Evaluation of High-Resolution Mass Spectrometry for the Quantitative Analysis of Mycotoxins in Complex Feed Matrices


 , ,
, ,  ,
,
Abstract

1. Introduction
2. Results and Discussion
2.1. LC Optimization
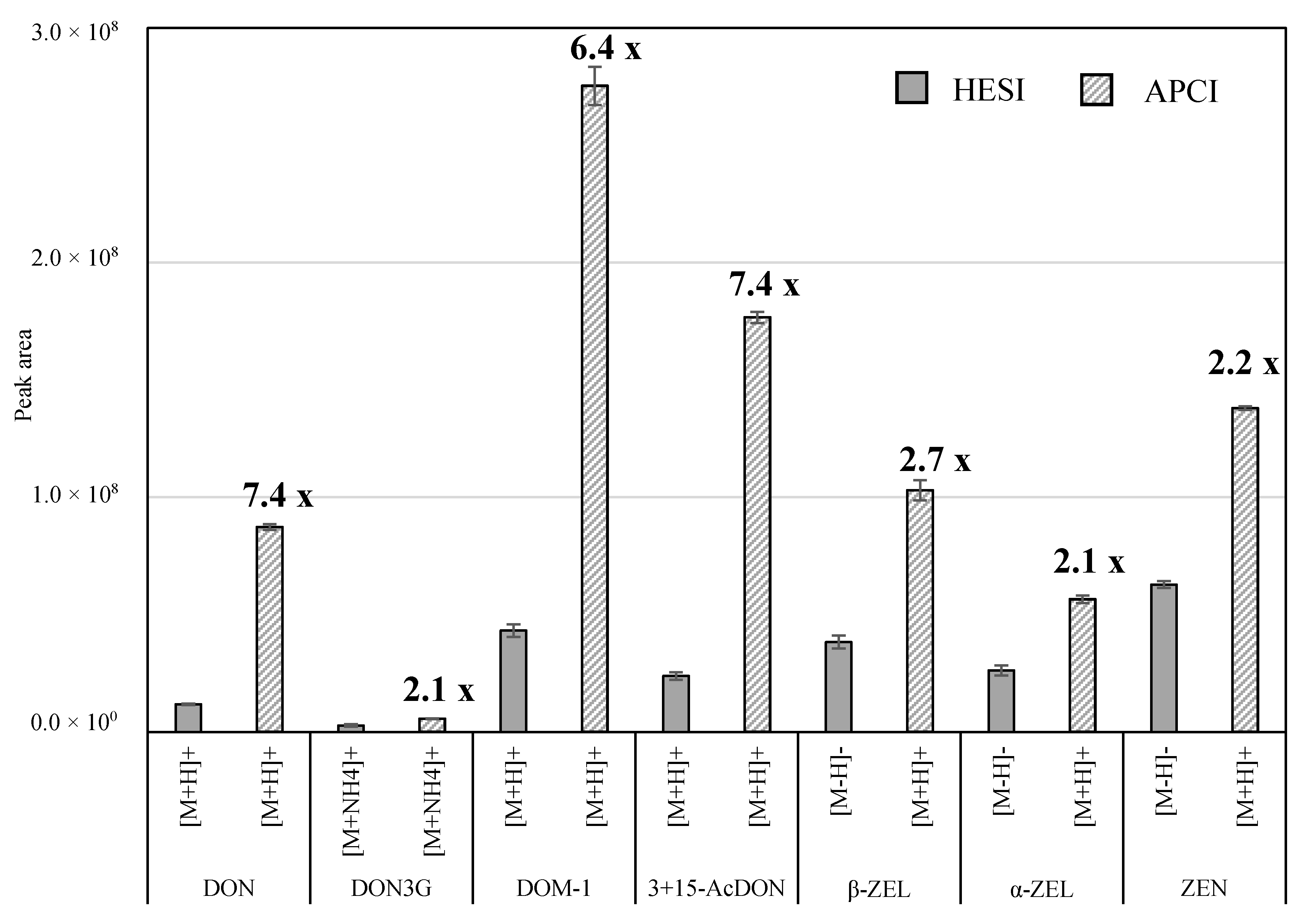
2.2. Selection of Ionization Mode
2.3. Resolving Power Requirements
2.4. Validation of the HRMS Method
2.5. Application of the HRMS Method to Real Samples
3. Conclusions
4. Materials and Methods
4.1. Chemicals
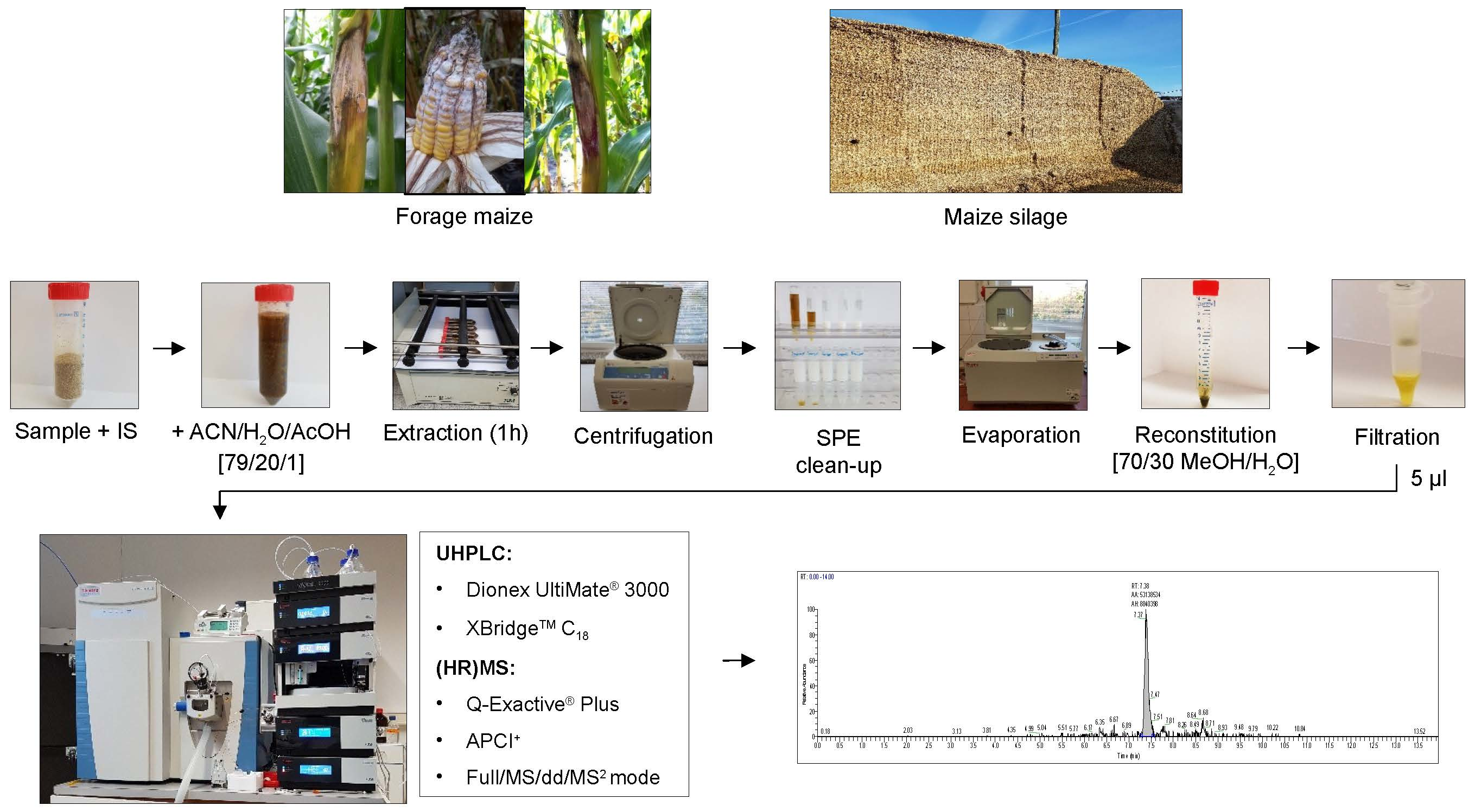
4.2. Sample Preparation
4.3. Samples
4.4. LC-HRMS
4.5. Method Validation
4.6. Data Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Righetti, L.; Paglia, G.; Galaverna, G.; Dall’Asta, C. Recent advances and future challenges in modified mycotoxin analysis: Why HRMS has become a key instrument in food contaminant research. Toxins 2016, 8, 361. [Google Scholar] [CrossRef] [PubMed]
- Streit, E.; Schwab, C.; Sulyok, M.; Naehrer, K.; Krska, R.; Schatzmayr, G. Multi-mycotoxin screening reveals the occurrence of 139 different secondary metabolites in feed and feed ingredients. Toxins 2013, 5, 504–523. [Google Scholar] [CrossRef]
- Alshannaq, A.; Yu, J.-H. Occurrence, toxicity, and analysis of major mycotoxins in food. Int. J. Environ. Res. Public Health 2017, 14, 632. [Google Scholar] [CrossRef]
- Van Egmond, H.P.; Schothorst, R.C.; Jonker, M.A. Regulations relating to mycotoxins in food: Perspectives in a global and European context. Anal. Bioanal. Chem. 2007, 389, 147–157. [Google Scholar] [CrossRef] [PubMed]
- Zachariasova, M.; Cajka, T.; Godula, M.; Malachova, A.; Veprikova, Z.; Hajslova, J. Analysis of multiple mycotoxins in beer employing (ultra)-high-resolution mass spectrometry. Rapid Commun. Mass Spectrom. 2010, 24, 3357–3367. [Google Scholar] [CrossRef]
- Lehner, S.M.; Neumann, N.K.N.; Sulyok, M.; Lemmens, M.; Krska, R.; Schuhmacher, R. Evaluation of LC-high-resolution FT-Orbitrap MS for the quantification of selected mycotoxins and the simultaneous screening of fungal metabolites in food. Food Addit. Contam. Part A 2011, 28, 1457–1468. [Google Scholar] [CrossRef]
