Role of Shiga Toxins in Cytotoxicity and Immunomodulatory Effects of Escherichia coli O157:H7 during Host-Bacterial Interactions in vitro

Abstract
1. Introduction
2. Results
2.1. Expression of Stx2 Subunits by 293T Cells
2.2. Survival of Human Cell Lines after Infection with EHEC
2.3. Survival of E. coli O157:H7 after Incubation with Human Cell Lines
2.4. Stx2 Activity in Supernatants from Infected Macrophages and HCT-8 Cells
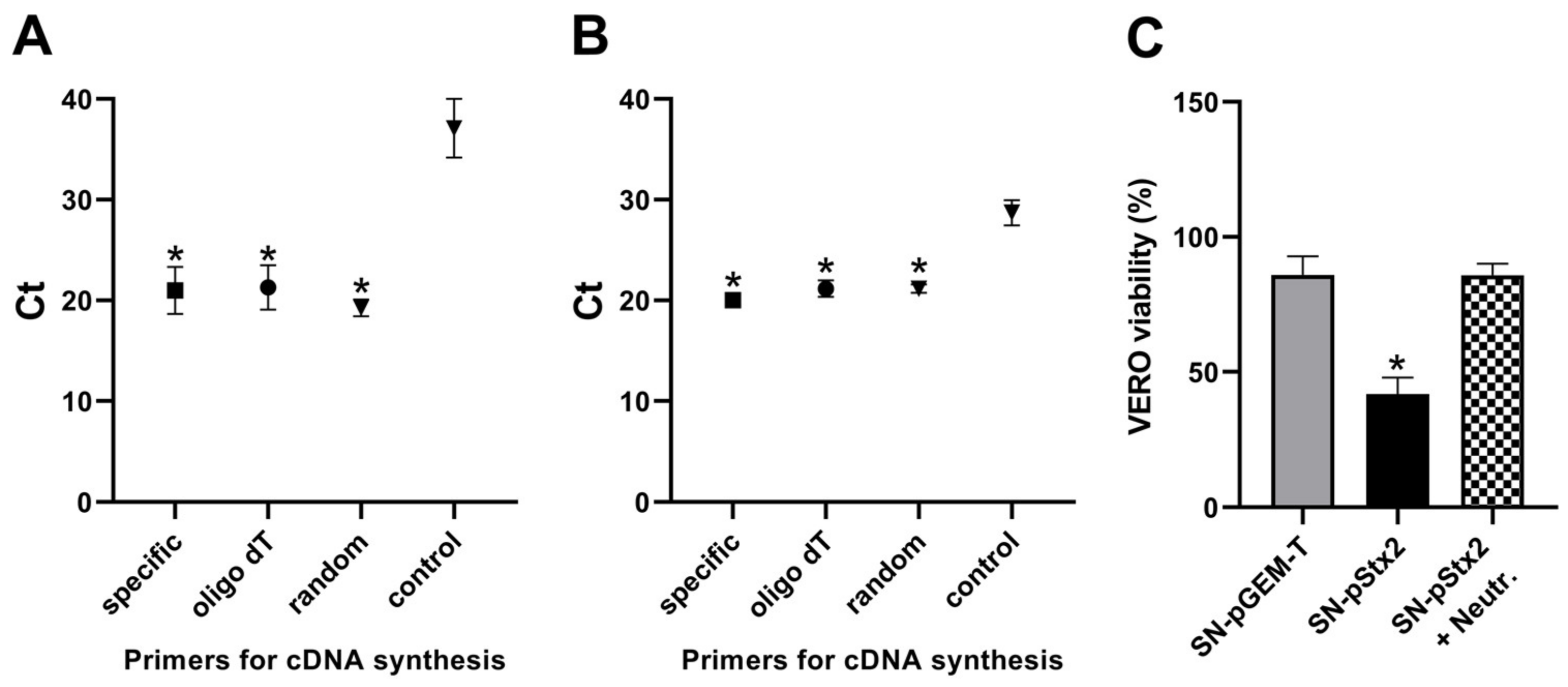
2.5. Analysis of Stx2-gene Expression
2.6. Mechanisms of Stx2-dependent Cytotoxicity on Infected Macrophages
2.7. Intereleukin-1 Betha (IL-1β) Secretion upon Macrophage Infection
2.8. Intereleukin-8 (IL-8) Secretion upon HCT-8 Infection
3. Discussion
4. Materials and Methods
4.1. Bacterial Strains
4.2. Cell Lines and Cell Culture
4.3. Infection Assay
4.4. Adhesion Assay
4.5. Stx2- cytotoxic Activity on Vero Cells
4.6. Cell Viability Assay
4.7. Cytokine Assay
4.8. Lactate Dehydrogenase (LDH) Assay
4.9. Plasmid Construction
4.10. Transfection Assay
4.11. Quantitative RT-qPCR
4.12. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Tarr, P.I.; Gordon, C.A.; Chandler, W.L. Shiga toxin-producing Escherichia coli and hemolytic uraemic syndrome. Lancet 2005, 365, 1073–1086. [Google Scholar] [CrossRef] [PubMed]
- Karmali, M.A. Infection by verocytotoxin-producing Escherichia coli. Clin. Microbiol. Rev. 1989, 2, 15–38. [Google Scholar] [CrossRef] [PubMed]
- Hunt, J.M. Shiga toxin-producing Escherichia coli (STEC). Clin. Lab. Med. 2010, 30, 21–45. [Google Scholar] [CrossRef] [PubMed]
- Davis, T.K.; Van De Kar, N.C.; Tarr, P.I. Shiga Toxin/Verocytotoxin-Producing Escherichia coli Infections: Practical Clinical Perspectives. Microbiol. Spectr. 2014, 2. [Google Scholar] [CrossRef]
- Rivas, M.; Chinen, I.; Miliwebsky, E.; Masana, M. Risk Factors for Shiga Toxin-Producing Escherichia coli-Associated Human Diseases. Micro. Biol. Spectr. 2014, 2, 381–402. [Google Scholar] [CrossRef]
- Phillips, A.D.; Navabpour, S.; Hicks, S.; Dougan, G.; Wallis, T.; Frankel, G. Enterohaemorrhagic Escherichia coli O157:H7 target Peyer’s patches in humans and cause attaching/effacing lesions in both human and bovine intestine. Gut 2000, 47, 377–381. [Google Scholar] [CrossRef]
- Bielaszewska, M.; Rüter, C.; Bauwens, A.; Greune, L.; Jarosch, K.A.; Steil, D.; Zhang, W.; He, X.; Lloubes, E.; Fruth, A.; et al. Host cell interactions of outer membrane vesicle-associated virulence factors of enterohemorrhagic Escherichia coli O157: Intracellular delivery, trafficking and mechanisms of cell injury. PLoS Pathog. 2017, 13, e1006159. [Google Scholar] [CrossRef]
