Plastics in Cyanobacterial Blooms—Genotoxic Effects of Binary Mixtures of Cylindrospermopsin and Bisphenols in HepG2 Cells



Abstract

1. Introduction
2. Results and Discussion
2.1. The Influence of CYN, BPs, and Their Combinations on Cell Viability
2.2. Induction of DNA Double-Strand Breaks by CYN, BPs, and Their Combinations
2.3. Gene Deregulation in Response to CYN/BPs Exposure
2.4. Xenobiotic Metabolism—CYP1A1
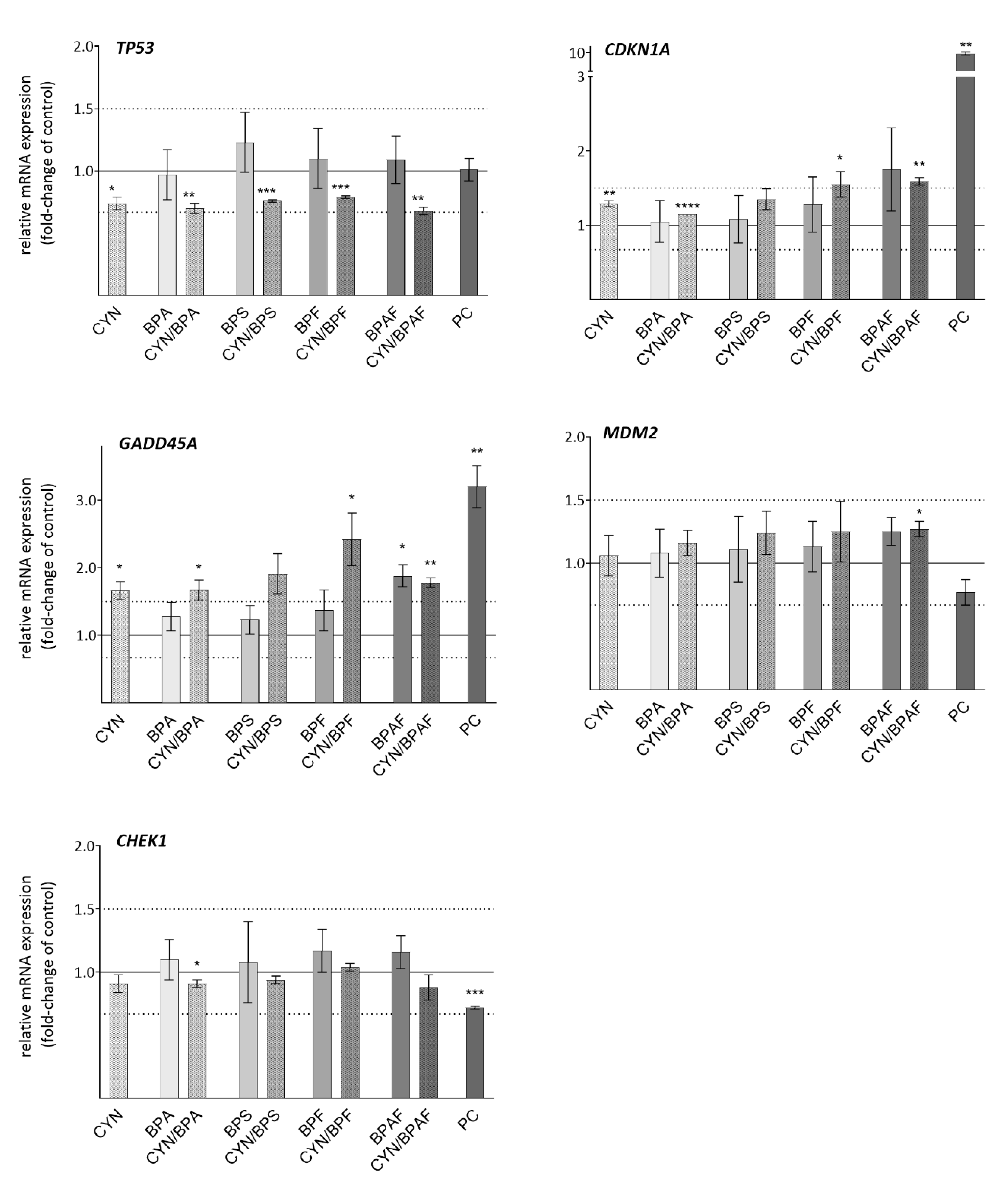
2.5. DNA Damage Response Genes
2.6. Oxidative Stress Response Genes
3. Conclusions
4. Materials and Methods
4.1. Chemicals
4.2. Cell Culture
4.3. Cell Viability—MTS Assay
4.4. Analyses of the Induction of DNA DSB by the γ-H2AX Assay
4.5. Real-Time Quantitative PCR (qPCR) Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Huisman, J.; Codd, G.A.; Paerl, H.W.; Ibelings, B.W.; Verspagen, J.M.H.; Visser, P.M. Cyanobacterial blooms. Nat. Rev. Microbiol. 2018, 16, 471–483. [Google Scholar] [CrossRef] [PubMed]
- Chiswell, R.K.; Shaw, G.R.; Eaglesham, G.; Smith, M.J.; Norris, R.L.; Seawright, A.A.; Moore, M.R. Stability of cylindrospermopsin, the toxin from the cyanobacterium, cylindrospermopsis raciborskii: Effect of pH, temperature, and sunlight on decomposition. Environ. Toxicol. 1999, 14, 155–161. [Google Scholar] [CrossRef]
- Wormer, L.; Cirés, S.; Carrasco, D.; Quesada, A. Cylindrospermopsin is not degraded by co-occurring natural bacterial communities during a 40-day study. Harmful Algae 2008, 7, 206–213. [Google Scholar] [CrossRef]
- Rücker, J.; Stüken, A.; Nixdorf, B.; Fastner, J.; Chorus, I.; Wiedner, C. Concentrations of particulate and dissolved cylindrospermopsin in 21 Aphanizomenon-dominated temperate lakes. Toxicon 2007, 50, 800–809. [Google Scholar] [CrossRef] [PubMed]
- Rzymski, P.; Poniedziałek, B. In search of environmental role of cylindrospermopsin: A review on global distribution and ecology of its producers. Water Res. 2014, 66, 320–337. [Google Scholar] [CrossRef]
- Antunes, J.T.; Leão, P.N.; Vasconcelos, V.M. Cylindrospermopsis raciborskii: Review of the distribution, phylogeography, and ecophysiology of a global invasive species. Front. Microbiol. 2015, 6, 473. [Google Scholar] [CrossRef]
- Szlag, D.C.; Sinclair, J.L.; Southwell, B.; Westrick, J.A. Cyanobacteria and cyanotoxins occurrence and removal from five high-risk conventional treatment drinking water plants. Toxins (Basel) 2015, 7, 2198–2220. [Google Scholar] [CrossRef]
- Yen, H.K.; Lin, T.F.; Liao, P.C. Simultaneous detection of nine cyanotoxins in drinking water using dual solid-phase extraction and liquid chromatography-mass spectrometry. Toxicon 2011, 58, 209–218. [Google Scholar] [CrossRef]
- Lei, L.; Peng, L.; Huang, X.; Han, B.P. Occurrence and dominance of Cylindrospermopsis raciborskii and dissolved cylindrospermopsin in urban reservoirs used for drinking water supply, South China. Environ. Monit. Assess. 2014, 186, 3079–3090. [Google Scholar] [CrossRef]
- Gutiérrez-Praena, D.; Jos, Á.; Pichardo, S.; Moreno, I.M.; Cameán, A.M. Presence and bioaccumulation of microcystins and cylindrospermopsin in food and the effectiveness of some cooking techniques at decreasing their concentrations: A review. Food Chem. Toxicol. 2013, 53, 139–152. [Google Scholar] [CrossRef]