- Gómez-Ramos, M.M.; Ferrer, C.; Malato, O.; Agüera, A.; Fernández-Alba, A.R. Liquid chromatography-high-resolution mass spectrometry for pesticide residue analysis in fruit and vegetables: Screening and quantitative studies. J. Chromatogr. A 2013, 1287, 24–37. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Pérez, M.L.; Romero-González, R.; Martínez Vidal, J.L.; Garrido Frenich, A. Analysis of veterinary drug and pesticide residues in animal feed by high-resolution mass spectrometry: Comparison between time-of-flight and Orbitrap. Food Addit. Contam. Part A 2015, 32, 1637–1646. [Google Scholar] [CrossRef]
- Kumar, P.; Rúbies, A.; Centrich, F.; Granados, M.; Cortés-Francisco, N.; Caixach, J.; Companyó, R. Targeted analysis with benchtop quadrupole-orbitrap hybrid mass spectrometer: Application to determination of synthetic hormones in animal urine. Anal. Chim. Acta 2013, 780, 65–73. [Google Scholar] [CrossRef]
- Wang, J.; Chow, W.; Chang, J.; Wong, J.W. Ultrahigh-performance liquid chromatography electrospray ionization Q-Orbitrap mass spectrometry for the analysis of 451 pesticide residues in fruits and vegetables: Method development and validation. J. Agric. Food Chem. 2014, 62, 10375–10391. [Google Scholar] [CrossRef]
- Lattanzio, V.M.T.; Della Gatta, S.; Godula, M.; Visconti, A. Quantitative analysis of mycotoxins in cereal foods by collision cell fragmentation-high-resolution mass spectrometry: Performance and comparison with triple-stage quadrupole detection. Food Addit. Contam. Part A 2011, 28, 1424–1437. [Google Scholar] [CrossRef] [PubMed]
- Rubert, J.; Dzuman, Z.; Vaclavikova, M.; Zachariasova, M.; Soler, C.; Hajslova, J. Analysis of mycotoxins in barley using ultra high liquid chromatography high resolution mass spectrometry: Comparison of efficiency and efficacy of different extraction procedures. Talanta 2012, 99, 712–719. [Google Scholar] [CrossRef] [PubMed]