- Kolling, G.L.; Matthews, K.R. Export of virulence genes and Shiga toxin by membrane vesicles of Escherichia coli O157:H7. Appl. Environ. Microbiol. 1999, 65, 1843–1848. [Google Scholar] [CrossRef]
- Kaper, J.B.; Nataro, J.P.; Mobley, H.L. Pathogenic Escherichia coli. Nat. Rev. Microbiol. 2004, 2, 123–140. [Google Scholar] [CrossRef]
- Herold, S.; Karch, K.; Schmidt, H. Shiga toxin-encoding bacteriophages—genomes in motion. Int. J. Med. Microbiol. 2004, 294, 115–121. [Google Scholar] [CrossRef]
- Wagner, P.L.; Neely, M.N.; Zhang, X.; Acheson, D.W.; Waldor, M.K.; Friedman, D.I. Role for a phage promoter in Shiga toxin 2 expression from a pathogenic Escherichia coli strain. J. Bacteriol. 2001, 183, 2081–2085. [Google Scholar] [CrossRef] [PubMed]
- Shaikh, N.; Tarr, P.I. Escherichia coli O157:H7 Shiga toxin-encoding bacteriophages: integrations, excisions, truncations, and evolutionary implications. J. Bacteriol. 2003, 185, 3596–3605. [Google Scholar] [CrossRef] [PubMed]
- Laing, C.R.; Zhang, Y.; Gilmour, M.W.; Allen, V.; Johnson, R.; Thomas, J.E.; Gannon, V.P. A comparison of Shiga-toxin 2 bacteriophage from classical enterohemorrhagic Escherichia coli serotypes and the German E. coli O104:H4 outbreak strain. Plos One 2012, 7, e37362. [Google Scholar] [CrossRef]
- Schmidt, H. Shiga-toxin-converting bacteriophages. Res. Microbiol. 2001, 152, 687–695. [Google Scholar] [CrossRef]
- Loś, J.M.; Loś, M.; Węgrzyn, G. Bacteriophages carrying Shiga toxin genes: genomic variations, detection and potential treatment of pathogenic bacteria. Future Microbiol. 2011, 6, 909–924. [Google Scholar] [CrossRef]
- Imamovic, L.; Jofre, J.; Schmidt, H.; Serra-Moreno, R.; Muniesa, M. Phage-Mediated Shiga Toxin 2 Gene Transfer in Food and Water. Appl. Environ. Microbiol. 2009, 75, 1764–1768. [Google Scholar] [CrossRef]
- Allison, H.E. Stx-phages: drivers and mediators of the evolution of STEC and STEC-like pathogens. Future Microbiol. 2007, 2, 165–174. [Google Scholar] [CrossRef]
- Bauwens, A.; Kunsmann, L.; Marejková, M.; Zhang, W.; Karch, H.; Bielaszewska, M.; Mellmann, A. Intrahost milieu modulates production of outer membrane vesicles, vesicle-associated Shiga toxin 2a and cytotoxicity in Escherichia coli O157:H7 and O104:H4. Environ. Microbiol. Rep. 2017, 9, 626–634. [Google Scholar] [CrossRef]
- Bentancor, L.V.; Bilen, M.F.; Mejías, M.P.; Fernández-Brando, R.J.; Panek, C.A.; Ramos, M.V.; Fernández, G.C.; Isturiz, M.; Ghiringhelli, P.D.; Palermo, M.S.; et al. Functional Capacity of Shiga-Toxin Promoter Sequences in Eukaryotic Cells. PLoS One 2013, 8, e57128. [Google Scholar] [CrossRef]