- Mohamed, Z.A.; Bakr, A. Concentrations of cylindrospermopsin toxin in water and tilapia fish of tropical fishponds in Egypt, and assessing their potential risk to human health. Environ. Sci. Pollut. Res. 2018, 25, 36287–36297. [Google Scholar] [CrossRef] [PubMed]
- Ohtani, I.; Moore, R.E.; Runnegar, M.T.C. Cylindrospermopsin: A Potent Hepatotoxin from the Blue-Green Alga Cylindrospermopsis raciborskii. J. Am. Chem. Soc. 1992, 114, 7941–7942. [Google Scholar] [CrossRef]
- Evans, D.M.; Hughes, J.; Jones, L.F.; Murphy, P.J.; Falfushynska, H.; Horyn, O.; Sokolova, I.M.; Christensen, J.; Coles, S.J.; Rzymski, P. Elucidating cylindrospermopsin toxicity via synthetic analogues: An in vitro approach. Chemosphere 2019, 234, 139–147. [Google Scholar] [CrossRef]
- Froscio, S.M.; Humpage, A.R.; Burcham, P.C.; Falconer, I.R. Cylindrospermopsin-induced protein synthesis inhibition and its dissociation from acute toxicity in mouse hepatocytes. Environ. Toxicol. 2003, 18, 243–251. [Google Scholar] [CrossRef]
- Runnegar, M.T.; Xie, C.; Snider, B.B.; Wallace, G.A.; Weinreb, S.M.; Kuhlenkamp, J. In vitro hepatotoxicity of the cyanobacterial alkaloid cyclindrospermopsin and related synthetic analogues. Toxicol. Sci. 2002, 67, 81–87. [Google Scholar] [CrossRef]
- Terao, K.; Ohmori, S.; Igarashi, K.; Ohtani, I.; Watanabe, M.F.; Harada, K.I.; Ito, E.; Watanabe, M. Electron microscopic studies on experimental poisoning in mice induced by cylindrospermopsin isolated from blue-green alga Umezakia natans. Toxicon 1994, 32, 833–843. [Google Scholar] [CrossRef]
- Puerto, M.; Prieto, A.I.; Maisanaba, S.; Gutiérrez-Praena, D.; Mellado-García, P.; Jos, Á.; Cameán, A.M. Mutagenic and genotoxic potential of pure Cylindrospermopsin by a battery of in vitro tests. Food Chem. Toxicol. 2018, 121, 413–422. [Google Scholar] [CrossRef]
- Štraser, A.; Filipič, M.; Žegura, B. Cylindrospermopsin induced transcriptional responses in human hepatoma HepG2 cells. Toxicol. Vitr. 2013, 27, 1809–1819. [Google Scholar] [CrossRef]
- Štraser, A.; Filipič, M.; Gorenc, I.; Žegura, B. The influence of cylindrospermopsin on oxidative DNA damage and apoptosis induction in HepG2 cells. Chemosphere 2013, 92, 24–30. [Google Scholar] [CrossRef]
- Pichardo, S.; Cameán, A.M.; Jos, A. In vitro toxicological assessment of cylindrospermopsin: A review. Toxins (Basel) 2017, 9, 402. [Google Scholar] [CrossRef]
- Young, F.M.; Micklem, J.; Humpage, A.R. Effects of blue-green algal toxin cylindrospermopsin (CYN) on human granulosa cells in vitro. Reprod. Toxicol. 2008, 25, 374–380. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Hernández, S.E.; Swift, S.; Singhal, N. Estrogenic activity of cylindrospermopsin and anatoxin-a and their oxidative products by FeIII-B*/H2O2. Water Res. 2018, 132, 309–319. [Google Scholar] [CrossRef] [PubMed]
- Žegura, B.; Štraser, A.; Filipič, M. Genotoxicity and potential carcinogenicity of cyanobacterial toxins—A review. Mutat. Res. Rev. Mutat. Res. 2011, 727, 16–41. [Google Scholar] [CrossRef] [PubMed]
- Bazin, E.; Mourot, A.; Humpage, A.R.; Fessard, V. Genotoxicity of a freshwater cyanotoxin, cylindrospermopsin, in two human cell lines: Caco-2 and HepaRG. Environ. Mol. Mutagen. 2010, 51, 251–259. [Google Scholar] [CrossRef]
- Štraser, A.; Filipič, M.; Žegura, B. Genotoxic effects of the cyanobacterial hepatotoxin cylindrospermopsin in the HepG2 cell line. Arch. Toxicol. 2011, 85, 1617–1626. [Google Scholar] [CrossRef]
