Biomonitoring of Enniatin B1 and Its Phase I Metabolites in Human Urine: First Large-Scale Study

 ,
,  , , and
, , and 
Abstract
1. Introduction
2. Results and Discussion
2.1. Evaluation of UHPLC-Q-Orbitrap HRMS Conditions
2.2. Method Performance
2.3. Occurrence of Enniatin B1 in Human Urines
2.4. Urinary Excretion Pattern of Enn B1 Phase I Metabolites
3. Conclusions
4. Materials
4.1. Chemicals, Reagents and Materials
4.2. Sampling
4.3. Sample Preparation
4.4. UHPLC-Q-Orbitrap HRMS Analysis
4.5. Metabolomic Data Processing
4.6. Method Validation
4.7. Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ferrigo, D.; Raiola, A.; Causin, R. Fusarium toxins in cereals: Occurrence, legislation, factors promoting the appearance and their management. Molecules 2016, 21, 627. [Google Scholar] [CrossRef]
- Commission Regulation (EC) No 1881/2006 of 19 December 2006 setting maximum levels for certain contaminants in foodstuffs. Off. J. Eur. Union 2006, 364, 324–365.
- Prosperini, A.; Berrada, H.; Ruiz, M.J.; Caloni, F.; Coccini, T.; Spicer, L.J.; Perego, M.C.; Lafranconi, A. A review of the mycotoxin enniatin B. Front. Public Health 2017, 5, 304. [Google Scholar] [CrossRef]
- Fraeyman, S.; Croubels, S.; Devreese, M.; Antonissen, G. Emerging Fusarium and Alternaria mycotoxins: Occurrence, toxicity and toxicokinetics. Toxins 2017, 9, 228. [Google Scholar] [CrossRef]
- Meca, G.; Zinedine, A.; Blesa, J.; Font, G.; Mañes, J. Further data on the presence of Fusarium emerging mycotoxins enniatins, fusaproliferin and beauvericin in cereals available on the Spanish markets. Food Chem. Toxicol. 2010, 48, 1412–1416. [Google Scholar] [CrossRef]
- Gruber-Dorninger, C.; Novak, B.; Nagl, V.; Berthiller, F. Emerging mycotoxins: Beyond traditionally determined food contaminants. J. Agric. Food Chem. 2016, 65, 7052–7070. [Google Scholar] [CrossRef] [PubMed]
- Jestoi, M.; Rokka, M.; Jarvenpaa, E.; Peltonen, K. Determination of Fusarium mycotoxins beauvericin and enniatins (A, A1, B, B1) in eggs of laying hens using liquid chromatography–tandem mass spectrometry (LC–MS/MS). Food Chem. 2009, 115, 1120–1127. [Google Scholar] [CrossRef]
- Johny, A.; Fæste, C.; Bogevik, A.S.; Berge, G.M.; Fernandes, J.M.O.; Ivanova, L. Development and Validation of a Liquid Chromatography High-Resolution Mass Spectrometry Method for the Simultaneous Determination of Mycotoxins and Phytoestrogens in Plant-Based Fish Feed and Exposed Fish. Toxins 2019, 11, 222. [Google Scholar] [CrossRef] [PubMed]
- EFSA. Scientific Opinion on the risks to human and animal health related to the presence of beauvericin and enniatins in food and feed. EFSA J. 2014, 12, 3802. [Google Scholar] [CrossRef]
- Maranghi, F.; Tassinari, R.; Narciso, L.; Tait, S.; Rocca, C.L.; Felice, G.D.; Butteroni, C.; Corinti, S.; Barletta, B.; Cordelli, E.; et al. In vivo toxicity and genotoxicity of beauvericin and enniatins. Combined approach to study in vivo toxicity and genotoxicity of mycotoxins beauvericin (BEA) and enniatin B (ENNB). EFSA Support. Publ. 2018, 15, 1406E. [Google Scholar] [CrossRef]
