Liao, C.H. Mechanism of Action of Botulinum Toxin A in Treatment of Functional Urological Disorders. Toxins 2020, 12, 129. [Google Scholar] [CrossRef]
- Palomar, F.J.; Mir, P. Neurophysiological changes after intramuscular injection of botulinum toxin. Clin. Neurophysol. 2012, 123, 54–60. [Google Scholar] [CrossRef]
- Navarro, X. Functional evaluation of peripheral nerve regeneration and target reinnervation in animal models: A critical overview. Eur. J. Neurosci. 2016, 43, 271–286. [Google Scholar] [CrossRef]
- Colloca, L.; Ludman, T.; Bouhassira, D.; Baron, R.; Dickenson, A.H.; Yarnitsky, D.; Freeman, R.; Truini, A.; Attal, N.; Finnerup, N.B.; et al. Neuropathic Pain. Nat. Rev. Dis. Primers 2017, 3, 17002. [Google Scholar] [CrossRef] [PubMed]
- Klein, A.W. The therapeutic potential of botulinum toxin. Dermatol. Surg. 2004, 30, 452–455. [Google Scholar] [PubMed]
- Bach-Rojecky, L.; Relja, M.; Lacković, Z. Botulinum toxin type A in experimental neuropathic pain. J. Neural. Transm. 2005, 112, 215–219. [Google Scholar] [CrossRef]
- Park, H.J.; Lee, Y.; Lee, J.; Park, C.; Moon, D.E. The effects of botulinum toxin A on mechanical and cold allodynia in a rat model of neuropathic pain. Can. J. Anaesth. 2006, 53, 470–477. [Google Scholar] [CrossRef]
- Luvisetto, S.; Marinelli, S.; Cobianchi, S.; Pavone, F. Anti-allodynic efficacy of botulinum neurotoxin A in a model of neuropathic pain. Neuroscience 2007, 14, 1–4. [Google Scholar] [CrossRef]
- Burma, N.E.; Leduc-Pessah, H.; Fan, C.Y.; Trang, T. Animal models of chronic pain: Advances and challenges for clinical translation. J. Neurosci. Res. 2017, 95, 1242–1256. [Google Scholar] [CrossRef] [PubMed]
- Marinelli, S.; Luvisetto, S.; Cobianchi, S.; Makuch, W.; Obara, I.; Mezzaroma, E.; Caruso, M.; Straface, E.; Przewlocka, B.; Pavone, F. Botulinum neurotoxin type A counteracts neuropathic pain and facilitates functional recovery after peripheral nerve injury in animal models. Neuroscience 2010, 171, 316–328. [Google Scholar] [CrossRef] [PubMed]
- Azarnia Tehran, D.; Zanetti, G.; Leka, O.; Lista, F.; Fillo, S.; Binz, T.; Shone, C.C.; Rossetto, O.; Montecucco, C.; Paradisi, C.; et al. A Novel Inhibitor Prevents the Peripheral Neuroparalysis of Botulinum Neurotoxins. Sci. Rep. 2015, 5, 17513. [Google Scholar] [CrossRef] [PubMed]
- Field, M.; Splevins, A.; Picaut, P.; van der Schans, M.; Langenberg, J.; Noort, D.; Snyder, D.; Foster, K. AbobotulinumtoxinA (Dysport®), OnabotulinumtoxinA (Botox®), and IncobotulinumtoxinA (Xeomin®) Neurotoxin Content and Potential Implications for Duration of Response in Patients. Toxins 2018, 10, 535. [Google Scholar] [CrossRef] [PubMed]
- Baptista, A.F.; Gomes, J.R.; Oliveira, J.T.; Santos, S.M.; Vannier-Santos, M.A.; Martinez, A.M. A new approach to assess function after sciatic nerve lesion in the mouse—Adaption of the sciatic static index. J. Neurosci. Meth. 2007, 161, 259–264. [Google Scholar] [CrossRef]