- Žegura, B.; Gajski, G.; Štraser, A.; Garaj-Vrhovac, V. Cylindrospermopsin induced DNA damage and alteration in the expression of genes involved in the response to DNA damage, apoptosis and oxidative stress. Toxicon 2011, 58, 471–479. [Google Scholar] [CrossRef]
- Hercog, K.; Maisanaba, S.; Filipič, M.; Jos, Á.; Cameán, A.M.; Žegura, B. Genotoxic potential of the binary mixture of cyanotoxins microcystin-LR and cylindrospermopsin. Chemosphere 2017, 189, 319–329. [Google Scholar] [CrossRef]
- Štraser, A.; Filipič, M.; Novak, M.; Žegura, B. Double strand breaks and cell-cycle arrest induced by the cyanobacterial toxin cylindrospermopsin in HepG2 cells. Mar. Drugs 2013, 11, 3077–3090. [Google Scholar]
- Falconer, I.R.; Humpage, A.R. Preliminary evidence for in vivo tumour initiation by oral administration of extracts of the blue-green alga cylindrospermopsis raciborskil containing the toxin cylindrospermopsin. Environ. Toxicol. 2001, 16, 192–195. [Google Scholar] [CrossRef]
- Shen, X.; Lam, P.K.S.; Shaw, G.R.; Wickramasinghe, W. Genotoxicity investigation of a cyanobacterial toxin, cylindrospermopsin. Toxicon 2002, 40, 1499–1501. [Google Scholar] [CrossRef]
- Díez-Quijada, L.; Llana-Ruiz-Cabello, M.; Cătunescu, G.M.; Puerto, M.; Moyano, R.; Jos, A.; Cameán, A.M. In vivo genotoxicity evaluation of cylindrospermopsin in rats using a combined micronucleus and comet assay. Food Chem. Toxicol. 2019, 132, 110664. [Google Scholar] [CrossRef] [PubMed]
- Geyer, R.; Jambeck, J.R.; Law, K.L. Production, use, and fate of all plastics ever made. Sci. Adv. 2017, 3, e1700782. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, E.; Yamashita, N.; Taniyasu, S.; Lam, J.; Lam, P.K.S.; Moon, H.-B.; Jeong, Y.; Kannan, P.; Achyuthan, H.; Munuswamy, N.; et al. Bisphenol A and other bisphenol analogues including BPS and BPF in surface water samples from Japan, China, Korea and India. Ecotoxicol. Environ. Saf. 2015, 122, 565–572. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.; Chen, Q.; Crawford, S.E.; Song, L.; Chen, W.; Hammers-Wirtz, M.; Strauss, T.; Seiler, T.B.; Schäffer, A.; Hollert, H. Cyanobacterial blooms act as sink and source of endocrine disruptors in the third largest freshwater lake in China. Environ. Pollut. 2019, 245, 408–418. [Google Scholar] [CrossRef]
- Park, J.C.; Lee, M.-C.; Yoon, D.-S.; Han, J.; Kim, M.; Hwang, U.-K.; Jung, J.-H.; Lee, J.-S. Effects of bisphenol A and its analogs bisphenol F and S on life parameters, antioxidant system, and response of defensome in the marine rotifer Brachionus koreanus. Aquat. Toxicol. 2018, 199, 21–29. [Google Scholar] [CrossRef]
- Lehmler, H.-J.; Liu, B.; Gadogbe, M.; Bao, W. Exposure to Bisphenol A, Bisphenol F, and Bisphenol S in U.S. Adults and Children: The National Health and Nutrition Examination Survey 2013–2014. ACS Omega 2018, 3, 6523–6532. [Google Scholar] [CrossRef]
- Chen, D.; Kannan, K.; Tan, H.; Zheng, Z.; Feng, Y.L.; Wu, Y.; Widelka, M. Bisphenol Analogues Other Than BPA: Environmental Occurrence, Human Exposure, and Toxicity—A Review. Environ. Sci. Technol. 2016, 50, 5438–5453. [Google Scholar] [CrossRef]
- Corrales, J.; Kristofco, L.A.; Steele, W.B.; Yates, B.S.; Breed, C.S.; Williams, E.S.; Brooks, B.W. Global Assessment of Bisphenol A in the Environment: Review and Analysis of Its Occurrence and Bioaccumulation. Dose-Response 2015, 13, 1559325815598308. [Google Scholar] [CrossRef]