- Jia, W.; Chu, X.; Ling, Y.; Huang, J.; Chang, J. Multi-mycotoxin analysis in dairy products by liquid chromatography coupled to quadrupole orbitrap mass spectrometry. J. Chromatogr. A 2014, 1345, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Dzuman, Z.; Zachariasova, M.; Veprikova, Z.; Godula, M.; Hajslova, J. Multi-analyte high performance liquid chromatography coupled to high resolution tandem mass spectrometry method for control of pesticide residues, mycotoxins, and pyrrolizidine alkaloids. Anal. Chim. Acta 2015, 863, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Liao, C.-D.; Wong, J.W.; Zhang, K.; Hayward, D.G.; Lee, N.S.; Trucksess, M.W. Multi-mycotoxin analysis of finished grain and nut products using high-performance liquid chromatography-triple-quadrupole mass spectrometry. J. Agric. Food Chem. 2015, 61, 4771–4782. [Google Scholar] [CrossRef] [PubMed]
- Tamura, M.; Mochizuki, N.; Nagatomi, Y.; Harayama, K.; Toriba, A.; Hayakawa, K. A method for simultaneous determination of 20 Fusarium toxins in cereals by high-resolution liquid chromatography-Orbitrap mass spectrometry with a pentafluorophenyl column. Toxins 2015, 7, 1664–1682. [Google Scholar] [CrossRef] [PubMed]
- Kellmann, M.; Muenster, H.; Zomer, P.; Mol, H. Full scan MS in comprehensive qualitative and quantitative residue analysis in food and feed matrices: How much resolving power is required? J. Am. Soc. Mass Spectrom. Chem. 2009, 20, 1464–1476. [Google Scholar] [CrossRef]
- Dzuman, Z.; Zachariasova, M.; Lacina, O.; Veprikova, Z.; Slavikova, P.; Hajslova, J. A rugged high-throughput analytical approach for the determination and quantification of multiple mycotoxins in complex feed matrices. Talanta 2014, 121, 263–272. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, R.R.; Storm, I.M.L.D.; Rasmussen, P.H.; Smedsgaard, J.; Nielsen, K.F. Multi-mycotoxin analysis of maize silage by LC-MS/MS. Anal. Bioanal. Chem. 2010, 397, 765–776. [Google Scholar] [CrossRef]
- Driehius, F. Silage and the safety and quality of dairy food: A review. Agric. Food Sci. 2013, 22, 16–34. [Google Scholar] [CrossRef]