- Cobianchi, S.; Marinelli, S.; Florenzano, F.; Pavone, F.; Luvisetto, S. Short- but not long-lasting treadmill running reduces allodynia and improves functional recovery after peripheral nerve injury. Neuroscience 2010, 168, 273–287. [Google Scholar] [CrossRef] [PubMed]
- Nakazato-Imasato, E.; Kurebayashi, Y. Pharmacological characteristics of the hind paw weight bearing difference induced by chronic constriction injury of the sciatic nerve in rats. Life Sci. 2009, 84, 622–626. [Google Scholar] [CrossRef] [PubMed]
- Mika, J.; Korostynski, M.; Kaminska, D.; Wawrzczak-Bargiela, A.; Osikowicz, M.; Makuch, W.; Przewlocki, R.; Przewlocka, B. Interleukin-1α has antiallodynic and antihyperalgesic activities in a rat neuropathic pain model. Pain 2008, 138, 587–597. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.; Rummler, L.S.; Palispis, W.; Truong, L.; Chao, T.; Rowshan, K.; Mozaffar, T.; Steward, O. Local down-regulation of myelin-associated glycoprotein permits axonal sprouting with chronic nerve compression injury. Exp. Neurol. 2006, 200, 418–429. [Google Scholar] [CrossRef]
- De la Hoz, C.L.; Oliveira, A.L.; Queiroz, L.S.; Langone, F. Wallerian degeneration in C57BL/6J and A/J mice: Differences in time course of neurofilament and myelin breakdown, macrophage recruitment and iNOS expression. J. Anat. 2003, 203, 567–578. [Google Scholar] [CrossRef]
- Pines, J. Four-dimensional control of the cell cycle. Nat. Cell Biol. 1999, 1, 73–79. [Google Scholar] [CrossRef]
- Manes, T.; Zheng, D.Q.; Tognin, S.; Woodard, A.S.; Marchisio, P.C.; Languino, L.R. Alpha(v)beta3 integrin expression up-regulates Cdc2, which modulates cell migration. J. Cell Biol. 2003, 161, 817–826. [Google Scholar] [CrossRef]
- Han, I.S.; Seo, T.B.; Kim, K.H.; Yoon, J.H.; Yoon, S.J.; Namgung, U. Cdc2-mediated Schwann cell migration during peripheral nerve regeneration. J. Cell Sci. 2007, 120, 246–255. [Google Scholar] [CrossRef]
- Bhatheja, K.; Field, J. Schwann cells: Origins and role in axonal maintenance and regeneration. Int. J. Biochem. Cell Biol. 2006, 38, 1995–1999. [Google Scholar] [CrossRef]
- Hayashi, A.; Koob, J.W.; Liu, D.Z.; Tong, A.Y.; Hunter, D.A.; Parsadanian, A.; Mackinnon, S.E.; Myckatyn, T.M. A double-transgenic mouse used to track migrating Schwann cells and regenerating axons following engraftment of injured nerves. Exp. Neurol. 2007, 207, 128–138. [Google Scholar] [CrossRef]
- Jessen, K.R.; Mirsky, R. The origin and development of glial cells in peripheral nerves. Nat. Rev. Neurosci. 2005, 6, 671–682. [Google Scholar] [CrossRef] [PubMed]
- Triolo, D.; Dina, G.; Lorenzetti, I.; Malaguti, M.; Morana, P.; del Carro, U.; Comi, G.; Messing, A.; Quattrini, A.; Previtali, S.C. Loss of glial fibrillary acidic protein (GFAP) impairs Schwann cell proliferation and delays nerve regeneration after damage. J. Cell Sci. 2006, 119, 3981–3993. [Google Scholar] [CrossRef] [PubMed]
- Marinelli, S.; Vacca, V.; Ricordy, R.; Uggenti, C.; Tata, A.M.; Luvisetto, S.; Pavone, F. The analgesic effect on neuropathic pain of retrogradely transported botulinum neurotoxin A involves Schwann cells and astrocytes. PLoS ONE 2012, 7, e47977. [Google Scholar] [CrossRef] [PubMed]