- Bentancor, L.V.; Mejías, M.P.; Pinto, A.; Bilen, M.F.; Meiss, R.; Rodriguez-Galán, M.C.; Baez, N.; Pedrotti, L.P.; Goldstein, J.; Ghiringhelli, P.D.; et al. Promoter sequence of Shiga toxin 2 (Stx2) is recognized in vivo, leading to production of biologically active Stx2. MBio. 2013, 4, e00501-13. [Google Scholar] [CrossRef]
- Poirier, K.; Faucher, S.P.; Béland, M.; Brousseau, R.; Gannon, V.; Martin, C.; Harel, J.; Daigle, F. Escherichia coli O157:H7 survives within human macrophages: Global gene expression profile and involvement of the Shiga toxins. Infect. Immunol. 2008, 76, 4814–4822. [Google Scholar] [CrossRef] [PubMed]
- Brandelli, J.R.; Griener, T.P.; Laing, A.; Mulvey, G.; Armstrong, G.D. The Effects of Shiga Toxin 1, 2 and Their Subunits on Cytokine and Chemokine Expression by Human Macrophage-Like THP-1 Cells. Toxins (Basel). 2015, 7, 4054–4066. [Google Scholar] [CrossRef] [PubMed]
- Van de Kar, N.C.; Monnens, L.A.; Karmali, M.A.; van Hinsbergh, V.W. Tumor necrosis factor and interleukin-1 induce expression of the verocytotoxin receptor globotriaosylceramide on human endothelial cells: implications for the pathogenesis of the hemolytic uremic syndrome. Blood 1992, 80, 2755–2764. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Rogers, T.J.; Paton, J.C.; Paton, A.W. Differential effects of Escherichia coli subtilase cytotoxin and Shiga toxin 2 on chemokine and proinflammatory cytokine expression in human macrophage, colonic epithelial, and brain microvascular endothelial cell lines. Infect. Immun. 2014, 82, 3567–3579. [Google Scholar] [CrossRef]
- Celli, J.; Olivier, M.; Finlay, B.B. Enteropathogenic Escherichia coli mediates antiphagocytosis through the inhibition of PI 3-kinase-dependent pathways. EMBO J. 2001, 20, 1245–1258. [Google Scholar] [CrossRef]
- Quitard, S.; Dean, P.; Maresca, M.; Kenny, B. The enteropathogenic Escherichiacoli EspF effector molecule inhibits PI-3 kinase-mediated uptake independently of mitochondrial targeting. Cell. Microbiol. 2006, 8, 972–981. [Google Scholar] [CrossRef][Green Version]
- Tahoun, A.; Siszler, G.; Spears, K.; McAteer, S.; Tree, J.; Paxton, E.; Gillespie, T.L.; Martinez-Argudo, I.; Jepson, M.A.; Shaw, D.J.; et al. Comparative analysis of EspF variants in inhibition of Escherichia coli phagocytosis by macrophages and inhibition of E. coli translocation through human- and bovine-derived M cells. Infect. Immun. 2011, 79, 4716–4729. [Google Scholar] [CrossRef]
- Iizumi, Y.; Sagara, H.; Kabe, Y.; Azuma, M.; Kume, K.; Ogawa, M.; Nagai, T.; Gillespie, P.G.; Sasakawa, C.; Handa, H.; et al. The enteropathogenic E. coli effector EspB facilitates microvillus effacing and antiphagocytosis by inhibiting myosin function. Cell Host Microbe 2007, 2, 383–392. [Google Scholar] [CrossRef]