- Weinberg, Z. New trends and opportunities in the development and use of inoculants for silage. FEMS Microbiol. Rev. 1996, 19, 53–68. [Google Scholar] [CrossRef]
- De Boevre, M.; Landschoot, S.; Audenaert, K.; Maene, P.; Di Mavungu, D.; Eeckhout, M.; Haesaert, G.; De Saeger, S. Occurrence and within field variability of Fusarium mycotoxins and their masked forms in maize crops in Belgium. World Mycotoxin J. 2014, 7, 91–102. [Google Scholar] [CrossRef]
- Malysheva, S.V.; Di Diana Mavungu, J.; Boonen, J.; de Spiegeleer, B.; Goryacheva, I.Y.; Vanhaecke, L.; De Saeger, S. Improved positive electrospray ionization of patulin by adduct formation: Usefulness in liquid chromatography-tandem mass spectrometry multi-mycotoxin analysis. J. Chromatogr. A 2012, 1270, 334–339. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Biselli, S.; Hummert, C. Development of a multicomponent method for Fusarium toxins using LC-MS/MS and its application during a survey for the content of T-2 toxin and deoxynivalenol in various feed and food samples. Food Addit. Contam. 2005, 22, 752–760. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, A.; Butcher, P.; Maden, K.; Walker, S.; Widmer, M. Comprehensive comparison of liquid chromatography selectivity as provided by two types of liquid chromatography detectors (high resolution mass spectrometry and tandem mass spectrometry): “where is the crossover point?”. Anal. Chim. Acta 2010, 673, 60–72. [Google Scholar] [CrossRef] [PubMed]
- Mol, H.G.J.; Tienstra, M.; Zomer, P. Evaluation of gas chromatography—electron ionization—full scan high resolution Orbitrap mass spectrometry for pesticide residue analysis. Anal. Chim. Acta 2016, 935, 161–172. [Google Scholar] [CrossRef] [PubMed]
- European Commission. Guidance Document on Identification of Mycotoxins in Food and Feed (SANTE/12089/2016), Implemented by 01/01/2017. Available online: https://ec.europa.eu/food/sites/food/files/safety/docs/cs_contaminants_sampling_guid-doc-ident-mycotoxins.pdf (accessed on 5 April 2018).