- Brookes, J.P. Assays for cholinergic properties in cultured rat Schwann cells. Proc. R. Soc. Lond Biol. Sci. 1984, 222, 121–134. [Google Scholar]
- Loreti, S.; Ricordy, R.; De Stefano, M.E.; Augusti-Tocco, G.; Tata, A.M. Acetylcholine inhibits cell cycle progression in rat Schwann cells by activation of the M2 receptor subtype. Neuron Glia Biol. 2007, 3, 269–279. [Google Scholar] [CrossRef]
- Syed, N.; Reddy, K.; Yang, D.P.; Taveggia, C.; Salzer, J.L.; Maurel, P.; Kim, H.A. Soluble Neuregulin 1 has bifunctional concentration dependent effects on Schwann cell myelination. J. Neurosci. 2010, 30, 6122–6131. [Google Scholar] [CrossRef]
- Barden, J.A.; Cotte, L.J.; Bennett, M.R. Vesicle-associated proteins and P2X receptor clusters at single sympathetic varoicosities in mouse vas deferens. J. Neurocytol. 1999, 28, 469–480. [Google Scholar] [CrossRef]
- Caleo, M.; Restani, L. Direct central nervous system effects of botulinum neurotoxin. Toxicon 2018, 147, 68–72. [Google Scholar] [CrossRef]
- Matak, I.; Riederer, P.; Lacković, Z. Botulinum toxin’s axonal transport from periphery to the spinal cord. Neurochem. Int. 2012, 61, 236–239. [Google Scholar] [CrossRef]
- Cobianchi, S.; Jaramillo, J.; Luvisetto, S.; Pavone, F.; Navarro, X. (2017) Botulinum neurotoxin A promotes functional recovery after peripheral nerve injury by increasing regeneration of myelinated fibers. Neuroscience 2017, 359, 82–91. [Google Scholar] [CrossRef]
- Pamphlett, R. Early terminal and nodal sprouting of motor axons after botulinum toxin. J. Neurol. Sci. 1989, 92, 181–192. [Google Scholar] [CrossRef]
- Lu, L.; Atchabahian, A.; Mackinnon, S.E.; Hunter, D.A. Nerve injection injury with botulinum toxin. Plast. Reconstr. Surg. 1988, 101, 1875–1880. [Google Scholar] [CrossRef] [PubMed]
- Hogan, Q.H. Pathophysiology of Peripheral Nerve Injury during Regional Anesthesia. Reg. Anesth. Pain Med. 2008, 33, 435–441. [Google Scholar] [CrossRef]
- Meyer-Frießem, C.H.; Eitner, L.B.; Kaisler, M.; Maier, C.; Vollert, J.; Westermann, A.; Zahn, P.K.; Avila González, C.A. Perineural injection of botulinum toxin-A in painful peripheral nerve injury-a case series: Pain relief, safety, sensory profile and sample size recommendation. Curr. Med. Res. Opin. 2019, 35, 1793–1803. [Google Scholar] [CrossRef]
- Irintchev, M.; Guntinas-Lichius, O.; Irintchev, A. Botulinum neurotoxin application to the severed femoral nerve modulates spinal synaptic responses to axotomy and enhances motor recovery in rats. Neural Plast. 2018, 2018, 7975013. [Google Scholar] [CrossRef]
- Franz, C.K.; Puritz, A.; Jordan, L.A.; Chow, J.; Ortega, J.A.; Kiskinis, E.; Heckman, C.J. Botulinum Toxin Conditioning Enhances Motor Axon Regeneration in Mouse and Human Preclinical Models. Neurorehabil. Neural. Repair 2018, 32, 735–745. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, P.N. A conditioning lesion induces changes in gene expression and axonal transport that enhance regeneration by increasing the intrinsic growth state of axons. Exp. Neurol. 2010, 223, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Mackinnon, S. Donor Distal, Recipient Proximal and Other Personal Perspectives on Nerve Transfers. Hand Clin. 2016, 32, 141–151. [Google Scholar] [CrossRef] [PubMed]