- Huang, C.H.; Wang, F.T.; Chan, W.H. Enniatin B1 exerts embryotoxic effects on mouse blastocysts and induces oxidative stress and immunotoxicity during embryo development. Environ. Toxicol. 2019, 34, 48–59. [Google Scholar] [CrossRef] [PubMed]
- Devreese, M.; Broekaert, N.; De Mil, T.; Fraeyman, S.; De Backer, P.; Croubels, S. Pilot toxicokinetic study and absolute oral bioavailability of the Fusarium mycotoxin enniatin B1 in pigs. Food Chem. Toxicol. 2014, 63, 161–165. [Google Scholar] [CrossRef] [PubMed]
- Devreese, M.; De Baere, S.; De Backer, P.; Croubels, S. Quantitative determination of the Fusarium mycotoxins beauvericin, enniatin A, A1, B and B1 in pig plasma using high performance liquid chromatography-tandem mass spectrometry. Talanta 2013, 106, 212–219. [Google Scholar] [CrossRef] [PubMed]
- Fraeyman, S.; Devreese, M.; Antonissen, G.; De Baere, S.; Rychlik, M.; Croubels, S. Comparative Oral Bioavailability, Toxicokinetics, and Biotransformation of Enniatin B1 and Enniatin B in Broiler Chickens. J. Agric. Food Chem. 2016, 64, 7259–7264. [Google Scholar] [CrossRef] [PubMed]
- Ivanova, L.; Uhlig, S.; Devreese, M.; Croubels, S.; Fæste, C.K. Biotransformation of the mycotoxin enniatin B1 in pigs: A comparative in vitro and in vivo approach. Food Chem. Toxicol. 2017, 105, 506–517. [Google Scholar] [CrossRef] [PubMed]
- Ivanova, L.; Denisov, I.G.; Grinkova, Y.V.; Sligar, S.G.; Fæste, C. Biotransformation of the Mycotoxin Enniatin B1 by CYP P450 3A4 and Potential for Drug-Drug Interactions. Metabolites 2019, 9, 158. [Google Scholar] [CrossRef]
- Ivanova, L.; Fæste, C.; Uhlig, S. In vitro phase I metabolism of the depsipeptide enniatin B. Anal. Bioanal. Chem. 2011, 400, 2889–2901. [Google Scholar] [CrossRef]
- Rodríguez-Carrasco, Y.; Izzo, L.; Gaspari, A.; Graziani, G.; Mañes, J.; Ritieni, A. Urinary levels of enniatin B and its phase I metabolites: First human pilot biomonitoring study. Food Chem. Toxicol. 2018, 118, 454–459. [Google Scholar] [CrossRef]
- European Commission. Commission Decision of 12 August 2002 implementing Council Directive 96/23/EC concerning the performance of analytical methods and the interpretation of results. Off. J. Eur. Communities 2002, 221, 8–36. [Google Scholar]
- Serrano, A.B.; Capriotti, A.L.; Cavaliere, C.; Piovesana, S.; Samperi, R.; Ventura, S.; Laganà, A. Development of a Rapid LC-MS/MS Method for the Determination of Emerging Fusarium mycotoxins Enniatins and Beauvericin in Human Biological Fluids. Toxins 2015, 7, 3554–3571. [Google Scholar] [CrossRef]
- Escrivá, L.; Manyes, L.; Font, G.; Berrada, H. Mycotoxin Analysis of Human Urine by LC-MS/MS: A Comparative Extraction Study. Toxins 2017, 9, 330. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Zhao, X.; Wu, L.; Zhou, S.; Gong, Z.; Zhao, Y.; Wu, Y. Development of a Sensitive and Reliable UHPLC-MS/MS Method for the Determination of Multiple Urinary Biomarkers of Mycotoxin Exposure. Toxins 2020, 12, 193. [Google Scholar] [CrossRef] [PubMed]
- FAO. Food Balance Sheets. Available online: http://www.fao.org/faostat/en/#data/ (accessed on 24 March 2020).