- Marchès, O.; Covarelli, V.; Dahan, S.; Cougoule, C.; Bhatta, P.; Frankel, G.; Caron, E. EspJ of enteropathogenic and enterohaemorrhagic Escherichia coli inhibitsopsono-phagocytosis. Cell. Microbiol. 2008, 10, 1104–1115. [Google Scholar] [CrossRef]
- Dong, N.; Liu, L.; Shao, F. A bacterial effector targets host DH-PH domain RhoGEFs and antagonizes macrophage phagocytosis. EMBO J. 2010, 29, 1363–1376. [Google Scholar] [CrossRef]
- Etienne-Mesmin, L.; Chassaing, B.; Sauvanet, P.; Denizot, J.; Blanquet-Diot, S.; Darfeuille-Michaud, A.; Pradel, N.; Livrelli, V. Interactions with M cells and macrophages as key steps in the pathogenesis of enterohemorrhagic Escherichia coli infections. PLoS One 2011, 6, e23594. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, T.; Ohta, Y.; Tsutsuki, H.; Noda, M. Construction of a novel bioluminescent reporter system for investigating Shiga toxin expression of enterohemorrhagic Escherichia coli. Gene 2011, 478, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Falguières, T.; Mallard, F.; Baron, C.; Hanau, D.; Lingwood, C.; Goud, B.; Salamero, J.; Johannes, L. Targeting of Shiga toxin B-subunit to retrograde transport route in association with detergent-resistant membranes. Mol. Biol. Cell. 2001, 12, 2453–2468. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.S.; Cherla, R.P.; Jenson, M.H.; Leyva-Illades, D.; Martinez-Moczygemba, M.; Tesh, V.L. Shiga toxins induce autophagy leading to differential signalling pathways in toxin-sensitive and toxin-resistant human cells. Cell Microbiol. 2011, 13, 1479–1496. [Google Scholar] [CrossRef]
- Harrison, L.M.; van Haaften, W.C.E.; Tesh, V.L. Regulation of Proinflammatory Cytokine Expression by Shiga Toxin 1 and/or Lipopolysaccharides in the Human Monocytic Cell Line THP-1. Infect. Immun. 2004, 72, 2618–2627. [Google Scholar] [CrossRef]
- Levya-Illades, D.; Cherla, R.P.; Lee, M.S.; Tesh, V.L. Regulation of cytokine and chemokine expression by the ribotoxic stress response elicited by Shiga toxin type 1 in human macrophage-like THP-1 cells. Infect. Immun. 2012, 80, 2109–2120. [Google Scholar] [CrossRef]
- Mejías, M.P.; Hiriart, Y.; Lauché, C.; Fernández-Brando, R.J.; Pardo, R.; Bruballa, A.; Ramos, M.V.; Goldbaum, F.A.; Palermo, M.S.; Zylberman, V.; et al. Development of camelid single chain antibodies against Shiga toxin type 2 (Stx2) with therapeutic potential against Hemolytic Uremic Syndrome (HUS). Sci. Rep. 2016, 6, e24913. [Google Scholar] [CrossRef]
- Tesh, V.L. Induction of apoptosis by Shiga toxins. Future Microbiol. 2010, 5, 431–453. [Google Scholar] [CrossRef]