- Rooke, J.A.; Hatfield, R.D. Biochemistry of Ensiling; Silage Science and Technology, Agronomy Monograph 42; American Society of Agronomy, Inc., Crop Science Society of America, Inc., Soil Science Society of America, Inc.: Madison, WI, USA, 2003. [Google Scholar]
- Najdekr, L.; Friedecký, D.; Tautenhahn, R.; Pluskal, T.; Wang, J.; Huang, Y.; Adam, T. Influence of mass resolving power in orbital ion-trap mass spectrometry-based metabolomics. Anal. Chem. 2016, 88, 11429–11435. [Google Scholar] [CrossRef] [PubMed]
- European Commission. Commission decision (2002/ 657/EC) of 12 August 2002, Implementing council directive 96/23/EC concerning the performance of analytical methods and the interpretation of results. Off. J. Eur. Comm. 2002, L221, 8–36. [Google Scholar]
- De Saeger, S. (Ed.) Determining Mycotoxins and Mycotoxigenic Fungi in Food and Feed; Woodhead Publishing: Oxford, UK, 2011. [Google Scholar]
- De Boevre, M.; Di Mavungu, J.D.; Maene, P.; Audenaert, K.; Deforce, D.; Haesaert, G.; Eeckhout, M.; Callebaut, A.; Berthiller, F.; van Peteghem, C.; et al. Development and validation of an LC-MS/MS method for the simultaneous determination of deoxynivalenol, zearalenone, T-2-toxin and some masked metabolites in different cereals and cereal-derived food. Food Addit. Contam. Part A 2012, 29, 819–835. [Google Scholar] [CrossRef] [PubMed]
- Njumbe Ediage, E.; van Poucke, C.; De Saeger, S. A multi-analyte LC-MS/MS method for the analysis of 23 mycotoxins in different sorghum varieties: The forgotten sample matrix. Food Chem. 2015, 177, 397–404. [Google Scholar] [CrossRef]
- European Commission. Commission Regulation (EC) No. 401/2006 of 23 February 2006 laying down the methods of sampling and analysis for the official control of the levels of mycotoxins in foodstuffs. Off. J. Eur. Union 2006, 70, 12–34. [Google Scholar]
- European Commission. Commission Recommendation (2006/576/EC) of 17 August 2006 on the presence of deoxynivalenol, zearalenone, ochratoxin A, T-2 and HT- 2 and fumonisins in products intended for animal feeding. Off. J. Eur. Union 2006, L229, 7–9. [Google Scholar]