- Balabanič, D.; Filipič, M.; Krivograd Klemenčič, A.; Žegura, B. Raw and biologically treated paper mill wastewater effluents and the recipient surface waters: Cytotoxic and genotoxic activity and the presence of endocrine disrupting compounds. Sci. Total Environ. 2017, 574, 78–89. [Google Scholar] [CrossRef]
- Usman, A.; Ahmad, M. From BPA to its analogues: Is it a safe journey? Chemosphere 2016, 158, 131–142. [Google Scholar] [CrossRef]
- Kwan, C.S.; Takada, H. Release of Additives and Monomers from Plastic Wastes. In Handbook of Environmental Chemistry; Springer Verlag: Heidelberg, Germany, 2019; Volume 78, pp. 51–70. [Google Scholar]
- Huang, R.P.; Liu, Z.H.; Yuan, S.F.; Yin, H.; Dang, Z.; Wu, P.X. Worldwide human daily intakes of bisphenol A (BPA) estimated from global urinary concentration data (2000–2016) and its risk analysis. Environ. Pollut. 2017, 230, 143–152. [Google Scholar] [CrossRef] [PubMed]
- Vandenberg, L.N.; Hauser, R.; Marcus, M.; Olea, N.; Welshons, W.V. Human exposure to bisphenol A (BPA). Reprod. Toxicol. 2007, 24, 139–177. [Google Scholar] [CrossRef] [PubMed]
- Fic, A.; Žegura, B.; Sollner Dolenc, M.; Filipič, M.; Peterlin Mašič, L. Mutagenicity and DNA damage of bisphenol a and its structural analogues in HepG2 cells. Arh. Hig. Rada Toksikol. 2013, 64, 189–200. [Google Scholar] [CrossRef]
- Xin, L.; Lin, Y.; Wang, A.; Zhu, W.; Liang, Y.; Su, X.; Hong, C.; Wan, J.; Wang, Y.; Tian, H. Cytogenetic evaluation for the genotoxicity of bisphenol-A in Chinese hamster ovary cells. Environ. Toxicol. Pharmacol. 2015, 40, 524–529. [Google Scholar] [CrossRef]
- Hercog, K.; Maisanaba, S.; Filipič, M.; Sollner-Dolenc, M.; Kač, L.; Žegura, B. Genotoxic activity of bisphenol A and its analogues bisphenol S, bisphenol F and bisphenol AF and their mixtures in human hepatocellular carcinoma (HepG2) cells. Sci. Total Environ. 2019, 687, 267–276. [Google Scholar] [CrossRef]
- Tiwari, D.; Kamble, J.; Chilgunde, S.; Patil, P.; Maru, G.; Kawle, D.; Bhartiya, U.; Joseph, L.; Vanage, G. Clastogenic and mutagenic effects of bisphenol A: An endocrine disruptor. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2012, 743, 83–90. [Google Scholar] [CrossRef]
- Santovito, A.; Cannarsa, E.; Schleicherova, D.; Cervella, P. Clastogenic effects of bisphenol A on human cultured lymphocytes. Hum. Exp. Toxicol. 2018, 37, 69–77. [Google Scholar] [CrossRef]
- Kittler, K.; Hurtaud-Pessel, D.; Maul, R.; Kolrep, F.; Fessard, V. In vitro metabolism of the cyanotoxin cylindrospermopsin in HepaRG cells and liver tissue fractions. Toxicon 2016, 110, 47–50. [Google Scholar] [CrossRef]
- Wu, L.-H.; Zhang, X.-M.; Wang, F.; Gao, C.-J.; Chen, D.; Palumbo, J.R.; Guo, Y.; Zeng, E.Y. Occurrence of bisphenol S in the environment and implications for human exposure: A short review. Sci. Total Environ. 2018, 615, 87–98. [Google Scholar] [CrossRef]
- Izzotti, A.; Kanitz, S.; D’Agostini, F.; Camoirano, A.; De Flora, S. Formation of adducts by bisphenol A, an endocrine disruptor, in DNA in vitro and in liver and mammary tissue of mice. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2009, 679, 28–32. [Google Scholar] [CrossRef]
- Sonavane, M.; Gassman, N.R. Bisphenol A co-exposure effects: A key factor in understanding BPA’s complex mechanism and health outcomes. Crit. Rev. Toxicol. 2019, 49, 371–386. [Google Scholar] [CrossRef] [PubMed]