- Zhang, X.; Cheng, Y.; Xiong, Y.; Ye, C.; Zheng, H.; Sun, H.; Zhao, H.; Ren, Z.; Xu, J. Enterohemorrhagic Escherichia coli specific enterohemolysin induced IL-1β in human macrophages and EHEC-induced IL-1β required activation of NLRP3 inflammasome. PLoS One 2012, 7, e50288. [Google Scholar] [CrossRef]
- Evavold, C.L.; Ruan, J.; Tan, Y.; Xia, S.; Wu, H.; Kagan, J.C. The Pore-Forming Protein Gasdermin D Regulates Interleukin-1 Secretion from Living Macrophages. Immunity 2018, 48, 35–44.e6. [Google Scholar] [CrossRef]
- Lee, M.S.; Kwon, H.; Lee, E.Y.; Kim, D.J.; Park, J.H.; Tesh, V.L.; Oh, T.K.; Kim, M.H. Shiga toxins activate the NLRP3 inflammasome pathway to promote both production of the proinflammatory cytokine interleukin-1β and apoptotic cell death. Infect. Immun. 2015, 84, 172–186. [Google Scholar] [CrossRef]
- Platnich, J.M.; Chung, H.; Lau, A.; Sandall, C.F.; Bondzi-Simpson, A.; Chen, H.M.; Komada, T.; Trotman-Grant, A.C.; Brandelli, J.R.; Chun, J.; et al. Shiga Toxin/Lipopolysaccharide Activates Caspase-4 and Gasdermin D to Trigger Mitochondrial Reactive Oxygen Species Upstream of the NLRP3 Inflammasome. Cell Rep. 2018, 25, 1525–1536.e7. [Google Scholar] [CrossRef] [PubMed]
- Zumbrun, S.D.; Hanson, L.; Sinclair, J.F.; Freedy, J.; Melton-Celsa, A.R.; Rodriguez-Canales, J.; Hanson, J.C.; O’Brien, A.D. Human intestinal tissue and cultured colonic cells contain globotriaosylceramide synthase mRNA and the alternate Shiga toxin receptor globotetraosylceramide. Infect Immun. 2010, 78, 4488–4499. [Google Scholar] [CrossRef] [PubMed]
- Kouzel, I.U.; Pohlentz, G.; Schmitz, J.S.; Steil, D.; Humpf, H.U.; Karch, H.; Müthing, J. Shiga Toxin Glycosphingolipid Receptors in Human Caco-2 and HCT-8 Colon Epithelial Cell Lines. Toxins (Basel). 2017, 9, 338. [Google Scholar] [CrossRef] [PubMed]
- Karve, S.S.; Pradhan, S.; Ward, D.V.; Weiss, A.A. Intestinal organoids model human responses to infection by commensal and Shiga toxin producing Escherichia coli. PLoS One. 2017, 12, e0178966. [Google Scholar] [CrossRef]
- Bielaszewska, M.; Marejková, M.; Bauwens, A.; Kunsmann-Prokscha, L.; Mellmann, A.; Karch, H. Enterohemorrhagic Escherichia coli O157 outer membrane vesicles induce interleukin 8 production in human intestinal epithelial cells by signaling via Toll-like receptors TLR4 and TLR5 and activation of the nuclear factor NF-κB. Int. J. Med. Microbiol. 2018, 308, 882–889. [Google Scholar] [CrossRef]
- Thorpe, C.M.; Smith, W.E.; Hurley, B.P.; Acheson, D.W. Shiga toxins induce, superinduce, and stabilize a variety of C-X-C chemokine mRNAs in intestinal epithelial cells, resulting in increased chemokine expression. InfectImmun. 2001, 69, 6140–6147. [Google Scholar] [CrossRef]