- Van Pamel, E.; Verbeken, A.; Vlaemynck, G.; de Boever, J.; Daeseleire, E. Ultrahigh-performance liquid chromatographic-tandem mass spectrometric multimycotoxin method for quantitating 26 mycotoxins in maize silage. J. Agric. Food Chem. 2011, 59, 9747–9755. [Google Scholar] [CrossRef] [PubMed]
- Dagnac, T.; Latorre, A.; Fernández Lorenzo, B.; Llompart, M. Validation and application of a liquid chromatography-tandem mass spectrometry based method for the assessment of the co-occurrence of mycotoxins in maize silages from dairy farms in NW Spain. Food Addit. Contam. Part A 2016, 33, 1850–1863. [Google Scholar] [CrossRef] [PubMed]
- Sulyok, M.; Berthiller, F.; Krska, R.; Schuhmacher, R. Development and validation of a liquid chromatography/tandem mass spectrometric method for the determination of 39 mycotoxins in wheat and maize. Rapid Commun. Mass Spectrom. 2006, 20, 2649–2659. [Google Scholar] [CrossRef] [PubMed]
- Schenzel, J.; Forrer, H.-R.; Vogelgsang, S.; Bucheli, T.D. Development, validation and application of a multi-mycotoxin method for the analysis of whole wheat plants. Mycotoxin Res. 2012, 28, 135–147. [Google Scholar] [CrossRef] [PubMed]
- Monbaliu, S.; van Poucke, C.; Detavernier, C.L.; Dumoulin, F.; van de Velde, M.; Schoeters, E.; van Dyck, S.; Averkieva, O.; van Peteghem, C.; De Saeger, S. Occurrence of mycotoxins in feed as analyzed by a multi-mycotoxin LC-MS/MS method. J. Agric. Food Chem. 2010, 58, 66–71. [Google Scholar] [CrossRef]
- ISO 21748. Guidance for the Use of Repeatability, Reproducibility and Trueness Estimates in Measurement Uncertainty Estimation; International Organization for Standardization: Geneva, 2017. [Google Scholar]



{kind=link}
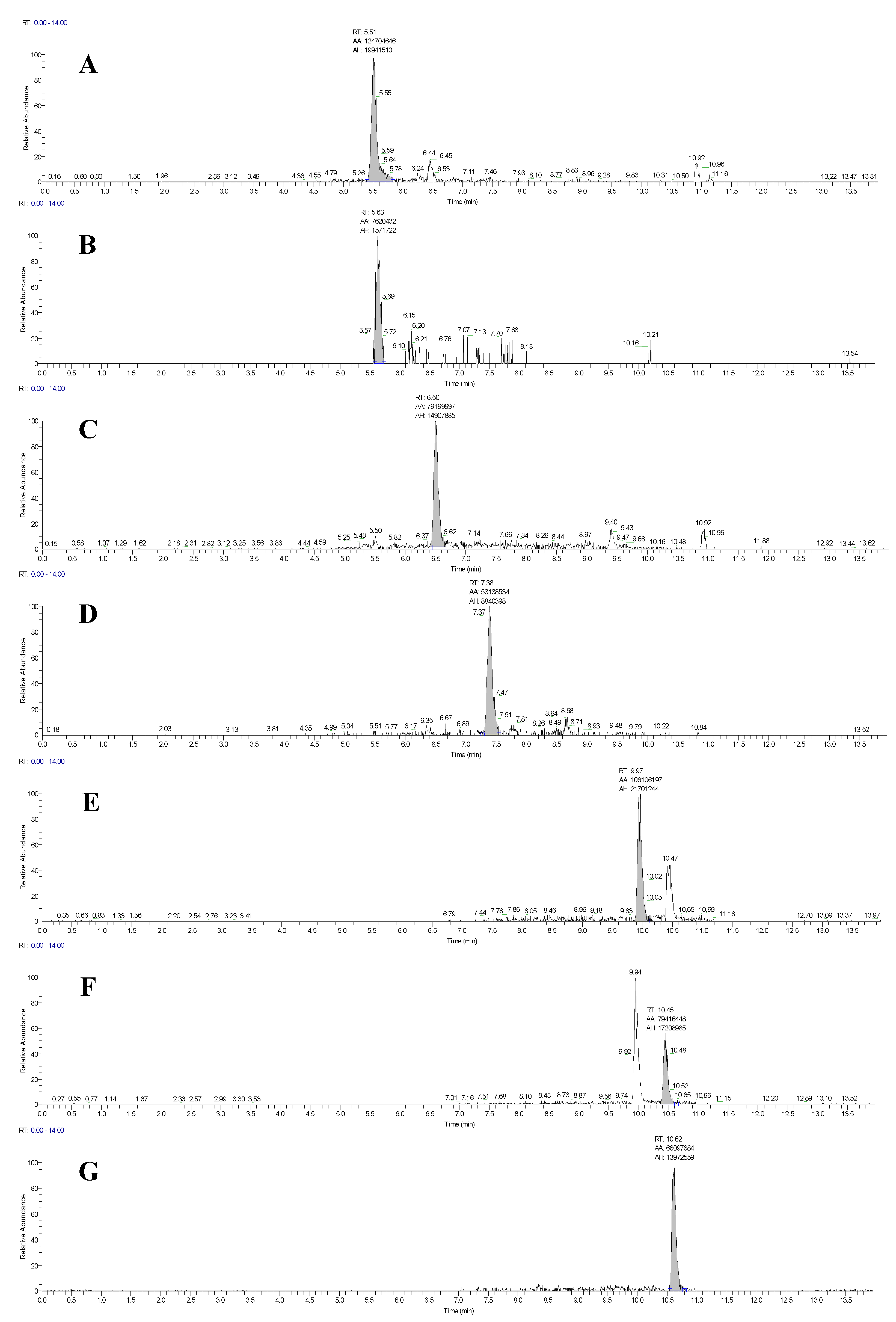{kind=link}
{kind=link}
| Concentration Level (µg/kg) | Resolving Power | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 17.500 FWHM a | 35.000 FWHM a | 70.000 FWHM a | 140.000 FWHM a | |||||||||||||
| <2 ppm | 2–5 ppm | 5–10 ppm | >10 ppm | <2 ppm | 2–5 ppm | 5–10 ppm | >10 ppm | <2 ppm | 2–5 ppm | 5–10 ppm | >10 ppm | <2 ppm | 2–5 ppm | 5–10 ppm | >10 ppm | |
| Forage maize | ||||||||||||||||