- OECD. No. 487: In Vitro Mammalian Cell Micronucleus Test. In OECD Guidelines for the Testing of Chemicals, Section 4; OECD iLibrary: Paris, France, 2014. [Google Scholar] [CrossRef]
- Westerink, W.M.A.; Schoonen, W.G.E.J. Cytochrome P450 enzyme levels in HepG2 cells and cryopreserved primary human hepatocytes and their induction in HepG2 cells. Toxicol. Vitr. 2007, 21, 1581–1591. [Google Scholar] [CrossRef] [PubMed]
- Nohmi, T. Thresholds of genotoxic and non-genotoxic carcinogens. Toxicol. Res. 2018, 34, 281–290. [Google Scholar] [CrossRef] [PubMed]
- Chan, G.K.Y.; Kleinheinz, T.L.; Peterson, D.; Moffat, J.G. A Simple High-Content Cell Cycle Assay Reveals Frequent Discrepancies between Cell Number and ATP and MTS Proliferation Assays. PLoS ONE 2013, 8, e63583. [Google Scholar] [CrossRef] [PubMed]
- Russo, G.; Capuozzo, A.; Barbato, F.; Irace, C.; Santamaria, R.; Grumetto, L. Cytotoxicity of seven bisphenol analogues compared to bisphenol A and relationships with membrane affinity data. Chemosphere 2018, 201, 432–440. [Google Scholar] [CrossRef]
- Rogakou, E.P.; Pilch, D.R.; Orr, A.H.; Ivanova, V.S.; Bonner, W.M. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J. Biol. Chem. 1998, 273, 5858–5868. [Google Scholar] [CrossRef]
- Rogakou, E.P.; Boon, C.; Redon, C.; Bonner, W.M. Megabase chromatin domains involved in DNA double-strand breaks in vivo. J. Cell Biol. 1999, 146, 905–916. [Google Scholar] [CrossRef]
- Kopp, B.; Khoury, L.; Audebert, M. Validation of the γH2AX biomarker for genotoxicity assessment: A review. Arch. Toxicol. 2019, 93, 2103–2114. [Google Scholar] [CrossRef]
- Audebert, M.; Dolo, L.; Perdu, E.; Cravedi, J.-P.; Zalko, D. Use of the γH2AX assay for assessing the genotoxicity of bisphenol A and bisphenol F in human cell lines. Arch. Toxicol. 2011, 85, 1463. [Google Scholar] [CrossRef]
- Humpage, A.R.; Fontaine, F.; Froscio, S.; Burcham, P.; Falconer, I.R. Cylindrospermopsin Genotoxicity and Cytotoxicity: Role Of Cytochrome P-450 and Oxidative Stress. J. Toxicol. Environ. Heal. Part A 2005, 68, 739–753. [Google Scholar] [CrossRef]
- Norris, R.L.; Seawright, A.; Shaw, G.; Senogles, P.; Eaglesham, G.; Smith, M.; Chiswell, R.; Moore, M. Hepatic xenobiotic metabolism of cylindrospermopsin in vivo in the mouse. Toxicon 2002, 40, 471–476. [Google Scholar] [CrossRef]
- Gramec Skledar, D.; Peterlin Mašič, L. Bisphenol A and its analogs: Do their metabolites have endocrine activity? Environ. Toxicol. Pharmacol. 2016, 47, 182–199. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.H.; Katayama, Y.; Kondo, F. Biodegradation or metabolism of bisphenol A: From microorganisms to mammals. Toxicology 2006, 217, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Hanioka, N.; Jinno, H.; Nishimura, T.; Ando, M. Suppression of male-specific cytochrome P450 isoforms by bisphenol A in rat liver. Arch. Toxicol. 1998, 72, 387–394. [Google Scholar] [CrossRef]
- Niwa, T.; Tsusui, M.; Kishimoto, K.; Yabusaki, Y.; Ishibashi, F.; Katagiri, M. Inhibition of Drug-Metabolizing Enzyme Activity in Human Hepatic Cytochrome P450s by Bisphenol A. Biol. Pharm. Bull. 2000, 23, 498–501. [Google Scholar] [CrossRef][Green Version]