- Kesty, N.C.; Mason, K.M.; Reedy, M.; Miller, S.E.; Kuehn, M.J. Enterotoxigenic Escherichia coli vesicles target toxin delivery into mammalian cells. EMBO J. 2004, 23, 4538–4549. [Google Scholar] [CrossRef]
- Ellis, T.N.; Kuehn, M.J. Virulence and immunomodulatory roles of bacterial outer membrane vesicles. Microbiol. Mol. Biol. Rev. 2010, 74, 81–94. [Google Scholar] [CrossRef]
- Amano, A.; Takeuchi, H.; Furuta, N. Outer membrane vesicles function as offensive weapons in host-parasite interactions. Microbes Infect. 2010, 12, 791–798. [Google Scholar] [CrossRef]
- Yokoyama, K.; Horii, T.; Yamashino, T.; Hashikawa, S.; Barua, S.; Hasegawa, T.; Watanabe, H.; Ohta, M. Production of Shiga toxin by Escherichia coli measured with reference to the membrane vesicle-associated toxins. FEMS Microbiol. Lett. 2000, 192, 139–144. [Google Scholar] [CrossRef] [PubMed]
- Rivas, M.; Miliwebsky, E.; Chinen, I.; Roldán, C.D.; Balbi, L.; García, B.; Fiorilli, G.; Sosa-Estani, S.; Kincaid, J.; Rangel, J.; et al. Case-Control Study Group. Characterization and epidemiologic subtyping of Shiga toxin-producing Escherichia coli strains isolated from hemolytic uremic syndrome and diarrhea cases in Argentina. Foodborne Pathog. Dis. 2006, 3, 88–96. [Google Scholar] [CrossRef] [PubMed]
- Brando, R.J.; Miliwebsky, E.; Bentancor, L.; Deza, N.; Baschkier, A.; Ramos, M.V.; Fernandez, G.C.; Meiss, R.; Rivas, M.; Palermo, M.S.; et al. Renal damage and death in weaned mice after oral infection with Shiga toxin 2-producing Escherichia coli strains. Clin. Exp. Immunol. 2008, 153, 297–306. [Google Scholar] [CrossRef] [PubMed]
- Amigo, N.; Mercado, E.; Bentancor, A.; Singh, P.; Vilte, D.; Gerhardt, E.; Zotta, E.; Ibarra, C.; Manning, S.D.; Larzábal, M.; et al. Clade 8 and Clade 6 Strains of Escherichia coli O157:H7 from Cattle in Argentina have Hypervirulent-Like Phenotypes. PLoS One 2015, 10, e0127710. [Google Scholar] [CrossRef] [PubMed]
- Albanese, A.; Gerhardt, E.; García, H.; Amigo, N.; Cataldi, A.; Zotta, E.; Ibarra, C. Inhibition of water absorption and selective damage to human colonic mucosa induced by Shiga toxin-2 are enhanced by Escherichia coli O157:H7 infection. Int. J. Med. Microbiol. 2015, 305, 348–354. [Google Scholar] [CrossRef] [PubMed]
- Del Cogliano, M.E.; Pinto, A.; Goldstein, J.; Zotta, E.; Ochoa, F.; Fernández-Brando, R.J.; Muniesa, M.; Ghiringhelli, P.D.; Palermo, M.S.; Bentancor, L.V.; et al. Relevance of Bacteriophage 933W in the Development of Hemolytic Uremic Syndrome (HUS). Front. Microbiol. 2018, 9, e3104. [Google Scholar] [CrossRef]