| 100 | 3 | 2 | 2 | 2 | 3 | 6 | 0 | 0 | 9 | 0 | 0 | 0 | 9 | 0 | 0 | 0 |
| 200 | 2 | 3 | 3 | 1 | 6 | 3 | 0 | 0 | 9 | 0 | 0 | 0 | 9 | 0 | 0 | 0 |
| 300 | 6 | 1 | 2 | 0 | 9 | 0 | 0 | 0 | 9 | 0 | 0 | 0 | 9 | 0 | 0 | 0 |
| Maize silage | ||||||||||||||||
| 100 | 1 | 2 | 3 | 3 | 3 | 0 | 5 | 1 | 9 | 0 | 0 | 0 | 9 | 0 | 0 | 0 |
| 200 | 1 | 5 | 0 | 3 | 5 | 2 | 1 | 0 | 9 | 0 | 0 | 0 | 9 | 0 | 0 | 0 |
| 300 | 3 | 3 | 2 | 1 | 8 | 1 | 0 | 0 | 9 | 0 | 0 | 0 | 9 | 0 | 0 | 0 |
| Mycotoxin | Spiked Concentration (µg/kg) | Forage Maize | Maize Silage | ||||||
|---|---|---|---|---|---|---|---|---|---|
| RA (%) | RSDr (%) | RSDR (%) | U (%) | RA (%) | RSDr (%) | RSDR (%) | U (%) | ||
| DON | 200 | 98 | 11 | 11 | 24 | 98 | 13 | 2 | 26 |
| 400 * | 106 | 7 | 5 | 15 | 101 | 6 | 7 | 20 | |
| 800 | 102 | 6 | 5 | 11 | 101 | 5 | 2 | 10 | |
| DON3G | 200 | 103 | 15 | 10 | 36 | 103 | 16 | 8 | 42 |
| 400 * | 106 | 11 | 9 | 29 | 94 | 11 | 2 | 24 | |
| 800 | 104 | 9 | 11 | 24 | 96 | 14 | 5 | 22 | |
| DOM-1 | 50 | 96 | 10 | 3 | 23 | 99 | 12 | 3 | 15 |
| 100 * | 108 | 7 | 4 | 17 | 95 | 5 | 4 | 13 | |
| 200 | 99 | 4 | 2 | 8 | 101 | 3 | 2 | 7 | |
| 3+15-AcDON | 50 | 101 | 9 | 8 | 13 | 100 | 6 | 3 | 13 |
| 100 * | 104 | 6 | 5 | 16 | 98 | 5 | 3 | 12 | |
| 200 | 97 | 5 | 4 | 13 | 101 | 3 | 2 | 7 | |
| β-ZEL | 200 | 105 | 11 | 8 | 22 | 96 | 9 | 6 | 21 |
| 400 * | 106 | 5 | 6 | 13 | 96 | 3 | 3 | 8 | |
| 800 | 100 | 6 | 3 | 12 | 99 | 2 | 3 | 7 | |
| α-ZEL | 250 | 95 | 8 | 12 | 31 | 104 | 16 | 7 | 41 |
| 500 * | 104 | 9 | 7 | 22 | 103 | 5 | 7 | 18 | |
| 1000 | 98 | 2 | 3 | 8 | 102 | 4 | 2 | 8 | |
| ZEN | 100 | 97 | 11 | 8 | 25 | 100 | 7 | 6 | 18 |
| 200 * | 105 | 8 | 6 | 20 | 98 | 6 | 3 | 14 | |
| 400 | 99 | 3 | 3 | 8 | 100 | 5 | 2 | 10 | |
| Mycotoxin | Range (µg/kg) | Forage Maize | Maize Silage | ||||
|---|---|---|---|---|---|---|---|
| R2 (mean) | CCα (µg/kg) | CCβ (µg/kg) | R2 (mean) | CCα (µg/kg) | CCβ (µg/kg) | ||
| DON | 200–800 | 0.9736 | 75 | 141 | 0.9865 | 47 | 82 |
| DON3G | 200–800 | 0.9768 | 17 | 29 | 0.9845 | 63 | 94 |
| DOM-1 | 50–200 | 0.9809 | 16 | 31 | 0.9790 | 15 | 31 |
| 3+15-AcDON | 50–200 | 0.9764 | 18 | 28 | 0.9839 | 11 | 20 |