- Pfeiffer, E.; Metzler, M. Effect of bisphenol A on drug metabolising enzymes in rat hepatic microsomes and precision-cut rat liver slices. Arch. Toxicol. 2004, 78, 369–377. [Google Scholar] [CrossRef]
- Runnegar, M.T.; Kong, S.-M.; Zhong, Y.-Z.; Lu, S.C. Inhibition of reduced glutathione synthesis by cyanobacterial alkaloid cylindrospermopsin in cultured rat hepatocytes. Biochem. Pharmacol. 1995, 49, 219–225. [Google Scholar] [CrossRef]
- Cabaton, N.; Zalko, D.; Rathahao, E.; Canlet, C.; Delous, G.; Chagnon, M.-C.; Cravedi, J.-P.; Perdu, E. Biotransformation of bisphenol F by human and rat liver subcellular fractions. Toxicol. Vitr. 2008, 22, 1697–1704. [Google Scholar] [CrossRef]
- Jaeg, J.P.; Perdu, E.; Dolo, L.; Debrauwer, L.; Cravedi, J.-P.; Zalko, D. Characterization of New Bisphenol A Metabolites Produced by CD1 Mice Liver Microsomes and S9 Fractions. J. Agric. Food Chem. 2004, 52, 4935–4942. [Google Scholar] [CrossRef]
- Atkinson, A.; Roy, D. In Vitro Conversion of Environmental Estrogenic Chemical Bisphenol A to DNA Binding Metabolite(s). Biochem. Biophys. Res. Commun. 1995, 210, 424–433. [Google Scholar] [CrossRef]
- Kolšek, K.; Mavri, J.; Sollner Dolenc, M. Reactivity of bisphenol A-3,4-quinone with DNA. A quantum chemical study. Toxicol. Vitr. 2012, 26, 102–106. [Google Scholar] [CrossRef]
- Xu, H.; Zhang, X.; Ye, Y.; Li, X. Bisphenol A affects estradiol metabolism by targeting CYP1A1 and CYP19A1 in human placental JEG-3 cells. Toxicol. Vitr. 2019, 61, 104615. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Zhang, C.; Liu, A.-L.; Xie, S.-H.; Chen, X.-M.; Lu, W.-Q. PCB126 enhanced the genotoxicity of BaP in HepG2 cells by modulating metabolic enzyme and DNA repair activities. Toxicol. Lett. 2009, 189, 91–95. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Lu, W.-Q.; Zou, Y.-L.; Wei, W.; Zhang, C.; Xie, H.; Chen, X.-M. Influence of aroclor 1254 on benzo(a)pyrene-induced DNA breakage, oxidative DNA damage, and cytochrome P4501A activity in human hepatoma cell line. Environ. Toxicol. 2009, 24, 327–333. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Lu, W.; Mersch-Sundermann, V. Benzo(a)pyrene induced micronucleus formation was modulated by persistent organic pollutants (POPs) in metabolically competent human HepG2 cells. Toxicol. Lett. 2003, 144, 143–150. [Google Scholar] [CrossRef]
- Ellinger-Ziegelbauer, H.; Stuart, B.; Wahle, B.; Bomann, W.; Ahr, H.J. Comparison of the expression profiles induced by genotoxic and nongenotoxic carcinogens in rat liver. Mutat. Res. Mol. Mech. Mutagen. 2005, 575, 61–84. [Google Scholar] [CrossRef]
- Hreljac, I.; Zajc, I.; Lah, T.; Filipič, M. Effects of model organophosphorous pesticides on DNA damage and proliferation of HepG2 cells. Environ. Mol. Mutagen. 2008, 49, 360–367. [Google Scholar] [CrossRef]
- Petković, J.; Žegura, B.; StevanoviĆ, M.; Drnovšek, N.; UskokoviĆ, D.; Novak, S.; FilipiČ, M. DNA damage and alterations in expression of DNA damage responsive genes induced by TiO 2 nanoparticles in human hepatoma HepG2 cells. Nanotoxicology 2011, 5, 341–353. [Google Scholar] [CrossRef]
- Pezdirc, M.; Žegura, B.; Filipič, M. Genotoxicity and induction of DNA damage responsive genes by food-borne heterocyclic aromatic amines in human hepatoma HepG2 cells. Food Chem. Toxicol. 2013, 59, 386–394. [Google Scholar] [CrossRef]