- Lemming, E.W.; Montes, A.M.; Schmidt, J.; Cramer, B.; Humpf, H.-U.; Moraeus, L.; Olsen, M. Mycotoxins in blood and urine of Swedish adolescents-possible associations to food intake and other background characteristics. Mycotoxin Res. 2020, 36, 193–206. [Google Scholar] [CrossRef] [PubMed]
- Jestoi, M.; Somma, M.; Kouva, M.; Veijalainen, P.; Rizzo, A.; Ritieni, A.; Peltonen, K. Levels of mycotoxins and sample cytotoxicity of selected organic and conventional grain-based products purchased from Finnish and Italian markets. Mol. Nutr. Food Res. 2004, 48, 299–307. [Google Scholar] [CrossRef]
- Wallin, S.; Gambacorta, L.; Kotova, N.; Warensjö Lemming, E.; Nälsén, C.; Solfrizzo, M.; Olsen, M. Biomonitoring of concurrent mycotoxin exposure among adults in Sweden through urinary multi-biomarker analysis. Food Chem. Toxicol. 2015, 83, 133–139. [Google Scholar] [CrossRef]
- Ciasca, B.; Pascale, M.; Altieri, V.; Longobardi, F.; Suman, M.; Catellani, D.; Lattanzio, V. In house validation and small scale collaborative study to evaluate analytical performances of multi-mycotoxin screening methods based on liquid chromatography-high resolution mass spectrometry: Case study on Fusarium toxins in wheat. J. Mass. Spectrom. 2018, 53, 743–752. [Google Scholar] [CrossRef]
- Rodríguez-Carrasco, Y.; Moltó, J.C.; Mañes, J.; Berrada, H. Development of a GC–MS/MS strategy to determine 15 mycotoxins and metabolites in human urine. Talanta 2014, 128, 125–131. [Google Scholar] [CrossRef]
- Ali, N.; Degen, G.H. Urinary biomarkers of exposure to the mycoestrogen zearalenone and its modified forms in German adults. Arch. Toxicol. 2018, 92, 2691–2700. [Google Scholar] [CrossRef]
- Li, C.; Deng, C.; Zhou, S.; Zhao, Y.; Wang, D.; Wang, X.; Gong, Y.Y.; Wu, Y. High-throughput and sensitive determination of urinary zearalenone and metabolites by UPLC-MS/MS and its application to a human exposure study. Anal. Bioanal. Chem. 2018, 410, 5301–5312. [Google Scholar] [CrossRef]
- Fæste, C.K.; Ivanova, L.; Uhlig, S. In Vitro Metabolism of the Mycotoxin Enniatin B in Different Species and Cytochrome P450 Enzyme Phenotyping by Chemical Inhibitors. Drug Metab. Dispos. 2011, 39, 1768–1776. [Google Scholar] [CrossRef]