| β-ZEL | 200–800 | 0.9676 | 73 | 108 | 0.9864 | 46 | 90 |
| α-ZEL | 250–1000 | 0.9814 | 16 | 26 | 0.9684 | 88 | 125 |
| ZEN | 100–400 | 0.9765 | 36 | 60 | 0.9740 | 40 | 61 |
| DON | DON3G | DOM-1 | 3+15-AcDON | β-ZEL | α-ZEL | ZEN | |
|---|---|---|---|---|---|---|---|
| Forage maize (n = 21) | |||||||
| Incidence (%) | 100 | 100 | 0 | 100 | 81 | 95 | 100 |
| Mean (µg/kg) a | 2794 | 574 | n.a. | 609 | 149 | 110 | 991 |
| Min (µg/kg) a | 466 | 119 | n.d. | 29 | 135 | 28 | 66 |
| Max (µg/kg) a | 10972 | 1240 | n.d. | 1832 | 163 | 423 | 1725 |
| CCβ | 141 | 29 | 31 | 28 | 60 | 26 | 108 |
| Maize Silage (n = 27) | |||||||
| Incidence (%) | 100 | 22 | 0 | 100 | 85 | 89 | 97 |
| Mean (µg/kg) a | 2051 | n.a. | n.a. | 50 | - | 221 | 527 |
| Min (µg/kg) a | 265 | <CCβ | n.d. | 21 | <CCβ | 178 | 63 |
| Max (µg/kg) a | 5401 | <CCβ | n.d. | 149 | <CCβ | 339 | 1596 |
| CCβ | 82 | 94 | 31 | 20 | 61 | 125 | 90 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jensen, T.; de Boevre, M.; Preußke, N.; de Saeger, S.; Birr, T.; Verreet, J.-A.; Sönnichsen, F.D. Evaluation of High-Resolution Mass Spectrometry for the Quantitative Analysis of Mycotoxins in Complex Feed Matrices. Toxins 2019, 11, 531. https://doi.org/10.3390/toxins11090531
Jensen T, de Boevre M, Preußke N, de Saeger S, Birr T, Verreet J-A, Sönnichsen FD. Evaluation of High-Resolution Mass Spectrometry for the Quantitative Analysis of Mycotoxins in Complex Feed Matrices. Toxins. 2019; 11(9):531. https://doi.org/10.3390/toxins11090531
Chicago/Turabian StyleJensen, Tolke, Marthe de Boevre, Nils Preußke, Sarah de Saeger, Tim Birr, Joseph-Alexander Verreet, and Frank D. Sönnichsen. 2019. "Evaluation of High-Resolution Mass Spectrometry for the Quantitative Analysis of Mycotoxins in Complex Feed Matrices" Toxins 11, no. 9: 531. https://doi.org/10.3390/toxins11090531
APA StyleJensen, T., de Boevre, M., Preußke, N., de Saeger, S., Birr, T., Verreet, J.-A., & Sönnichsen, F. D. (2019). Evaluation of High-Resolution Mass Spectrometry for the Quantitative Analysis of Mycotoxins in Complex Feed Matrices. Toxins, 11(9), 531. https://doi.org/10.3390/toxins11090531