- White, C.M.; Greensmith, L.; Vrbová, G. Repeated stimuli for axonal growth causes motoneuron death in adult rats: The effect of botulinum toxin followed by partial denervation. Neuroscience 1999, 95, 1101–1109. [Google Scholar] [CrossRef]
- Finocchiaro, A.; Marinelli, S.; De Angelis, F.; Vacca, V.; Luvisetto, S.; Pavone, F. Botulinum Toxin B Affects Neuropathic Pain but Not Functional Recovery after Peripheral Nerve Injury in a Mouse Model. Toxins 2018, 10, 128. [Google Scholar] [CrossRef] [PubMed]
- Wilson, J.R.; Cadotte, D.W.; Fehlings, M.G. Clinical predictors of neurological outcome, functional status, and survival after traumatic spinal cord injury: A systematic review. J. Neurosurg. Spine 2012, 17 (Suppl. 1), 11–26. [Google Scholar] [CrossRef]
- Alizadeh, A.; Dyck, S.M.; Karimi-Abdolrezaee, S. Traumatic Spinal Cord Injury: An Overview of Pathophysiology, Models and Acute Injury Mechanisms. Front. Neurol. 2019, 10, 282. [Google Scholar] [CrossRef]
- Kim, Y.H.; Ha, K.Y.; Kim, S. Spinal cord injury and related clinical trials. CiOS Clin. Orthop. Surg. 2017, 9, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Horner, P.J.; Gage, F.H. Regenerating the damaged central nervous system. Nature 2000, 407, 963–970. [Google Scholar] [CrossRef]
- Hutson, T.H.; Di Giovanni, S. The translational landscape in spinal cord injury: Focus on neuroplasticity and regeneration. Nat. Rev. Neurol. 2019, 15, 732–745. [Google Scholar] [CrossRef] [PubMed]
- Fakhoury, M. Spinal cord injury: Overview of experimental approaches used to restore locomotor activity. Rev. Neurosci. 2015, 26, 397–405. [Google Scholar] [CrossRef]
- Thuret, S.; Moon, L.D.; Gage, F.H. Therapeutic interventions after spinal cord injury. Nat. Rev. Neurosci. 2006, 7, 628–643. [Google Scholar] [CrossRef]
- Lipson, A.C.; Horner, P.J. Potent possibilities: Endogenous stem cells in the adult spinal cord. Prog. Brain Res. 2002, 137, 283–297. [Google Scholar] [PubMed]
- Barton, M.J.; John, J.S.; Clarke, M.; Wright, A.; Ekberg, J. The Glia Response after Peripheral Nerve Injury: A Comparison between Schwann Cells and Olfactory Ensheathing Cells and Their Uses for Neural Regenerative Therapies. Int. J. Mol. Sci. 2017, 18, 287. [Google Scholar] [CrossRef]
- Xiao, N.; Le, Q.T. Neurotrophic Factors and Their Potential Applications in Tissue Regeneration. Arch. Immunol. Ther. Exp. (Warsz) 2016, 64, 89–99. [Google Scholar] [CrossRef]
- Zuo, J.; Neubauer, D.; Dyess, K.; Ferguson, T.A.; Muir, D. Degradation of chondroitin sulfate proteoglycan enhances the neurite-promoting potential of spinal cord tissue. Exp. Neurol. 1998, 154, 654–662. [Google Scholar] [CrossRef] [PubMed]