- Žegura, B.; Zajc, I.; Lah, T.T.; Filipič, M. Patterns of microcystin-LR induced alteration of the expression of genes involved in response to DNA damage and apoptosis. Toxicon 2008, 51, 615–623. [Google Scholar] [CrossRef]
- Oren, M. Decision making by p53: Life, death and cancer. Cell Death Differ. 2003, 10, 431–442. [Google Scholar] [CrossRef] [PubMed]
- Lakin, N.D.; Jackson, S.P. Regulation of p53 in response to DNA damage. Oncogene 1999, 18, 7644–7655. [Google Scholar] [CrossRef] [PubMed]
- Cazzalini, O.; Scovassi, A.I.; Savio, M.; Stivala, L.A.; Prosperi, E. Multiple roles of the cell cycle inhibitor p21CDKN1A in the DNA damage response. Mutat. Res. Mutat. Res. 2010, 704, 12–20. [Google Scholar] [CrossRef]
- Zhan, Q. Gadd45a, a p53- and BRCA1-regulated stress protein, in cellular response to DNA damage. Mutat. Res. Mol. Mech. Mutagen. 2005, 569, 133–143. [Google Scholar] [CrossRef]
- Michael, D.; Oren, M. The p53–Mdm2 module and the ubiquitin system. Semin. Cancer Biol. 2003, 13, 49–58. [Google Scholar] [CrossRef]
- Ranuncolo, S.M.; Wang, L.; Polo, J.M.; Dell’Oso, T.; Dierov, J.; Gaymes, T.J.; Rassool, F.; Carroll, M.; Melnick, A. BCL6-mediated attenuation of DNA damage sensing triggers growth arrest and senescence through a p53-dependent pathway in a cell context-dependent manner. J. Biol. Chem. 2008, 283, 22565–22572. [Google Scholar] [CrossRef]
- Gassman, N.R. Induction of oxidative stress by bisphenol A and its pleiotropic effects. Environ. Mol. Mutagen. 2017, 58, 60–71. [Google Scholar] [CrossRef]
- Huc, L.; Lemarié, A.; Guéraud, F.; Héliès-Toussaint, C. Low concentrations of bisphenol A induce lipid accumulation mediated by the production of reactive oxygen species in the mitochondria of HepG2 cells. Toxicol. Vitr. 2012, 26, 709–717. [Google Scholar] [CrossRef]
- Mokra, K.; Woźniak, K.; Bukowska, B.; Sicińska, P.; Michałowicz, J. Low-concentration exposure to BPA, BPF and BPAF induces oxidative DNA bases lesions in human peripheral blood mononuclear cells. Chemosphere 2018, 201, 119–126. [Google Scholar] [CrossRef]
- Maćczak, A.; Cyrkler, M.; Bukowska, B.; Michałowicz, J. Bisphenol A, bisphenol S, bisphenol F and bisphenol AF induce different oxidative stress and damage in human red blood cells (in vitro study). Toxicol. Vitr. 2017, 41, 143–149. [Google Scholar] [CrossRef]
- Zangar, R.C.; Davydov, D.R.; Verma, S. Mechanisms that regulate production of reactive oxygen species by cytochrome P450. Toxicol. Appl. Pharmacol. 2004, 199, 316–331. [Google Scholar] [CrossRef] [PubMed]
- Matés, J.M. Effects of antioxidant enzymes in the molecular control of reactive oxygen species toxicology. Toxicology 2000, 153, 83–104. [Google Scholar] [CrossRef]
- Schafer, F.Q.; Buettner, G.R. Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free Radic. Biol. Med. 2001, 30, 1191–1212. [Google Scholar] [CrossRef]
- Griffith, O.W. Biologic and pharmacologic regulation of mammalian glutathione synthesis. In Proceedings of the Free Radical Biology and Medicine; Pergamon: Oxford, United Kingdom, 1999; Volume 27, pp. 922–935. [Google Scholar]
- Circu, M.L.; Aw, T.Y. Reactive oxygen species, cellular redox systems, and apoptosis. Free Radic. Biol. Med. 2010, 48, 749–762. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing. R Foundation forStatistical Computing, Vienna, Austria. 2018. Available online: https://www.R-project.org/ (accessed on 11 December 2019).