- Pérez, R.; Domenech, E.; Coscollà, C.; Yusa, V. Human Biomonitoring of food contaminants in Spanish children: Design, sampling and lessons learned. Int. J. Hyg. Environ. Health 2017, 220, 1768–1776. [Google Scholar] [CrossRef] [PubMed]
- INRAN. Dietary Guidelines for Healthy Eating; Revision 2018; National Institute for Food Research and Nutrition: Rome, Italy, 2018. [Google Scholar]
- Rodríguez-Carrasco, Y.; Moltó, J.C.; Mañes, J.; Berrada, H. Development of microextraction techniques in combination with GC–MS/MS for the determination of mycotoxins and metabolites in human urine. J. Sep. Sci. 2017, 40, 1572–1582. [Google Scholar] [CrossRef] [PubMed]


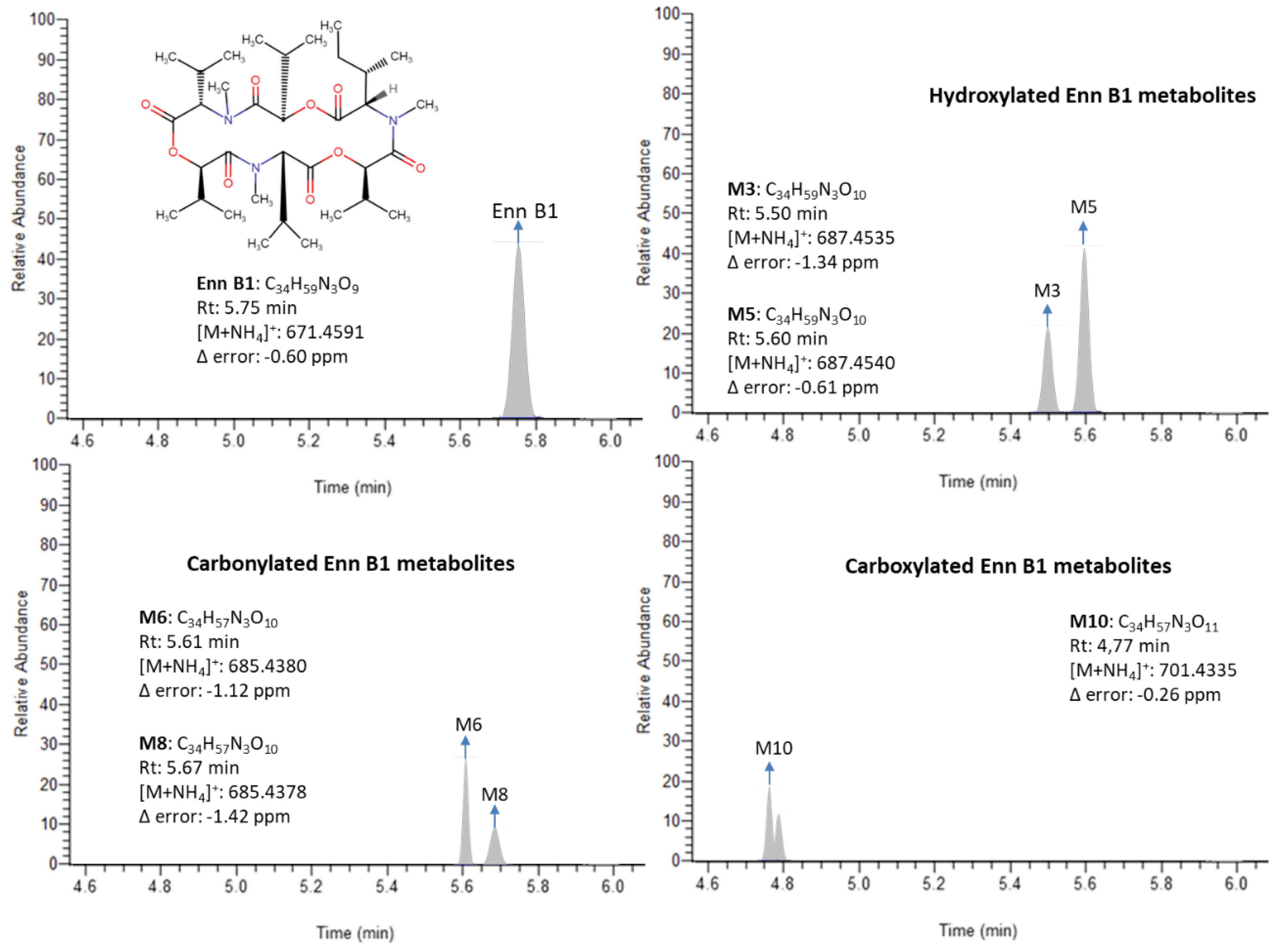{kind=link}
| Compound | Retention Time (min) | Molecular Formula | Observed Mass (M + NH4)+ | Accuracy (Δppm) |
|---|---|---|---|---|
| Enn B1 | 5.75 | C34H59N3O9 | 671.4591 | −0.60 |
| M1 | 4.76 | C33H55N3O11 | 687.4175 | −0.78 |
| M2 | 5.44 | C34H59N3O10 | 687.4532 | −1.78 |
| M3 | 5.5 | 687.4535 | −1.34 | |
| M4 | 5.55 | 687.4536 | −1.19 | |
| M5 | 5.6 | 687.4540 | −0.61 | |
| M6 | 5.61 | C34H57N3O10 | 685.4380 | −1.12 |
| M7 | 5.64 | 685.4384 | −0.54 | |
| M8 | 5.67 | 685.4378 | −1.42 | |
| M9 | 4.1 | C34H57N3O11 | 701.4328 | −1.26 |
| M10 | 4.77 | 701.4335 | −0.26 | |
| M11 | 4.79 | 701.4333 | −0.55 |
| Parameters | R2 | SSE (%) | Recovery, % (RSDR, %; n = 9) | LOD (ng/mL) | LOQ (ng/mL) | |||
|---|---|---|---|---|---|---|---|---|
| 5 ng/mL | 1 ng/mL | 0.5 ng/mL | 0.1 ng/mL | |||||
| Enn B1 | 0.9997 | 63 | 95 (7) | 88 (6) | 78 (7) | 84 (12) | 0.0005 | 0.001 |
| Determination | |||||||
|---|---|---|---|---|---|---|---|
| Urine Samples (n) | Origin | Positives Pamples (n, (%)) | Sample Preparation | Range (ng/mL) | Sensitivity (LOQ, ng/mL) | Detection Method | Reference |
| 10 | Italy | 1 (10) | SPE | <LOQ | 0.005 | QQQ (Thermo Fisher Scientific) ESI+ SRM mode | Serrano et al. (2015) [20] |
| 10 | Spain | 6 (60) | DLLME | <LOQ–0.34 | 0.1 | QQQ (Applied Biosystems) ESI+ SRM mode | Escrivá et al. (2017) [21] |