- Keirstead, H.S.; Morgan, S.V.; Wilby, M.J.; Fawcett, J.W. Enhanced axonal regeneration following combined demyelination plus Schwann cell transplantation therapy in the injured adult spinal cord. Exp. Neurol. 1999, 159, 680–683. [Google Scholar] [CrossRef] [PubMed]
- Brewer, K.I.; Bethea, J.R.; Yezierski, R.P. Neuroprotective effects of interleukin-10 following excitotoxic spinal cord injury. Exp. Neurol. 1999, 159, 484–493. [Google Scholar] [CrossRef]
- Jhang, J.F.; Kuo, H.C. Novel Applications of OnabotulinumtoxinA in Lower Urinary Tract Dysfunction. Toxins 2018, 10, 260. [Google Scholar] [CrossRef]
- Romo, P.G.B.; Smith, C.P.; Cox, A.; Averbeck, M.A.; Dowling, C.; Beckford, C.; Manohar, P.; Duran, S.; Cameron, A.P. Non-surgical urologic management of neurogenic bladder after spinal cord injury. World J. Urol. 2018, 36, 1555–1568. [Google Scholar] [CrossRef] [PubMed]
- Fried, G.W.; Fried, K.M. Spinal cord injury and use of botulinum toxin in reducing spasticity. Phys. Med. Rehabil. Clin. North Am. 2003, 14, 901–910. [Google Scholar] [CrossRef]
- Ward, A.B. Spasticity treatment with botulinum toxins. J. Neural. Trans. 2008, 115, 607–616. [Google Scholar] [CrossRef]
- Palazón-García, R.; Alcobendas-Maestro, M.; Esclarin-de Ruz, A.; Benavente-Valdepeñas, A.M. Treatment of spasticity in spinal cord injury with botulinum toxin. J. Spinal Cord Med. 2019, 42, 281–287. [Google Scholar] [CrossRef]
- Li, S.; Francisco, G.E. Spasticity. Handb. Exp. Pharmacol. 2019. [Google Scholar] [CrossRef]
- Richardson, D.; Edwards, S.; Sheean, G.L.; Greenwood, R.J.; Thompson, A.J. The effect of botulinum toxin on hand function after incomplete spinal cord injury at the level of C5/6: A case report. Clin. Rehabil. 1997, 11, 288–292. [Google Scholar] [CrossRef]
- Catz, A.; Barkol, H.; Steinberg, F.; Ronen, J.; Bluvshtein, V.; Keren, O. Repeated botulinum toxin injections can improve mobility in patients with spinal cord lesions. Eura. Medicophys. 2007, 43, 319–325. [Google Scholar]
- Naicker, A.S.; Roohi, S.A.; Chan, J.L. Botulinum toxin type A for rehabilitation after a spinal cord injury: A case report. J. Orthop. Surg. (Hong Kong) 2009, 17, 96–99. [Google Scholar] [CrossRef]
- Santamato, A.; Panza, F.; Ranieri, M.; Amoruso, M.T.; Amoruso, L.; Frisardi, V.; Solfrizzi, V.; Fiore, P. Effect of intrathecal baclofen, botulinum toxin type A and a rehabilitation programme on locomotor function after spinal cord injury: A case report. J. Rehabil. Med. 2010, 42, 891–894. [Google Scholar] [CrossRef]
- Venkata Krishnan, R. Restoring Motor functions in Spinal cord injury, Hemiplegic Cerebral Palsy, and Stroke by Botulinum toxin-induced Synaptic Competitive-Learning Therapy. J. Neurol. Disord. 2013, 1, 134. [Google Scholar]
- Siddall, P.J.; Mcclelland, J.M.; Rutkowski, S.B.; Cousins, M.J. A longitudinal study of the prevalence and characteristics of pain in the first 5 years following spinal cord injury. Pain 2003, 103, 249–257. [Google Scholar] [CrossRef]
- Chun, A.; Levy, I.; Yang, A.; Delgado, A.; Tsai, C.Y.; Leung, E.; Taylor, K.; Kolakowsky-Hayner, S.; Huang, V.; Escalon, M.; et al. Treatment of at-level spinal cord injury pain with botulinum toxin A. Spinal Cord Ser. Cases 2019, 5, 77. [Google Scholar] [CrossRef]