- Wickham, H. Reshaping data with the reshape package. J. Stat. Softw. 2007, 21, 1–20. [Google Scholar] [CrossRef]
- Pinheiro, J.; Bates, D.; DebRoy, S.; Sarkar, D. R Development Core Team. 2010. nlme: Linear and Nonlinear Mixed Effects Models. R Packag. version 3 2007, 186. Available online: https://www.R-project.org/ (accessed on 11 December 2019).
- Maisanaba, S.; Hercog, K.; Ortuño, N.; Jos, Á.; Žegura, B. Induction of micronuclei and alteration of gene expression by an organomodified clay in HepG2 cells. Chemosphere 2016, 154, 240–248. [Google Scholar] [CrossRef]
- Baebler, Š.; Svalina, M.; Petek, M.; Stare, K.; Rotter, A.; Pompe-Novak, M.; Gruden, K. QuantGenius: Implementation of a decision support system for qPCR-based gene quantification. BMC Bioinform. 2017, 18, 276. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Abbreviation | CAS N° | Chemical Structure | Formula | MW (g/mol) | |
|---|---|---|---|---|---|
| Cylindrospermopsin | CYN | 143545-90-8 |  | C15H21N5O7S | 415.4 |
| Bisphenol A | BPA | 080-05-7 |  | C15H16O2 | 228.29 |
| Bisphenol S | BPS | 080-09-1 |  | C12H10O4S | 250.27 |
| Bisphenol F | BPF | 620-92-8 |  | C13H12O2 | 200.23 |
| Bisphenol AF | BPAF | 1478-61-1 |  | C15H10F6O2 | 336.23 |
| Gene Symbol | Assay ID | Entrez Gene Name | Cellular Function |
|---|---|---|---|
| CYP1A1 | Hs01054797_g1 | Cytochrome P450, family 1, subfamily A, polypeptide 1 | Metabolism of xenobiotics, detoxification response |
| GCLC | Hs00155249_m1 | Glutamate-cysteine ligase, catalytic subunit | Oxidative stress response |
| GPX1 | Hs00829989_gH | Glutathione peroxidase 1 | Oxidative stress response |
| GSR | Hs00167317_m1 | Glutathione reductase | Oxidative stress response |
| SOD1A | Hs00533490_m1 | Superoxide dis-mutase 1 | Oxidative stress response |
| CAT | Hs00156308_m1 | Catalase | Oxidative stress response |
| CDKN1A | Hs00355782_m1 | Cyclin-dependent kinase inhibitor 1A (p21. Cip1) | DNA-damage response genes |
| GADD45A | Hs00169255_m1 | Growth arrest and DNA damage-inducible, alpha | DNA-damage response genes |
| MDM2 | Hs00234753_m1 | MDM2 proto-oncogen | DNA-damage response genes |
| TP53 | Hs01034249_m1 | Tumor protein P53 | DNA-damage response genes |
| CHEK1 | Hs00967506_m1 | Checkpoint kinase 1 | DNA-damage response genes |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hercog, K.; Štern, A.; Maisanaba, S.; Filipič, M.; Žegura, B. Plastics in Cyanobacterial Blooms—Genotoxic Effects of Binary Mixtures of Cylindrospermopsin and Bisphenols in HepG2 Cells. Toxins 2020, 12, 219. https://doi.org/10.3390/toxins12040219
Hercog K, Štern A, Maisanaba S, Filipič M, Žegura B. Plastics in Cyanobacterial Blooms—Genotoxic Effects of Binary Mixtures of Cylindrospermopsin and Bisphenols in HepG2 Cells. Toxins. 2020; 12(4):219. https://doi.org/10.3390/toxins12040219
Chicago/Turabian StyleHercog, Klara, Alja Štern, Sara Maisanaba, Metka Filipič, and Bojana Žegura. 2020. "Plastics in Cyanobacterial Blooms—Genotoxic Effects of Binary Mixtures of Cylindrospermopsin and Bisphenols in HepG2 Cells" Toxins 12, no. 4: 219. https://doi.org/10.3390/toxins12040219
APA StyleHercog, K., Štern, A., Maisanaba, S., Filipič, M., & Žegura, B. (2020). Plastics in Cyanobacterial Blooms—Genotoxic Effects of Binary Mixtures of Cylindrospermopsin and Bisphenols in HepG2 Cells. Toxins, 12(4), 219. https://doi.org/10.3390/toxins12040219