- Karmali, M.A.; Petric, M.; Lim, C.; Cheung, R.; Arbus, G.S. Sensitive method for detecting low numbers of verotoxin-producing Escherichia coli in mixed cultures by use of colony sweeps and polymyxin extraction of verotoxin. J. Clin. Microbiol. 1985, 22, 614–619. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J. Immunol. Methods. 1983, 65, 55–63. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sequence | Forward (5′–3′) | Reverse (5′–3′) |
|---|---|---|
| stx2-A subunit | TGGCGTTAATGGAGTTCAGTGG | ACACAGGAGCAGTTTCAGACAG |
| stx2-B subunit | AGTCGCTGGAATCTGCAACCGTTAC | TCAGCAAATCCGGAGCCTGATTCAC |
| SDHA | AAGTCCCTCCAATTAAACCAAACG | GTCTTCAGGTGCTTTAGGTCTCC |
| Sequence | [Mg2+](mM) | PCR Program |
|---|---|---|
| stx2-A subunit | 2.5 | 95 °C × 2 min, 40 cycles of: [95 °C × 15 sec, 60 °C × 20 sec, 68 °C × 20 sec] |
| stx2-B subunit | 2.5 | 95 °C × 2 min, 40 cycles of: [95 °C × 15 sec, 60 °C × 20 sec, 68 °C × 20 sec] |
| SDHA | 4 | 95 °C × 2 min, 40 cycles of: [95 °C × 15 sec, 60 °C × 20 sec, 68 °C × 20 sec] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bruballa, A.C.; Shiromizu, C.M.; Bernal, A.M.; Pineda, G.E.; Sabbione, F.; Trevani, A.S.; Bentancor, L.V.; Ramos, M.V.; Fernández-Brando, R.J.; Muñoz, M.J.; et al. Role of Shiga Toxins in Cytotoxicity and Immunomodulatory Effects of Escherichia coli O157:H7 during Host-Bacterial Interactions in vitro. Toxins 2020, 12, 48. https://doi.org/10.3390/toxins12010048
Bruballa AC, Shiromizu CM, Bernal AM, Pineda GE, Sabbione F, Trevani AS, Bentancor LV, Ramos MV, Fernández-Brando RJ, Muñoz MJ, et al. Role of Shiga Toxins in Cytotoxicity and Immunomodulatory Effects of Escherichia coli O157:H7 during Host-Bacterial Interactions in vitro. Toxins. 2020; 12(1):48. https://doi.org/10.3390/toxins12010048
Chicago/Turabian StyleBruballa, Andrea Cecilia, Carolina Maiumi Shiromizu, Alan Mauro Bernal, Gonzalo Ezequiel Pineda, Florencia Sabbione, Analia Silvina Trevani, Leticia Verónica Bentancor, María Victoria Ramos, Romina Jimena Fernández-Brando, Manuel Javier Muñoz, and et al. 2020. "Role of Shiga Toxins in Cytotoxicity and Immunomodulatory Effects of Escherichia coli O157:H7 during Host-Bacterial Interactions in vitro" Toxins 12, no. 1: 48. https://doi.org/10.3390/toxins12010048
APA StyleBruballa, A. C., Shiromizu, C. M., Bernal, A. M., Pineda, G. E., Sabbione, F., Trevani, A. S., Bentancor, L. V., Ramos, M. V., Fernández-Brando, R. J., Muñoz, M. J., & Palermo, M. S. (2020). Role of Shiga Toxins in Cytotoxicity and Immunomodulatory Effects of Escherichia coli O157:H7 during Host-Bacterial Interactions in vitro. Toxins, 12(1), 48. https://doi.org/10.3390/toxins12010048