- Moeini-Naghani, I.; Hashemi-Zonouz, T.; Jabbari, B. Botulinum Toxin Treatment of Spasticity in Adults and Children. Semin. Neurol. 2016, 36, 64–72. [Google Scholar] [CrossRef]
- Paolucci, S.; Martinuzzi, A.; Scivoletto, G.; Smania, N.; Solaro, C.; Aprile, I.; Armando, M.; Bergamaschi, R.; Berra, E.; Berto, G.; et al. Assessing and treating pain associated with stroke, multiple sclerosis, cerebral palsy, spinal cord injury and spasticity. Evidence and recommendations from the Italian Consensus Conference on Pain in Neurorehabilitation. Eur. J. Phys. Rehabil. Med. 2016, 52, 827–840. [Google Scholar]
- Marinelli, S.; Pavone, F.; Luvisetto, S.; Vacca, V. Therapeutic Use of the Botulinum Neurotoxin Serotype A. U.S. Patent US 2019/0224288A1, 25 July 2019. Available online: http://www.freepatentsonline.com/y2019/0224288.html (accessed on 1 March 2020).
- Vacca, V.; Madaro, L.; De Angelis, F.; Proietti, D.; Cobianchi, S.; Orsini, T.; Puri, P.L.; Luvisetto, S.; Pavone, F.; Marinelli, S. Revealing the therapeutic potential of Botulinum neurotoxin type A in counteracting paralysis and neuropathic pain in spinally injured mice. Toxins 2020. under review. [Google Scholar]
- Kundi, S.; Bicknell, R.; Ahmed, Z. Spinal cord injury: Current mammalian models. Am. J. Neurosci. 2014, 4, 1–12. [Google Scholar] [CrossRef]
- Cheriyan, T.; Ryan, D.J.; Weinreb, J.H.; Cheriyan, J.; Paul, J.C.; Lafage, V.; Kirsch, T.; Errico, T.J. Spinal cord injury models: A review. Spinal Cord 2014, 52, 588–595. [Google Scholar] [CrossRef]
- Marinelli, S.; Vacca, V.; De Angelis, F.; Pieroni, L.; Orsini, T.; Parisi, C.; Soligo, M.; Protto, V.; Manni, L.; Guerrieri, R.; et al. Innovative mouse model mimicking human-like features of spinal cord injury: Efficacy of Docosahexaenoic acid on acute and chronic phases. Sci. Rep. 2019, 9, 8883. [Google Scholar] [CrossRef]
- Basso, D.M.; Fisher, L.C.; Anderson, A.J.; Jakeman, L.B.; McTigue, D.M.; Popovich, P.G. Basso Mouse Scale for locomotion detects differences in recovery after spinal cord injury in five common mouse strains. J. Neurotrauma 2006, 23, 635–659. [Google Scholar] [CrossRef]


© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Luvisetto, S. Botulinum Toxin and Neuronal Regeneration after Traumatic Injury of Central and Peripheral Nervous System. Toxins 2020, 12, 434. https://doi.org/10.3390/toxins12070434
Luvisetto S. Botulinum Toxin and Neuronal Regeneration after Traumatic Injury of Central and Peripheral Nervous System. Toxins. 2020; 12(7):434. https://doi.org/10.3390/toxins12070434
Chicago/Turabian StyleLuvisetto, Siro. 2020. "Botulinum Toxin and Neuronal Regeneration after Traumatic Injury of Central and Peripheral Nervous System" Toxins 12, no. 7: 434. https://doi.org/10.3390/toxins12070434
APA StyleLuvisetto, S. (2020). Botulinum Toxin and Neuronal Regeneration after Traumatic Injury of Central and Peripheral Nervous System. Toxins, 12(7), 434. https://doi.org/10.3390/toxins12070434