| 60 | China | 0 (0) | SPE | - | 0.0002 | QQQ (AB SCIEX) ESI+ MRM mode | Liu et al. (2020) [22] |
| 300 | Italy | 283 (94) | SALLE | <LOQ–0.429 | 0.001 | Q-Orbitrap (Exactive, Thermo Fisher Scientific) ESI+ HRMS | This work |
| Compound/Group | Incidence (%) | Range (ng/mL) | Mean a (ng/mL) |
|---|---|---|---|
| Parent Compound | |||
| Enn B1 | 94.3 | 0.007–0.429 | 0.069 |
| Enn B1 Biotransformation Products | |||
| M1 | 5.3 | 0.007–0.177 | 0.035 |
| Demethylated and hydroxylated (M1) | 5.3 | 0.007–0.177 | 0.035 |
| M2 | 11.0 | 0.006–0.019 | 0.010 |
| M3 | 50.0 | 0.005–0.076 | 0.023 |
| M4 | 18.0 | 0.002–0.143 | 0.025 |
| M5 | 77.3 | 0.006–0.186 | 0.047 |
| Hydroxylated group (M2–M5) | 78.0 | 0.006–0.233 | 0.069 |
| M6 | 40.0 | 0.012–1.511 | 0.105 |
| M7 | 30.7 | 0.008–0.510 | 0.085 |
| M8 | 48.0 | 0.042–1.310 | 0.128 |
| Carbonylated group (M6–M8) | 66.0 | 0.012–1.763 | 0.196 |
| M9 | 0.7 | 0.019–0.045 | 0.032 |
| M10 | 21.0 | 0.008–0.241 | 0.047 |
| M11 | 14.0 | 0.002–0.451 | 0.053 |
| Carboxylated group (M9–M11) | 26.3 | 0.008–0.656 | 0.066 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodríguez-Carrasco, Y.; Narváez, A.; Izzo, L.; Gaspari, A.; Graziani, G.; Ritieni, A. Biomonitoring of Enniatin B1 and Its Phase I Metabolites in Human Urine: First Large-Scale Study. Toxins 2020, 12, 415. https://doi.org/10.3390/toxins12060415
Rodríguez-Carrasco Y, Narváez A, Izzo L, Gaspari A, Graziani G, Ritieni A. Biomonitoring of Enniatin B1 and Its Phase I Metabolites in Human Urine: First Large-Scale Study. Toxins. 2020; 12(6):415. https://doi.org/10.3390/toxins12060415
Chicago/Turabian StyleRodríguez-Carrasco, Yelko, Alfonso Narváez, Luana Izzo, Anna Gaspari, Giulia Graziani, and Alberto Ritieni. 2020. "Biomonitoring of Enniatin B1 and Its Phase I Metabolites in Human Urine: First Large-Scale Study" Toxins 12, no. 6: 415. https://doi.org/10.3390/toxins12060415
APA StyleRodríguez-Carrasco, Y., Narváez, A., Izzo, L., Gaspari, A., Graziani, G., & Ritieni, A. (2020). Biomonitoring of Enniatin B1 and Its Phase I Metabolites in Human Urine: First Large-Scale Study. Toxins, 12(6), 415. https://doi.org/10.3390/toxins12060415